

Abstract
Nerve-resident immune cells in the peripheral nervous system (PNS) are essential to maintaining neuronal integrity in a healthy nerve. The immune cells of the PNS are affected by injury and disease, affecting the nerve function and the capacity for regeneration. Neuronal immune cells are commonly analyzed by immunofluorescence (IF). While IF is essential for determining the location of the immune cells in the nerve, IF is only semi-quantitative and the method is limited to the number of markers that can be analyzed simultaneously and the degree of surface expression. In this study, flow cytometry was used for quantitative analysis of leukocyte infiltration into sciatic nerves or dorsal root ganglions (DRGs) of individual mice. Single cell analysis was performed using DAPI and several proteins were analyzed simultaneously for either surface or intracellular expression. Both sciatic nerves from one mouse that were treated according to this protocol generated ≥ 30,000 single nucleated events. The proportion of leukocytes in the sciatic nerves, determined by expression of CD45, was approximately 5% of total cell content in the sciatic nerve and approximately 5-10% in the DRG. Although this protocol focuses primarily on the immune cell population within the PNS, the flexibility of flow cytometry to measure a number of markers simultaneously means that the other cells populations present within the nerve, such as Schwann cells, pericytes, fibroblasts, and endothelial cells, can also be analyzed using this method. This method therefore provides a new means for studying systemic effects on the PNS, such as neurotoxicology and genetic models of neuropathy or in chronic diseases, such as diabetes.
Keywords: Neuroscience, Issue 130, Flow cytometry, mouse, sciatic nerve, macrophage, inflammation, leukocytes, neuropathy
Introduction
Immune cells which enter the PNS from the circulation, as defined by the expression of CD45 and CD11b, help to maintain the integrity of the nerve and play a role both in regeneration and degeneration1. Macrophages (defined by their expression of CD68 in mouse) can be skewed towards an inflammatory phenotype, expressing more MHC class II and CD86 on their surface (M1), or towards an anti-inflammatory phenotype, expressing more intracellular CD206 (M2)2. Skewing of the macrophage phenotype is a dynamic process regulated through Akt signaling3, reflecting the different tasks of macrophages in the defense against pathogens (M1) and the role in tissue regeneration (M2). Regeneration of an injured nerve first requires phagocytosis of myelin debris by macrophages in the nerve4,5, and anti-inflammatory (CD68+CD206+) M2 macrophages have been shown to promote axon outgrowth in the PNS6. Reduced recruitment or macrophages to the PNS, or impaired capacity for phagocytosis may result in impaired regeneration and maintenance of nerve integrity. Inflammatory M1 macrophages, expressing MHC class II, are less capable of phagocytosis than M2 macrophages, and neuronal inflammation is implicated in the pathogenesis of several neurodegenerative diseases7.
The changes that occur to the nerve resident immune system as a consequence of damage may be quantitative, manifesting in loss of CD45+ leukocytes (or increased infiltration of CD45+ leukocytes in the case inflammation), or qualitative, such as change of macrophage phenotype from M2 to M1 phenotype. The immune cells of the PNS have traditionally been analyzed by means of IF, using frozen sections or paraffin-embedded material8. IF is required for determining localization of the cells of interest. However, quantification using IF often relies on counting a relatively small number of cells in a narrow section of the tissue, making quantification unreliable and vulnerable to selection bias. For identification of specific subsets of immune cells, the simultaneous detection of extracellular and/or intracellular markers is required, whilst determination of the macrophage phenotype requires at least at least two markers, specifically CD206 and MHC class II. As most commonly available microscopes are limited to at least two-color channels, such as fluorescein isothiocyanate (FITC) and phycoerythrin (PE), the characterization of the specific subsets of immune cells by IF can be restrictive and incomplete, requiring the need to have multiple slides, derived from the same area of interest, which are stained and analyzed in parallel. This time-consuming aspect therefore does not necessary lend itself to the analysis of large sample sets. Furthermore, as most of the markers of interest are extracellular, the detection in tissue, which has either been embedded in paraffin or cryoconserved, can be problematic due to the disruption of membrane integrity and the masking of epitopes, as well as the loss of the antigens of interest from the use of solvents, such as acetone and methanol9.
In contrast, flow cytometry, which measures optical and fluorescence characteristics of single cells in suspension as they pass through a beam of light, provides a more practical and comprehensive means for analysis of the cell populations. Flow cytometry, rather than producing a digital image of the tissue, provides an automated quantification of set parameters, which include a cell's relative size and reflective index, referred to as the forward scatter (FSC), granularity/internal complexity or side-scatter (SSC), and relative fluorescence intensity, providing that the cell has been labeled with an appropriate fluorophore, such as a conjugated antibody. A typical flow cytometer consists of two, air-cooled lasers; an argon laser produces blue light at 488 nm and a helium-neon laser produces light at 633 nm. This combination allows for the detection and measurement, simultaneously, of at least five targets either on the surface or within the intracellular compartment. More advanced flow cytometers can consist of multiple lasers, which allow for the detection of up to eight different fluorochromes at once, providing that the peak emission wavelengths of the selected fluorochromes do not overlap significantly.
For analyzing by flow cytometry, the tissue of interest must first be enzymatically digested, generally with collagenase, to generate a single-cell suspension. The analysis of murine sciatic nerves has previously been difficult due to the small amount of tissue obtained from each mouse. In addition, the high fat content of the myelin around the axons hampers cell recovery and produces large amounts of debris. The method described herein for sciatic nerve preparation and digestion was adapted from the Schwann cell isolation protocol of Barrette et al.7, and aims to isolate enough cells from nerves of individual mice for flow cytometry analysis, in order to reduce variation between mice. Using DAPI for the identification of single cells in the raw tissue digest circumvents the need to remove axon debris, which commonly leads to cell loss. Washing several times with a detergent-rich buffer aids in the release of cells trapped within the fatty debris, thereby increasing the yield. Digestion of both full-length sciatic nerves from a single mouse according to this protocol generates ≥30,000 single nucleated events and at least 3 times that number was retrieved from the DRG. The proportion of CD45+ leukocytes was approximately 5% of total cell content in the sciatic nerve digest and approximately5-10% in the DRG digest. The majority of the CD45+ cells in the sciatic nerve expressed the macrophage markers, CD68 and CD206.
Protocol
Wild-type C57BL/6 mice (Males; 10-12 weeks) were kept in standard 12 h light/dark cycle and were provided free access to standard chow diet and water. All animal experiments were conducted in accordance to the relevant guidelines by the local Animal Care and Use Committee and approved by the local Animal Care and Use Committee at the regional authority in Karlsruhe, Germany (G216/10).
1. Perfusion of the Mouse
Sacrifice the mouse (C57BL/6; Males; 10-12weeks) using CO2 or in accordance with local regulations. NOTE: Retro-orbital bleeding can be performed to obtain control leukocytes, using EDTA-coated 1.5 mL tubes. This method of bleeding is only efficient immediately after the sacrifice. Further, peritoneal cavity lavage (PCL) can be performed to obtain control macrophages, using pre-heated PBS at 37 °C to avoid cold agglutination of the blood, which may hamper perfusion.
Disinfect the mouse by spraying the fur on the abdomen liberally with 70% v/v ethanol and repeatedly wiping with a tissue with cotton gauze. Using blunt scissors, cut open the abdominal skin by a longitudinal incision.
Expose the pleural cavity by first making an incision into the diaphragm using curved, blunt scissors and then extending the incision along the entire length of the rib cage. Make a similar cut on the contralateral side until the sternum can be carefully lifted away.
With a clear view of the heart and the major vessels, insert a butterfly 23¾ gauge needle, connected to a 50 mL syringe filled with ice-cold PBS, into the left chamber of the heart.
Make an incision to the right atrium of the heart using iris scissors to create as large of an outlet as possible without damaging the descending aorta, and then gently depress the syringe to push through approximately30 mL of PBS. The exudate should be running clear. The clearing of the liver and/or kidneys can be used as an indicator of a good perfusion.
2. Dissection of Sciatic Nerve and DRG
NOTE: For the dissection of the sciatic nerves, the skin above the gluteus maximus is removed and the mouse is positioned ventral side down whilst stretching out the lower limbs.
Bilaterally dissect the whole sciatic nerves by separating the muscle tissue from the upper dorsal thigh, and follow the nerve up along the spinal column. Cut the nerve where it exits from the spinal column and immediately above the knee at the other end. NOTE: Avoid excessive handling of the nerve such as crushing or pulling with forceps.
Using forceps, transfer the excised nerve(s), approximately 2 cm per nerve, to a 1.5 mL tube containing 1 mL of ice-cold PBS and store on ice. NOTE: The nerve can be stored overnight on ice. NOTE: For the DRG dissection, using forceps and a scalpel, detach the skin from the underlying soft tissue by gently peeling off the remaining back skin to expose the cervical, thoracic, and lumbar spinal regions. Remove the head by cutting at the base of the skull using a blunt pair of scissors.
Cut the ribs parallel with and close to the spinal column on both sides, before detaching the viscera connected to the anterior side of the spinal column. Cut the femurs and remove the spinal column by a transverse cut at the level of the femurs. Using iris scissors, cut away any muscle, fat, and other soft tissues from the spine.
Place the spinal column with the dorsal side facing up and open it, starting at the caudal end, by sliding one tip of the scissors into the vertebral canal at the three o'clock position and the bone snipped. Repeat the procedure at the nine o'clock position. NOTE: It is important to cut exactly in the center to avoid damaging the DRG.
Repeat the cutting from side-to-side until the rostral end is reached and the spinal cord is fully exposed. Using a dissection microscope, remove the spinal cord to expose the spinal nerves, which can be used to locate the DRGs within the spine.
Remove the lumbar DRGs by gently tugging on the dorsal root to pull the DRG into the vertebral canals, then clipping the nerves and connective tissue with iris scissors to remove the DRGs. NOTE: It is possible to restrict the collection to the L4-L6 DRG, which are contributing to the sciatic nerve.
Using forceps, transfer the DRGs to a 1.5 mL tube containing approximately 1 mL of ice-cold PBS, and store on ice. NOTE: The nerve can be stored overnight on ice.
3. Digestion of Sciatic Nerve and DRG
Prepare the digestion media (DMEM, 10 mM HEPES, 5 mg/mL BSA, 1.6 mg/mL collagenase Type 4) supplemented with 100 µg/mL of deoxyribonuclease, and store on ice. NOTE: The amount of digestion medium required per mouse (sciatic nerves + DRGs) is 1 mL. Once prepared, aliquots of the digestion media can be stored at -20 °C.
Transfer the sciatic nerves and DRGs from one mouse into separate 1.5 mL tubes containing 500 µL of digestion media. For the sciatic nerves, chop the material into 15-20 pieces using spring scissors and then incubate the tissue suspensions in a shaking incubator at 37 °C for 20 min with gentle agitation (≤ 450 rpm).
Repeatedly triturate the cell suspension through 1,000 µL pipette tip to mechanically dissociate any incompletely digested tissue. NOTE: If the suspension is difficult to titrate, then cut the pipette tip with scissors to create a blunt end. Incubate the tissue suspension at 37 °C for 20 min with gentle agitation (≤ 450 rpm).
Repeat the trituration through a 1,000 µL pipette tip and incubate for a further 10 min at 37 °C with gentle agitation (≤ 450 rpm).
Prepare the FACS buffer (PBS supplemented with 10% FCS and 1 mM EDTA) and store on ice. Rinse and soak a stainless-steel screen with a mesh size of 140 µm with FACS buffer in 60 x 15 mm Petri dish on ice. NOTE: Nylon screen filters should be avoided as it leads to a significant loss in the yield of nucleated cells.
Repeatedly pass the tissue suspensions through the 140 µm screen filter, as it is held over the Petri dish, with blunt forceps, on ice. Collect the cell suspension from the Petri dish and transfer it to a new 1.5 mL tube stored on ice.
Push any incompletely digested tissue that remains on the filter through by repeated trituration with a 1,000 µL pipette tip. Rinse the screen filter twice with 500 µL of FACS buffer and collect the resulting cell suspension in the Petri dish to combine with the cell suspension from step 3.6. NOTE: If the flow cytometer used is sensitive to debris, the cell suspension from step 3.7 can be passed through a 70 µm screen filter; however, this may lead to a loss in the yield of nucleated cells.
Centrifuge the cell suspensions at 300 x g for 5 min at 4 °C. Discard the supernatant and resuspend the cell pellet in 1 mL of FACS buffer. Repeat this washing step once.
Resuspend the cell pellet in 150-350 µL of FACS buffer, depending upon the number of staining that are required (150 µL FACS buffer per staining panel).
4. Staining of Sciatic Nerve and DRG with Fluorescently Labeled Antibodies for Flow Cytometry
Transfer 100 µL of the sciatic nerve or DRG cell suspension into a V-shaped well in a 96-well plate on ice. NOTE: Single-stained cells are necessary for all antibodies used as they will provide the necessary controls for setting up the flow cytometry with respect to photomultiplier tube (PMT) voltage gains and compensation, as well as to help distinguish specific from non-specific binding. Due to the limited material available from the sciatic nerve and DRGs, blood leukocytes and macrophages from the PCL (steps 1.1-1.2) can be used as the single-stained controls, the staining for which should be performed in parallel with the staining of the sciatic nerve and DRGs.
Pool the remaining cells suspensions (approximately 50 µL per sample) from either the sciatic nerves or DRG samples, to serve as an unstained control.
Centrifuge the plate at 400 x g for 5 min at 4 °C. Discard the supernatant by inverting the plate over a bin and applying a sharp flick of the wrist, and gently tap dry on paper towels.
For surface staining, resuspend each of the cell pellets in 20 µL of FACS buffer containing the antibodies of interest (CD45-A647, CD11b-PerCP/Cy5.5, and MHC Class II-Biotin) and incubate, protected from light, on ice for 30 min. NOTE: (1) The optimal antibody dilution should be determined prior to use by performing a dilution series according to the manufacturer's product information; (2) Non-specific stainings can be reduced by incubation with Fc-block at this step, according to the manufacturer's instructions; and (3) Isotype control stainings are generally not required when using primary antibodies directly conjugated with a fluorophore and/or when the antigen(s) of interest produce distinct populations.
Add 130 µL of FACS buffer to each well and centrifuge the plate at 400 x g for 5 min at 4 °C. Discard the supernatant as previously described (step 4.3) and wash the cell pellets twice with 150 µL of FACS buffer. NOTE: If using an unconjugated primary antibody or a primary antibody conjugated to biotin, then an appropriate secondary reagent, conjugated to a fluorophore, can be added at this point and steps 4.4-4.5 are repeated. In the case of this protocol, streptavidin-PE/Cy7 or Streptavidin-PerCP was used to detect MHC Class II in combination with CD45-A647/CD11b-PerCP/Cy5.5, or in combination with CD68-APC and F4/80-PE/Cy7, respectively.
For fixation, after the last washing step, resuspend the cell pellets in 100 µL of 4% paraformaldehyde in PBS (pH 7.4). Incubate the plate on ice for 10 min and then centrifuge at 400 x g for 5 min at 4 °C. Discard the supernatant as previously described (step 4.3) and wash the cell pellets once with 150 µL of FACS buffer. Caution: Paraformaldehyde is toxic; wear appropriate protection such as gloves and eyeglasses, etc. NOTE: Resuspend the cell pellets in 150 µL of FACS buffer and store protected from the light, at 4° C for up to two days. If no intracellular staining is required, then proceed to step 4.11 after centrifuging the plate at 400 x g for 5 min at 4 °C.
For intracellular staining, resuspend the cell pellets in 150 µL of FACS buffer+0.5% detergent, and incubate at room temperature, protected from the light for 15 min. Centrifuge the plate at 400 x g for 5 min at 4 °C. Discard the supernatant as previously described (step 4.3) and wash the cell pellet once with 150 µL of FACS buffer+0.5 % detergent. Resuspend the cell pellet in 20 µL of FACS buffer+0.5% detergent containing the antibodies of interest (CD68-APC, CD206-PE) and incubate, protected from light, at room temperature for 30 min.
Add 130 µL of FACS buffer+0.5% detergent to each well and centrifuge the plate at 400 x g for 5 min at 4 °C. Discard the supernatant as previously described (step 4.3) and wash the cell pellets twice with 150 µL of FACS buffer+0.5 % detergent. NOTE: If using an unconjugated primary antibody or a primary antibody conjugated to biotin, then an appropriate secondary reagent, conjugated to a fluorophore, can be added at this point and steps 4.9-4.10 repeated.
Resuspend the cell pellets in 150 µL of FACS buffer+0.5 % detergent and store, protected from the light, at 4 °C for up to two days.
Resuspend the cell pellets with 150 µL FACS buffer+0.5 % detergent supplemented with 5 µg/mL DAPI and incubate for 5 min at room temperature, protected from the light. Centrifuge the plate at 400 x g for 5 min at 4 °C. Discard the supernatant as previously described (step 4.3).
Resuspend the cell pellets in 150 µL of PBS and transfer to an appropriately labeled FACs tube. Store at room temperature, protected from the light, until ready for analysis. NOTE: After resuspension in PBS, the cells should be analyzed within hours, since the DAPI will slowly dissociate from the DNA, leading to a loss in signal.
5. Setup of Flow Cytometry and Running of Samples
Ensure that the flow cytometer is equipped with the appropriate lasers and/or filter sets to measure the fluorophores used to stain the cells. This can be determined under the default cytometer configuration on the system. In the case of this protocol, cells were analyzed using a flow cytometer equipped with three lasers (405 nm, 488 nm, and 633 nm) allowing for the detection of ≤ 5 fluorophores.
Select the appropriate channels for detection of the fluorophores of interest. All fluorescent signals are best detected with logarithmic amplification.
Select an appropriate flow rate for the samples. This parameter can vary depending upon the system used and should be determined empirically. In the case of this protocol, a flow rate of MED-to-HI (35-60 µL/min) was used to prevent clogging of the needle.
Select the total number of events/cells to be collected per sample. The minimum number of events should be 1,000,000.
Create scatter plots under a global worksheet of the SSC-A versus FSC-A, as well for each fluorophore of interest versus FCS-A. Create a statistical view that will display the number of events and the mean fluorescent intensity (MFI) for the selected fluorophores.
Set the voltage of the FSC-A and SSC-A channels, such that the smallest cells fit into the lower left 10% of the scatter plot. Determine the background fluorescence and minimum sample fluorescence from the unstained controls from either the sciatic nerves or DRGs. Determine the parent (P1) gate based upon the scatter plot from DAPI-only staining controls. NOTE: This is not a collection gate, due in part, to the heterogeneous nature of the DAPI+ population in the PNS.
Using the FSC scatter plots for each of the fluorophores of interest, adjust the voltages so that the cells of the unstained control fall within the lower quarter of their respective scatter plots. Gates are then created above this region to define those cells that are positive for each of the respective markers/fluorophores. The placement of a gate is confirmed by running of the single-stained control (step 4.1). NOTE: Compensation for overlapping emission spectra can be performed at this point or applied retrospectively with the appropriate analysis software.
Once the flow cytometer has been set up, as described, the samples can be run.
After the run, the remaining sample material can be discarded and the FCS files can be exported for further analysis.
Representative Results
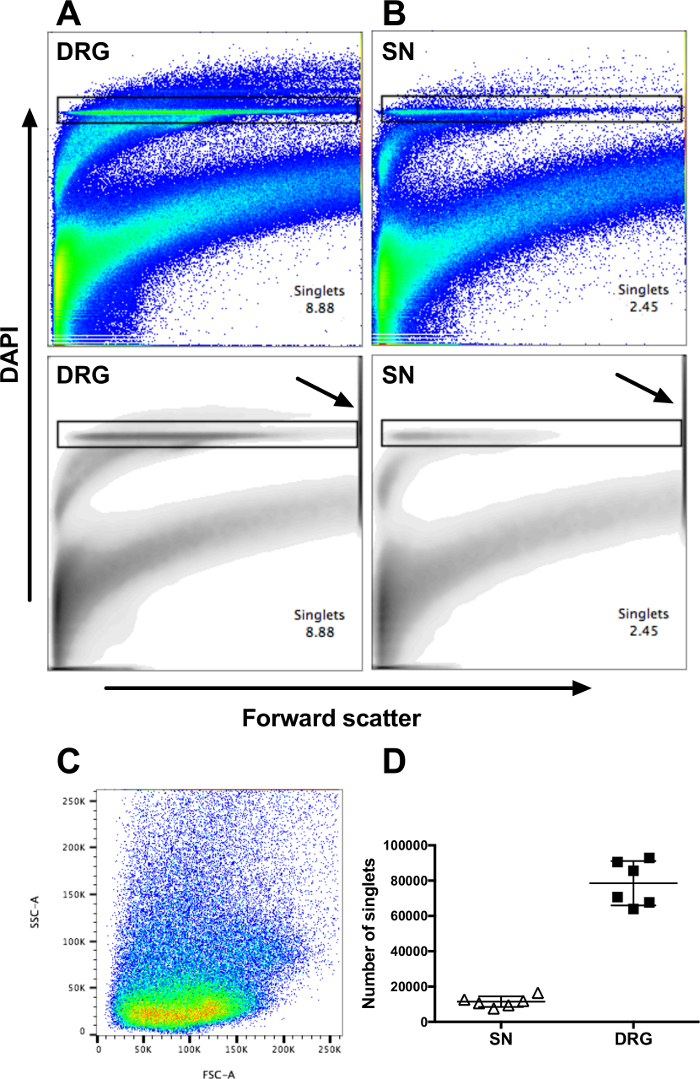
Cell suspensions from both full-length sciatic nerves and all major DRG were prepared, according to the protocol, from six healthy C57BL/6 mice and divided into three equal aliquots for staining. Counter staining with DAPI, which stains DNA, allows for the detection of a single cell population (Figure 1A-B, upper panel). Washing out the DAPI, prior to analysis by flow cytometry, decreased to an extent the fluorescent intensity of the non-nucleated debris, which allows for the selection of a discrete population over the FSC-axis of single-nucleated events or singlets. No collection gate is applied as the singlets from PNS are distributed over a large range of FSC and SSC (Figure 1C). The material on the high end of the FSC axis, which contains incompletely digested tissue, must be excluded in the selection of the singlet population (Figure 1A-B). Using a density plot (lower panel) to display the results may facilitate in the exclusion of the large debris (lower panel). A total of ≥ 30,000 singlets can be regularly obtained from two full sciatic nerves and ≥200,000 singlets can be obtained from DRGs (Figure 1D). From the sciatic nerves, approximately 5% of singlets expressed CD45 on their surface, whereas approximately 5-10% were found in the DRGs, thereby defining these cells as leukocytes of hematopoietic origin (Figure 2A).
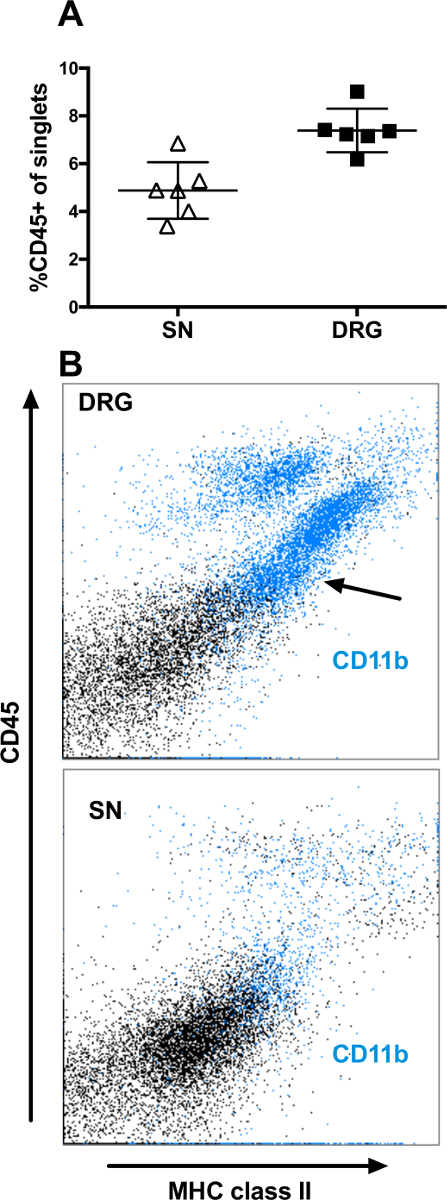
A majority of the CD45+ singlets also expressed MHC class II, in a gradual manner (Figure 2B). In the DRGs, CD11b can be used to detect microglial-like cells (CD45dim, MHCII+ and CD11b+) (Figure 2B, upper panel). However, this microglia population is absent from the sciatic nerves (Figure 2B, lower panel). A majority of the singlets expressing CD45 in the PNS are CD68+ macrophages. In the sciatic nerve and DRG, 2-5% of the singlets expressed CD68 (Figure 3A). All cells expressing CD68 also expressed CD45 (data not shown). In the DRGs, the CD68+ macrophages were more heterogeneous in their expression of both MHC class II and CD206, a marker for M2 macrophages, than in the sciatic nerves (Figure 3B-D). In the sciatic nerves, most macrophages co-expressed MHC class II and CD206 (Figure 3B-D, lower panels); typically, approximately 60-80% of the CD68+ macrophages expressed CD206, while a smaller proportion of the CD68+ co-expressed CD206 in the DRGs (Figure 3B). Interestingly, the M1 macrophage marker CD86 did not generate any positive staining neither on macrophages from nerve nor from PCL (data not shown). The macrophage marker, F4/80, showed a heterogeneous expression both in DRG and in sciatic nerves (Figure 3B-C), staining a proportion of the CD68- and MHCII- cells. Furthermore, F4/80 expression did not fully overlap with the CD68 expression.
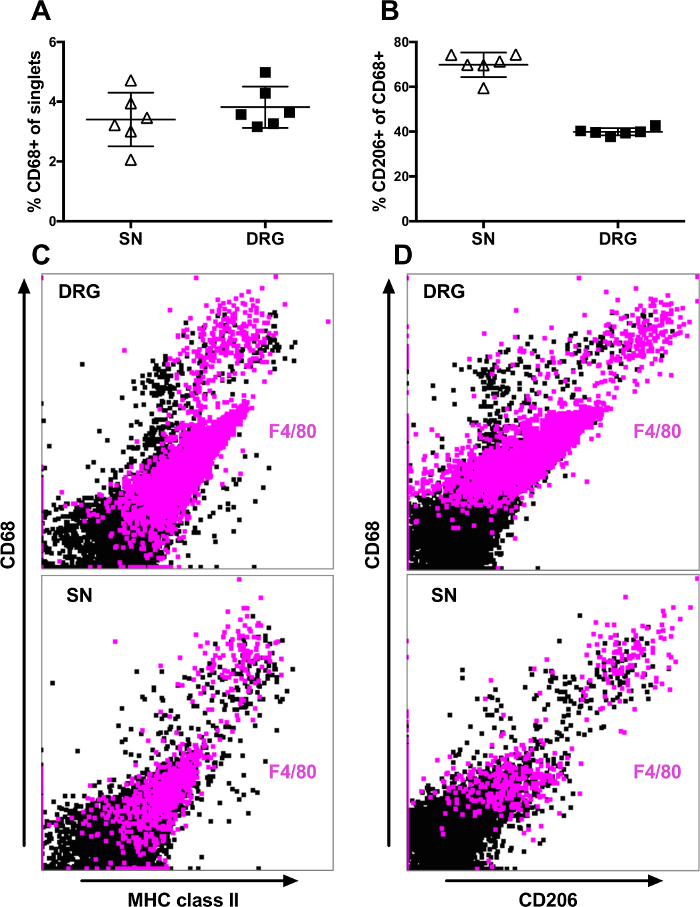
Figure 1: Representative data from flow cytometry analysis of DAPI-stained nucleated cells from DRG and sciatic nerve. All events collected, shown as scatterplots (upper panel) or density plots (lower panel), from (A) DRG or (B) sciatic nerve (SN) of one representative mouse. Arrows in the lower panels indicate the gates, excluding the material at the top the FCS axis. (C) Back-gating of the cells collected in the DAPI-gate (singlets), shown in panel (A) on SSC vs. FSC scatter plot. (D) Number of singlets from both SN and all major DRG from six healthy C57BL/6 mice. Data represent mean ± SD. Please click here to view a larger version of this figure.
Figure 2: Flow cytometry analysis of surface expression of CD45, MHC class II, and CD11b on singlets from DRG and sciatic nerve. Events selected in the DAPI-gate (singlets) shown in Figure 1 were analyzed. (A) Proportion of the singlets expressing CD45 in the sciatic nerve (SN) and DRG from six control mice. (B) Representative scatter plots of singlets from DRG (upper panel) and SN (lower panel). Cells expressing CD11b are indicated with blue color. Data represent mean ± SD. Please click here to view a larger version of this figure.
Figure 3: Flow cytometry analysis of surface expression of CD68, MHC class II, and CD206 on singlets from DRG and sciatic nerve. Events selected in the DAPI-gate (singlets) shown in Figure 1 were analyzed. (A) Proportion of the singlets expressing CD68 in sciatic nerve (SN) and DRGs from six healthy C57BL/6 mice. (B) Proportion of CD68+ macrophages co-expressing CD206 in SN and DRG from six healthy C57BL/6 mice. (C) Representative scatter plots of singlets from DRG (upper panel) and SN (lower panel) showing CD68 and MHC class II expression. (D) Representative scatter plots of singlets from DRG (upper panel) and SN (lower panel) showing CD68 and CD206 expression. Cells expressing F4/80 are indicated with magenta color. Data represent mean ± SD. Please click here to view a larger version of this figure.
Discussion
Sciatic nerves contain a large proportion of lipids, such as cholesterol, due to the content of myelin around the axons. Since the properties of lipids change with temperature, different results may be obtained at different temperatures. To ensure cell preservation, all steps in this protocol after the digestion were performed on ice. Whilst consistency is recommended for the sake of reproducibility of the results, it may be possible to increase yield by performing the steps 3.6 and 3.7 at room temperature. If the nerves at step 3.8 have not been sufficiently digested, small pieces of nerve will be visible on the mesh. In case of insufficient digestion, the material retained on the mesh can be re-digested by transferring to a new 1.5 mL tube containing 250 µL of digestion media and repeating steps 3.2-3.6. If the yield is poor, then a sample of the cell suspension, after passing through the mesh screen (step 3.6) should be viewed under a light microscope with trypan blue. Viable, non-blue, cells should be visible along with rod-like structures, which are fragments of axons. If no viable cells are present, then the digestion conditions and/or incorrect handing of the material has caused necrosis. If only large aggregates are visible, then it is recommended to collect the material by centrifugation and repeat the digestion (steps 3.2-3.6).
The method of flow cytometer analysis of murine sciatic nerves and DRGs is useful for questions involving whole nerves and several DRGs. Recently there has been an increased interest in the analysis of the PNS using flow cytometry11,12. Liu et al. have developed a method using papain and mechanical dissociation of the sciatic nerves and DRGs using 21G and 23G to specifically analyze as little as 1 cm section of the nerve, thereby allowing for the analysis of nerve material from specific sites of injury11. The method presented here is recommended for use when the effect on the PNS is expected to be systemic, as in neurotoxicology and genetic models of neuropathy or in chronic diseases, such as diabetes. Due to the low yield of nucleated cells obtained from sciatic nerve, it is not recommended to attempt to study small populations with ≤ 100 gated events from an individual mouse. However, pooling of material from several mice would facilitate the study of such small populations within the nerve. Nevertheless, it would be of interest to investigate whether the enzymatic tissue dissociation using papain and 21G/23G needles described by Liu et al.11 could be combined with the flow cytometry staining of nuclei, as described in this study, to enhance the yield of single cells. The ability to analyze very small amounts of tissue is crucial for performing comparison between ipsi- and contralateral DRGs following unilateral sciatic nerve axotomy. However, this is beyond the scope of this study. Interestingly, commonly used markers for identification of macrophages were found to have an unexpectedly heterogeneous expression in the PNS of mice. For instance, CD11b and F4/80 were both found to be expressed on a large proportion of non-hematopoietic, non-antigen presenting cells in both sciatic nerve and DRGs. Furthermore, in the sciatic nerves, not all CD68+ macrophages expressed CD11b and F4/80, as would have been expected from the study of other organs. It is therefore recommended that caution is taken when using these markers for the study of macrophages in the PNS and that other markers, such as CD163, should be investigated. Furthermore, the sensitivity of the different markers to the enzymatic dissociation should also be established as this maybe an explanation for the differences observed in expression, such as with CD11b or F4/80.
This protocol has previously been used to analyze the immune cells in the PNS after toxic challenge with streptozotocin (STZ)13, a chemical commonly used for the induction of diabetes in model rodent organisms. It was shown that STZ, which activates both TRPV1 and TRPA1, initially causes a depletion of immune cells in both sciatic nerve and DRGs, while not causing any inflammation in the nerve. Although the numbers of immune cells in the DRGs recovered, the immune cells and notably macrophages were still significantly depleted three weeks post toxic challenge. It was subsequently hypothesized that macrophage phagocytosis of nerve debris, resulting from injury by ion channel hyperexcitability may have led to the apoptosis of the macrophages, resulting in their depletion from the general cell population. It is not known how other peripheral nerve toxins affect nerve resident macrophages. If depletion of macrophages is a common result of such agents this may have implications for treatment.
It was found that only 5-10% of the nucleated cells from the nerve were leukocytes of hematopoietic origin. The remaining cells are presumably a mixture of the other nerve cell types, namely Schwann cells, pericytes, fibroblasts, and endothelial cells. Due to capacity of flow cytometry systems to measure multiple parameters and providing that there are specific markers and/or antibodies available, it conceivable that these cell types could also be analyzed using this method. This method has been described for murine material. However, with some modifications, it has also been used for the analysis of PNS material from pig and from human. It is anticipated that this method will be more useful for the analysis of material from species larger than mice, in the future.
Disclosures
The authors have no conflicts of interest in regard to this study.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft (DFG; SFB1118). The authors would like to thank Axel Erhardt for performing most of the dissection and helpfully transmitting this knowledge, and Dr. Volker Eckstein for assisting in the technical aspect of the flow cytometer.
References
- DeFrancesco-Lisowitz A, Lindborg JA, Niemi JP, Zigmond RE. The neuroimmunology of degeneration and regeneration in the peripheral nervous system. Neuroscience. 2014;302:174–203. doi: 10.1016/j.neuroscience.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Erreni M, Allavena P, Porta C. Macrophage polarization in pathology. Cell. Mol. Life Sci. 2015;72:4111–4126. doi: 10.1007/s00018-015-1995-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017;198:1006–1014. doi: 10.4049/jimmunol.1601515. [DOI] [PubMed] [Google Scholar]
- Perrin FE, Lacroix S, Avilés-Trieueros M, David S. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain. 2005;128:854–866. doi: 10.1093/brain/awh407. [DOI] [PubMed] [Google Scholar]
- Niemi JP, et al. A Critical Role for Macrophages Near Axotomized Neuronal Cell Bodies in Stimulating Nerve Regeneration. J Neurosci. 2013;33:16236–16248. doi: 10.1523/JNEUROSCI.3319-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokarram N, Merchant A, Mukhatyar V, Patel G, Bellamkonda RV. Effect of modulating macrophage phenotype on peripheral nerve repair. Biomaterials. 2012;33:8793–8801. doi: 10.1016/j.biomaterials.2012.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R, Willison H. Neuroinflammation in the peripheral nerve: Cause, modulator, or bystander in peripheral neuropathies? Glia. 2016;64:475–486. doi: 10.1002/glia.22899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriel V, Garzón I, Alaminos M, Cornelissen M. Histological assessment in peripheral nerve tissue engineering. Neural Regen. Res. 2014;9:1657–1660. doi: 10.4103/1673-5374.141798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong TY-M, Leong AS-Y. How does antigen retrieval work? Adv. Anat. Pathol. 2007;14:129–131. doi: 10.1097/PAP.0b013e31803250c7. [DOI] [PubMed] [Google Scholar]
- Barrette B, et al. Requirement of myeloid cells for axon regeneration. J. Neurosci. 2008;28:9363–9376. doi: 10.1523/JNEUROSCI.1447-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yin Y, Li F, Malhotra C, Cheng J. Flow cytometry analysis of inflammatory cells isolated from the sciatic nerve and DRG after chronic constriction injury in mice. J. Neurosci. Methods. 2017;284:47–56. doi: 10.1016/j.jneumeth.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Pannell M, et al. Adoptive transfer of M2 macrophages reduces neuropathic pain via opioid peptides. J. Neuroinflammation. 2016;13:262. doi: 10.1186/s12974-016-0735-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidmark AS, Nawroth PP, Fleming T. STZ causes depletion of immune cells in sciatic nerve and dorsal root ganglion in experimental diabetes. J. Neuroimmunol. 2017;306:76–82. doi: 10.1016/j.jneuroim.2017.03.008. [DOI] [PubMed] [Google Scholar]