

Abstract
Pulmonary involvement in amyloidosis is a distinct rarity. This clinical entity usually presents as tracheobronchial amyloidosis (TBA). A 32-year-old, never-smoker man presented with episodic dyspnoea and wheezing along with cough and mucoid sputum. The chest radiograph was suggestive of a middle lobe syndrome (MLS). High-resolution CT (HRCT) of the chest confirmed the presence of MLS. In addition, HRCT showed circumferential thickening of the trachea and the main bronchi, with thickening of the posterior membranous wall of trachea. Fibrebronchoscopy, done to evaluate MLS, visualised multiple small polypoidal lesions in the lower part of trachea and carina. Endobronchial biopsies showed homogeneous, acellular amorphous deposit in the subepithelial region, which was congophilic in nature. A diagnosis of TBA presenting as MLS was made. To the best of our knowledge, this is the first detailed report of MLS as a presentation of TBA in the English literature.
Keywords: Asthma, Radiology
Background
Amyloidosis is characterised by extracellular deposition of abnormal insoluble fibrillar protein in organs and tissues.1 It can be classified as primary or secondary, local or systemic, depending on their distribution. The term ‘secondary’ is used to describe patients with an underlying disorder while ‘primary’ refers to patients with no coexistent disease. In patients with ‘systemic’ amyloidosis, the abnormal protein deposition occurs in multiple anatomic sites while ‘localised’ disease involves a single anatomic site.2
Lesser,3 in 1877, first described amyloidosis of the lower respiratory tract in an autopsy specimen. Pulmonary involvement in amyloid manifests most commonly as tracheobronchial disease, the diagnosis of which is based on characteristic fibreoptic (FOB) findings and confirmed by histopathology.2
Graham et al4 in 1948 described middle lobe syndrome (MLS) as chronic or recurrent collapse of the right middle lobe. Recurrent collapse of the right middle lobe due to enlarged tuberculous lymph nodes was first described by Brock et al5 and is commonly known as ‘Brock’s syndrome’. Several respiratory diseases are known to cause MLS, a distinct but uncommon radiological presentation that still continues to fascinate clinicians.6–9
MLS as a presentation of tracheobronchial amyloidosis (TBA) was documented only once before in the German literature.10 The paucity of the literature on the subject prompted this report of a young man with history of asthma who presented with MLS. FOB done to evaluate MLS revealed a diagnosis of TBA.
Case presentation
A 32-year-old HIV-negative man, a never smoker, was referred to our institute for evaluation of breathlessness, wheezing, cough and mucoid sputum of 1-year duration. His clinical course was characterised by episodic dyspnoea and wheezing. A fortnight prior to presentation, he experienced streaky haemoptysis. A chest X-ray suggested MLS, which prompted the referral. There was no fever, chest pain, palpitations or pain in small joints. The patient had a respiratory count of 18 per minute and was afebrile on presentation. There was no cyanosis, clubbing or pallor. Diaphragmatic excursion was comparable on both sides. Vesicular breath sounds of equal intensity with prolonged expiration were audible bilaterally.
Investigations
Oxygen saturation at room air was 97% and the total leucocyte count was 15 400 cells/mm3 with neutrophils constituting 92%. The ECG, echocardiogram, urine analyses, blood sugar levels and renal and hepatic functions were within normal limits. On presentation, the chest radiograph was suggestive of loss of cardiac silhouette due to an ill-defined opacity in the right middle zone adjoining the cardiac border (figure 1A). A wedge-shaped density was visible on the right lateral view. The shadow extended from the hilum anteriorly and inferiorly along with loss of volume, which was suggestive of MLS (figure 1B). Contrast-enhanced high-resolution CT (HRCT) of the thorax (figure 2A: mediastinal window) revealed circumferential thickening of the trachea and the main bronchi along with thickening of the posterior membranous wall of trachea. In addition, HRCT confirmed MLS with the presence of a trapezoidal opacity with its base towards the hilum and approximating the right cardiac border (figure 2B: lung window). Sputum stains and cultures for Mycobacterium tuberculosis, fungi and other aerobic organisms were negative. On spirometry, the forced vital capacity (FVC) was 2.73 L (53% of predicted), forced expiratory volume in 1 s (FEV1) was 2.1 L (49% of predicted) and the FEV1/FVC ratio was 0.77 with no significant bronchodilator reversibility. This was indicative of severe restriction. Complete pulmonary function testing revealed a residual volume (RV) of 1.07 L (65% of predicted), total lung capacity (TLC) of 3.84 L (56% of predicted) and RV/TLC of 28% (116% of predicted). Diffusion capacity for carbon monoxide was 26.02 (77% of predicted). This was suggestive of severe restriction with impaired diffusion capacity. On 6 min walk test, he covered a distance of 340 m without desaturation. On pulmonary function testing, there was no evidence of asthma. Antinuclear antibody, cytoplasmic antineutrophil cytoplasmic antibodies and perinuclear antineutrophil cytoplasmic antibodies were not detected. In view of the MLS, the patient’s consent was taken for FOB, which visualised multiple small polypoidal lesions in the lower part of trachea and carina projecting from the mucosa into the lumen (figure 3). The mucosa, which bled on touch, was pale, oedematous and highly vascular. Stains and cultures of the bronchial aspirate were negative for M. tuberculosis and other aerobic organisms as well as pathogenic fungi. The aspirate was also negative for GeneXpert. Endobronchial biopsies from these lesions showed homogeneous, acellular amorphous deposit in the subepithelial region, which was congophilic in nature (figure 4A). Stains for amyloid (Congo red, thioflavin T and methyl violet) were positive (figure 4B,C). Immunohistochemistry (IH) stain was weakly positive for amyloid. Biopsy and IH were suggestive of amyloidosis. No evidence of granulomatous or neoplastic pathology was seen. There were no Bence Jones proteins in urine and the serum protein electrophoresis result was normal. The patient did not consent for a rectal biopsy.
Figure 1.
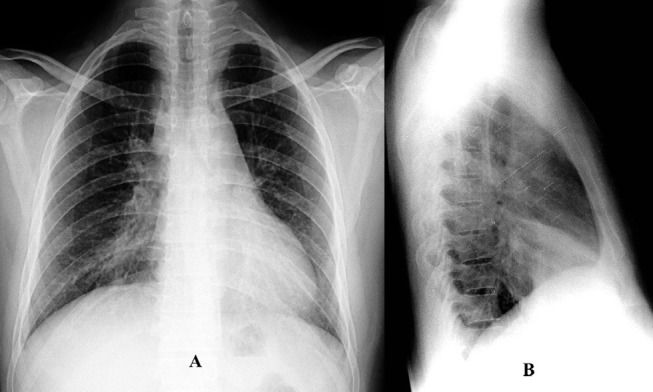
(A) Chest X-ray posteroanterior view showing ill-defined opacity in the right mid and lower zones adjoining the right cardiac border with loss of cardiac silhouette, suggestive of right middle lobe syndrome (MLS). (B) Chest X-ray lateral view showing wedge-shaped density extending from the hilum anteriorly and inferiorly along with loss of volume, suggestive of MLS.
Figure 2.
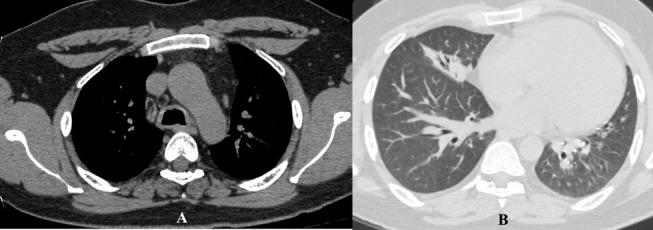
(A) High-resolution CT of the thorax (mediastinal window) showing circumferential thickening of the trachea along with thickening of the posterior membranous wall of trachea. (B) High-resolution CT of the thorax (lung window) showing a trapezoidal opacity with its base towards the hilum and contiguous with the right cardiac border, confirming the presence of middle lobe syndrome.
Figure 3.
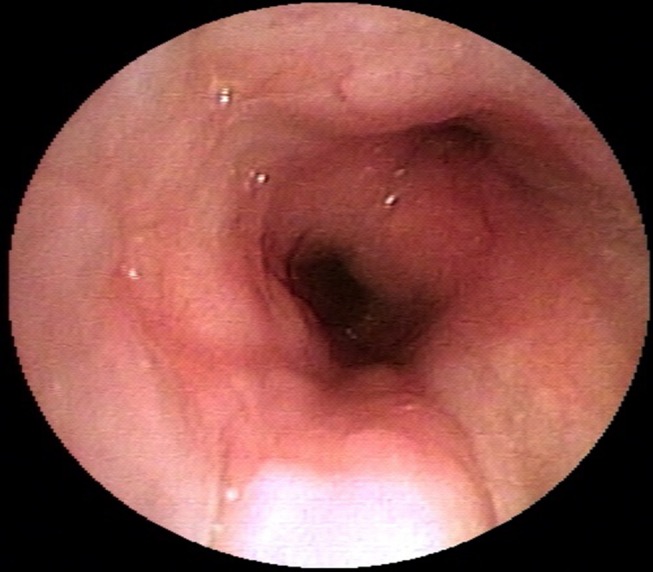
Fibreoptic bronchoscopic image showing multiple small polypoidal lesions in the lower part of trachea and carina projecting from the mucosa into the lumen.
Figure 4.

(A) Medium-power view (10×) of the patient’s biopsy specimen on H&E stain showing respiratory lining epithelium and the subepithelial stroma demonstrating homogeneous, eosinophilic, acellular material (white arrows). (B) Medium-power view (10×) of the patient’s biopsy specimen on Congo red stain showing orangophilia in the subepithelial deposits (black arrows). (C) Medium-power view (10×) of the patient’s biopsy specimen on Congo red stain under polaroid microscope shows apple green birefringence (white arrows).
Diagnosis
A diagnosis of TBA with MLS was made. This was based on (1) circumferential thickening of the trachea and main bronchi on HRCT; (2) FOB finding of polypoidal lesions in the lower part of the trachea, which was highly vascular; (3) histopathological finding of homogeneous, acellular amorphous deposit in the subepithelial region, which stained positive with Congo red stain; and (4) characteristic imaging presentation suggestive of right MLS.
Treatment
The patient was apprised of the treatment options. The patient refused to undergo bronchoscopic intervention. He was then initiated on symptomatic treatment in the form of combination of inhaled bronchodilators and steroids along with short bursts of oral prednisolone.
Outcome and follow-up
Within a fortnight, the patient experienced clinical improvement with control of symptoms. The patient came regularly for follow-up for a period of 6 months. He continued to be largely symptom-free during the follow-up period. However, radiologically there was no resolution of the MLS and was then lost to follow-up.
Discussion
In amyloidosis, pulmonary involvement can be a part of systemic disease or localised only to the lungs. The lungs are affected in nearly half the patients with amyloidosis,2 with primary pulmonary amyloidosis being the most common presentation. This can be further subclassified into five different types: tracheobronchial, nodular (solitary or multiple), senile pulmonary, mediastinal-hilar and diffuse interstitial.2 TBA is the most common form and is characterised by amyloid deposits as submucosal plaques and/or polypoid tumours in the trachea and large bronchi.11 This deposition of amyloid material in the airways can be localised, diffuse or multifocal. The three different patterns of airway involvement in TBA are (1) proximal, (2) mid-bronchial or main bronchial and (3) distal disease.12 Patients with proximal/upper tracheal disease usually present with features of upper airway obstruction. In patients with mid-tracheal or distal tracheal and main bronchial disease, lobar collapse or recurrent parenchymal infections are the presenting symptoms, while in those with distal disease (sparing the trachea) repeated pneumonia, cough and bronchiectasis are the the most common presentation.13 TBA is a slowly progressive disease usually seen after fifth decade with uncharacteristic symptoms such as exertional dyspnoea, wheezing, cough and haemoptysis. This non-specific presentation leads to a diagnostic delay and the disease is usually treated as asthma, recurrent pneumonia or tracheobronchitis.11 Our patient had TBA with distal tracheal involvement, which mimicked bronchial asthma and presented as MLS.
In patients with TBA, a normal chest radiograph is seen in nearly half of the patients,12 while other findings include lobar/segmental collapse, bronchiectasis and hilar lymphadenopathy. CT features are suggestive of nodules, endoluminal irregularities, circumferential thickening of the airways and tracheal stenosis and calcification.14 15 In addition, parenchymal abnormalities such as bronchiectasis and lobar collapse may be evident especially in distal disease.15 HRCT helps in defining the extent and degree of luminal narrowing and planning therapy.14 The HRCT in our patient also showed circumferential thickening of the airway wall, endoluminal irregularities and an MLS.
The patterns of amyloid deposition on FOB include (1) nodular disease and (2) diffuse submucosal disease. The lesions of TBA can be sessile, nodular or polypoidal in nature.12 The sessile lesions appear as hard yellow plaques and may cause significant airway stenosis especially when diffuse. The nodular or polypoidal lesions may appear as endoluminal malignant lesions.16 These submucosal nodules are firm and non-mobile with an overlying hypervascular mucosa. As a result, these lesions are highly friable and bleed on contact with the bronchoscope.12 The diagnosis is usually based on histopathological evaluation of bronchoscopic biopsy specimen. On light microscopy, amyloid appears as a homogeneous, acellular eosinophilic material and shows a characteristic apple green birefringence under polarised light when stained with Congo red.11 12 Histopathological evaluations in TBA shows submucosal amyloid deposits leading to the formation of irregular nodules or diffuse lamina covered with bronchial epithelium.12
A search of the literature on the subject using the PubMed, IndMED and other databases found mentions of a single case of TBA as a cause of MLS in the German literature.10 In addition, there is also a mention of a patient with nodular amyloidosis who presented with MLS. The presence of a tumorous amyloid deposit at the opening of the middle lobe caused the occurrence of MLS in this patient.17 MLS is generally caused by endobronchial tumours, lymphadenopathies, foreign body or granulomatous infection and benign inflammatory causes.18 19 The infrequent causes of MLS include allergic bronchopulmonary aspergillosis,6 8 bronchial anthracofibrosis,20 endobronchial tuberculosis7 and pulmonary hydatid.9
Imaging is the initial diagnostic modality for MLS, and it presents as an ill-defined opacity adjacent to the right cardiac border causing a loss of cardiac silhouette. The lateral view shows a characteristic wedge-shaped opacity extending anterior and inferior to the hilum. On CT, MLS appears as a trapezoidal opacity with its base towards the hilum and contiguous with the right cardiac border,18 as was seen in our patient.
TBA can be confused with tracheobronchopathia osteochondroplastica (TBO), which was our first impression during FOB. Other conditions, which would resemble TBA, include diffuse tracheal diseases like relapsing polychondritis (RP) and granulomatosis with polyangiitis. Tuberculosis of the tracheobronchial tree can also be considered. The posterior tracheal and bronchial membranes are characteristically spared in TBO and RP.11
To date, there has been no effective medical therapy for TBA, with therapeutic bronchoscopic procedures using neodymium:yttrium-aluminium-garnet (Nd:YAG) laser being the only effective intervention.21 22 External beam radiation therapy is another promising mode of treatment.23 Colchicine, oral glucocorticoids and melphalan have been tried without much success.11 12
Pulmonary amyloidosis is a distinct clinical entity and mimics asthma in its clinical presentation, leading to a delay in the diagnosis. Its presentation as MLS on imaging is a distinct rarity.
Learning points.
Tracheobronchial amyloidosis, a rare clinical entity, often mimics the presentation of bronchial asthma, leading to a diagnostic confusion and delay.
An array of respiratory disorders is known to cause middle lobe syndrome (MLS). The characteristic radiological presentation of MLS still continues to mesmerise the clinicians.
The diagnosis of tracheobronchial amyloidosis is based on characteristic bronchoscopic finding along with histopathological evidence of amyloid material when stained with Congo red, which is confirmatory.
Footnotes
Contributors: SK, AK and AS collected the clinical data and reviewed the literature. SD reviewed the pathological aspects. SK, SD, AK and AS drafted the manuscript. AS worked on the concept and is responsible for the genuineness of the data and is also the guarantor of the paper. All the authors have read and approved the final manuscript.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid 2010;17:101–4. 10.3109/13506129.2010.526812 [DOI] [PubMed] [Google Scholar]
- 2.Utz JP, Swensen SJ, Gertz MA. Pulmonary amyloidosis. the Mayo Clinic experience from 1980 to 1993. Ann Intern Med 1996;124:407–13. 10.7326/0003-4819-124-4-199602150-00004 [DOI] [PubMed] [Google Scholar]
- 3.Lesser A. Ein fall von enchondroma osteoides mixtum der Lunge mit partieller Amyloidentartung. Archiv für Pathologische Anatomie und Physiologie und für Klinische Medicin 1877;69:404–9. 10.1007/BF02326214 [DOI] [Google Scholar]
- 4.Graham EA, Burford TH, Mayer JH. Middle Lobe syndrome. Postgrad Med 1948;4:29–34. 10.1080/00325481.1948.11693655 [DOI] [PubMed] [Google Scholar]
- 5.Brock RC, Cann RJ, Dickinson JR. Tuberculous mediastinal lymphadenitis in childhood; secondary effects on the lungs. Guy’s Hospital Resp 1937;87:295. [Google Scholar]
- 6.Shah A, Behera S, Panjabi C. Middle lobe syndrome: a rare presentation of allergic bronchopulmonary aspergillosis. Eur Ann Allergy Clin Immunol 2014;46:147–51. [PubMed] [Google Scholar]
- 7.Garg T, Gera K, Shah A. Middle lobe syndrome: an extraordinary presentation of endobronchial tuberculosis. Pneumonol Alergol Pol 2015;83:387–91. 10.5603/PiAP.2015.0062 [DOI] [PubMed] [Google Scholar]
- 8.Shah A, Gera K, Panjabi C. Childhood allergic bronchopulmonary aspergillosis presenting as a middle lobe syndrome. Asia Pac Allergy 2016;6:67–9. 10.5415/apallergy.2016.6.1.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kunal S, Pilaniya V, Shah A. Middle lobe syndrome: a singularly rare presentation of complicated pulmonary hydatid disease. BMJ Case Rep 2016. 10.1136/bcr-2016-214670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rehbock T, Andresen R, Rehbock B, et al. [Tracheobronchial amyloidosis as the cause of a middle lobe syndrome]. Rontgenpraxis 1999;52:206–9. [Article in German]. [PubMed] [Google Scholar]
- 11.de Almeida RR, Zanetti G, Pereira E Silva JL, et al. Respiratory Tract Amyloidosis. State-of-the-Art Review with a focus on pulmonary involvement. Lung 2015;193:875–83. 10.1007/s00408-015-9791-x [DOI] [PubMed] [Google Scholar]
- 12.Berk JL, O’Regan A, Skinner M. Pulmonary and tracheobronchial amyloidosis. Semin Respir Crit Care Med 2002;23:155–66. 10.1055/s-2002-25304 [DOI] [PubMed] [Google Scholar]
- 13.O’Regan A, Fenlon HM, Beamis JF, et al. Tracheobronchial amyloidosis. the Boston University experience from 1984 to 1999. Medicine 2000;79:69–79. 10.1097/00005792-200003000-00001 [DOI] [PubMed] [Google Scholar]
- 14.Renapurkar RD, Kanne JP. Metabolic and storage lung diseases: spectrum of imaging appearances. Insights Imaging 2013;4:773–85. 10.1007/s13244-013-0289-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Czeyda-Pommersheim F, Hwang M, Chen SS, et al. Amyloidosis: modern Cross-sectional Imaging. Radiographics 2015;35:1381–92. 10.1148/rg.2015140179 [DOI] [PubMed] [Google Scholar]
- 16.Aggarwal AN, Gupta D, Joshi K, et al. Tracheobronchial amyloidosis: a report of two cases. Indian J Chest Dis Allied Sci 2000;42:115–8. [PubMed] [Google Scholar]
- 17.Kronschnabel EF, Landis FB. Primary amyloid tumor: cause of Middle Lobe syndrome. Archives of Otolaryngology - Head and Neck Surgery 1962;76:233–8. 10.1001/archotol.1962.00740050241008 [DOI] [Google Scholar]
- 18.Gudbjartsson T, Gudmundsson G. Middle lobe syndrome: a review of clinicopathological features, diagnosis and treatment. Respiration 2012;84:80–6. 10.1159/000336238 [DOI] [PubMed] [Google Scholar]
- 19.Wagner RB, Johnston MR. Middle Lobe syndrome. Ann Thorac Surg 1983;35:679–86. 10.1016/S0003-4975(10)61085-5 [DOI] [PubMed] [Google Scholar]
- 20.Kala J, Sahay S, Shah A. Bronchial anthracofibrosis and tuberculosis presenting as a middle lobe syndrome. Prim Care Respir J 2008;17:51–5. 10.3132/pcrj.2008.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madden BP, Lee M, Paruchuru P. Successful treatment of endobronchial amyloidosis using nd:yag laser therapy as an alternative to lobectomy. Monaldi Arch Chest Dis 2001;56:27–9. [PubMed] [Google Scholar]
- 22.Berraondo J, Novella L, Sanz F, et al. Management of tracheobronchial amyloidosis with therapeutic bronchoscopic techniques. Arch Bronconeumol 2013;49:207–9. 10.1016/j.arbres.2012.08.002 [DOI] [PubMed] [Google Scholar]
- 23.Monroe AT, Walia R, Zlotecki RA, et al. Tracheobronchial amyloidosis: a case report of successful treatment with external beam radiation therapy. Chest 2004;125:784–9. [DOI] [PubMed] [Google Scholar]