

Abstract
The full details of mechanistic investigation on enantioselective sulfenofunctionalization of alkenes under Lewis base catalysis are described. Solution spectroscopic identification of the catalytically active sulfenylating agent has been accomplished along with the spectroscopic identification of putative thiiranium ion intermediates generated in the enantiodetermining step. The structural insights gleaned from these studies informed the design of new catalyst architectures to improve enantioselectivity. In addition, structural modification of the sulfenylating agents had a significant and salutary effect on the enantioselectivity of sulfenofunctionalization which was demonstrated to be general for trans disubstituted alkenes. Whereas electronic modulation had little effect on the rate and selectivity, steric bulk on arylsulfenylphthalimides was very beneficial.
Keywords: sulfenofunctionalization, thiiranium ions, Lewis base catalysis, selenophosphoramides, sulfenylating agents
Introduction
The importance of organosulfur compounds[1,2] manifests itself in the myriad of constructive and functional manipulations involving these as building blocks as well as in the abundance of sulfur-containing natural products.[3] Among a variety of methods for the introduction of sulfur groups, the vicinal sulfenofunctionalization of alkenes represents a powerful approach. In this context, the role of thiiranium ions as versatile reactive intermediates has been extensively studied since the 1960’s[4] and a practical method for their generation is the addition of electrophilic sulfur(II)[5] reagents to alkenes (Scheme 1). Subsequent invertive capture with nucleophiles affords 1,2-difunctionalized products with defined relative configuration of the introduced functionalities. The applicability of this reaction has been demonstrated with a variety of sulfenylating agents and nucleophiles. [2] However, despite the sound understanding of the mechanism, catalytic asymmetric sulfenofunctionalizations have remained largely under-developed.
Scheme 1.

Sulfenofunctionalization of unactivated alkenes.
Only recently have catalytic, enantioselective sulfenylations of activated alkenes derived from aldehydes,[6] ketones,[7] and amides[8] been reported. In addition, the catalytic, enantioselective sulfenoetherification of unactivated alkenes under chiral Brønsted acid catalysis has been described, albeit with moderate enantioselectivities.[9] The necessity for generating enantioenriched thiiranium ions has been elegantly circumvented by asymmetric desymmetrization of meso-thiiranium ions, wherein ring-opening by a nucleophile is the stereodetermining step.[10,11] Notwithstanding this progress, there remains a dearth of methods that control the absolute stereochemical outcome of 1,2-sulfenofunctionalizations, and this pertains especially to isolated alkenes.
The difficulties in developing catalytic enantioselective processes originate, inter alia, from the inherent high reactivity of electrophilic sulfur reagents, which may account for a pronounced, racemic background reaction. In addition, even if the background reaction can be suppressed and the thiiranium ions can be generated enantioselectively in the first place, several mechanisms can erode the configurational integrity of this intermediate before the attack of a nucleophile at the carbon atom (Scheme 2). One plausible pathway for racemization leads through an open carbocation intermediate resulting in configurational instability of the thiiranium ion (Scheme 2a).[12] Furthermore, the Lewis acidic sulfur center itself can be attacked by the nucleophile, thus liberating an achiral sulfenyl transfer reagent that decreases the enantiomeric purity of thiiranium ions upon redelivering the sulfenyl cation to an alkene (Scheme 2b).[13] Finally, racemization via “olefin-to-olefin” transfer cannot be excluded at all, although detailed mechanistic studies revealed that for thiiranium ions this transfer proceeds only at ambient temperature (Scheme 2c).[14]
Scheme 2.

Racemization mechanisms of thiiranium ions.
As a consequence, for many years only two examples of direct enantioselective sulfenofunctionalizations were known, both employing chiral reagents in stoichiometric amounts. Rayner and coworkers synthesized a number of camphoric acid-derived, enantiomerically enriched thiosulfonium salts.[15] Their application in the sulfenocyclization reactions, however, afforded the corresponding products with poor enantioselectivity (Scheme 3a). The second, more successful example, was reported by Pasquato and coworkers using a binaphthyl-derived reagent (Scheme 3b).[16] Under optimized conditions the enantioselective sulfenylation followed by nucleophilic capture of the thiiranium ion furnished the sulfenofunctionalized product with an appreciable enantiomeric purity of 88% ee. If substoichiometric amounts of the chiral sulfenylating reagent were used at elevated temperature (−20 °C) a significant erosion of the enantiomeric composition was observed. Obviously, one of the above mentioned racemization mechanisms becomes prevalent under these conditions preventing the elaboration of a catalytic protocol.
Scheme 3.

Reagent controlled enantioselective sulfenofunctionalization.
Background
As part of the ongoing program to apply the concept of Lewis base activation of Lewis acids[17,18] to reactions of main group elements, we envisioned the possibility of generating reactive, ionic sulfenylating species from weakly electrophilic sulfur(II) sources (Scheme 4). On the one hand, this approach would allow for the utilization of sulfur reagents with attenuated reactivity and thus, reduced tendency for an uncatalyzed background reaction. On the other hand, the transfer of the sulfenium ion to the alkene double bond could be rendered enantioselective by the judicious choice of a chiral Lewis base catalyst. If reaction conditions can be optimized such that the aforementioned racemization pathways for the enantiomerically enriched thiiranium are suppressed, a catalytic, enantioselective process is feasible.
Scheme 4.
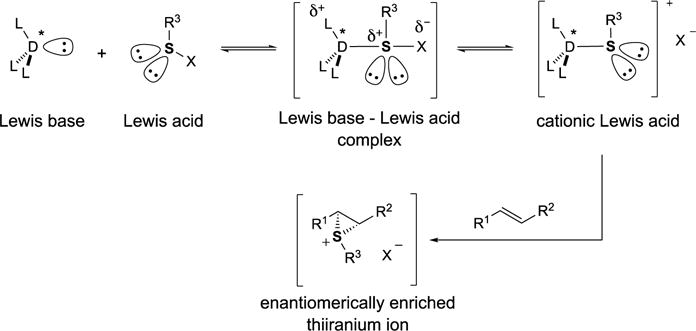
Lewis base catalyzed sulfenofunctionalization of unactivated alkenes.
At the outset, the general feasibility of a Lewis base catalyzed chalcogenofunctionalization of unactivated alkenes was examined. Previous investigations systematically investigated the stability of seleniranium[19,20] and thiiranium ions[14,21] to establish boundary conditions for catalytic, enantioselective chalcogenofunctionalizations. On the basis of these examinations the first enantioselective sulfenoetherification was realized using a selenophosphoramide catalyst and a phthalimide derivative as the sulfenylating reagent (upper, Scheme 5).[22] Moreover, a Brønsted acid is required as a co-activator for the otherwise unreactive sulfenylating agents, and its specific function has been examined in a recent study from these laboratories.[23] Under the optimized reaction conditions representative olefins are converted into the corresponding tetrahydropyrans with good to excellent site selectivities and high enantioselectivities. By changing the substitution pattern of the olefins, the constitutional isomers (i.e. tetrahydropyrans) are accessible albeit with decreased enantioselectivities. Preliminary attempts of capturing the thiiranium ions intermolecularly with an external nucleophile is also possible.
Scheme 5.
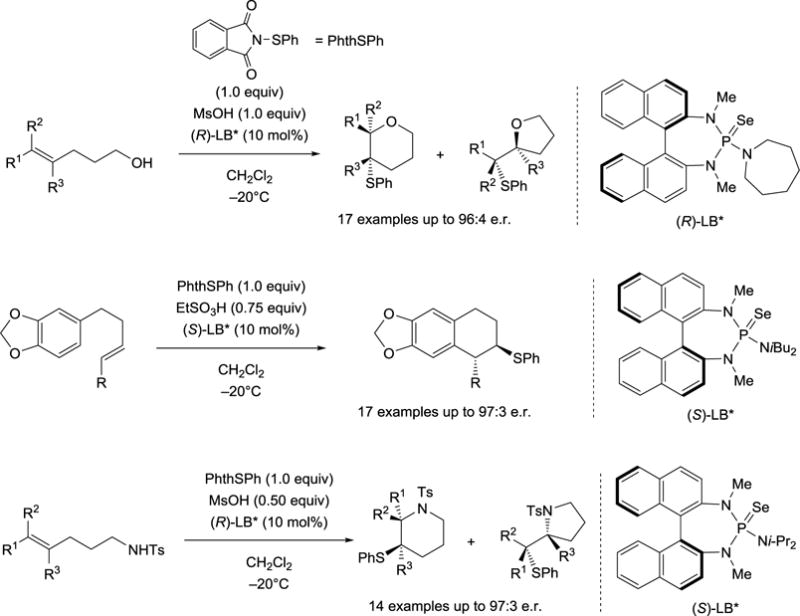
Lewis base catalyzed enantioselective sulfenofunctionalization.
The generality of this catalytic system has been demonstrated using various nucleophiles. In addition to the use of hydroxyl groups in sulfenoetherification reactions[22], electron-rich arenes have been deployed in carbosulfenylation of olefins affording trans-tetrahydronaphthalenes with complete diastereospecificity and high enantiomeric ratios (middle, Scheme 5).[24] The protocol has been applied to tosyl-protected amines that can be engaged in sulfenoamination reactions yielding enantioenriched nitrogen heterocycles (lower, Scheme 5).[25] More recently, the process has been extended to include sulfenocyclization reactions involving both phenols[26] and anilines[27] with great success. The catalytic, enantioselective sulfenylation of ketone-derived enoxysilanes has also been demonstrated (not shown).[28]
A catalytic cycle has been proposed on the basis of spectroscopic observations and kinetic studies (Scheme 6).[21, 29] The cycle commences with the sulfenylation of Lewis base mediated by MsOH to generate the catalytically active species which is the resting state of the catalyst with a diagnostic 31P NMR chemical shift at δ = 59.9 ppm. Subsequently, in the turnover-limiting and stereodetermining step, the arylsulfenyl group is transferred to the alkene forming the enantiomerically enriched thiiranium ion. Its stereospecific capture by an internal or external nucleophile delivers the sulfenofunctionalized product and regenerates the catalyst.
Scheme 6.

Proposed catalytic cycle.
Results from the kinetic study suggest that the catalytically active species is the sulfenylated catalyst [(R2N)3P=Se–SPh]+X− generated upon the MsOH mediated reaction of N-phenylsulfenylphthalimide with the Lewis base.[29] Its existence is supported by 31P NMR spectroscopy, in which the diagnostic signal for selenophosphoramide (80–90 ppm, depending on the catalyst structure) disappears and a new resonance at around 60 ppm is observed. This value is in accord with previously reported and structurally related compounds of the type [(R2N)3P=Se–YAr]+X−. [19,24,30] The proposed active species is assumed to transfer the sulfenium ion to an alkene thus forming a thiiranium ion. Since this event most likely constitutes the enantiodetermining step, detailed knowledge about the geometries of the active species structure could help understanding the trajectory of the alkene approach and thus the origin of enantioselectivity. A preliminary stereochemical model has been proposed by positing that the PhS-Se moiety of the catalytically active species is oriented toward the binaphthyl backbone.[24b] Thus, the approaching alkene faces steric interaction with one of the naphthyl subunits resulting in the discrimination of the enantiotopic faces of the alkene. This assumption is supported by DFT calculations,[29] however, no experimental evidence had been provided until this point. To obtain a quantitative insight into the 3-dimensional structure of species [(R2N)3P=Se–SPh]+X−, its isolation and crystallographic characterization was planned.
Initially, the focus was on the identification of suitable conditions for generating the active species [(R2N)3P=Se–SPh]+X− without any byproducts that would disrupt the crystallization process. The first approach relied on the reaction of a selenophosphoramide Lewis base with [PhS(SMe2)]SbCl6[31] which had been used as a transfer agent of the S-phenyl group to alkenes. As it turned out, this reagent was also competent in transferring the sulfenyl moiety to the catalyst forming the active species along with SMe2 as the sole byproduct (equation 1). The formation of the active species was confirmed by the appearance of a diagnostic signal at 60.4 ppm in the 31P NMR spectrum.[29] Unfortunately, during the crystallization attempts, a disproportionation reaction occurred reducing the sulfenyl group to PhS–SPh while oxidizing the selenophosphoramide to the dicationic dimer [(R2N)3P=Se–Se=P(NR2)3]2+ 2SbCl6− (equation 2). An X-ray crystal structure[29] of this species could be obtained providing a first glimpse into how the groups in the actual active agent might be oriented. However, the key objective was to crystallize a species that resembles the actual catalytically active sulfenylating agent as closely as possible. Therefore, further modification and optimization was required.
| (1) |
| (2) |
Project Goals
Despite the positive developments, further improvement of the catalyst performance of the Lewis base catalyzed sulfenofunctionalization is desirable for some of the substrates. For instance, efficient discrimination between enantiotopic faces of terminal, 1,1-disubstituted and trisubstituted double bonds still remains a challenge. Beyond an empirical attempt to solve this problem, the in-depth understanding of processes occurring during the sulfenofunctionalization as well as detailed knowledge about the structure of the active sulfenylating agent can contribute substantially to the rational design of a more potent catalytic system. Given the thiiranium ion as the common intermediate, a more enantioselective preparation thereof would be beneficial to all conceivable substrate classes of this transformation. Therefore, the project goals encompassed:
crystallographic characterization of the catalytically active species of the Lewis base catalyzed sulfenofunctionalization
rationalization of the stereochemical outcome of the reaction in order to advance and further generalize the methodology, and to enable the rational design of a more selective catalyst
synthesis of new Lewis base catalysts (in an empirical approach) and their evaluation in the sulfenofunctionalization reaction
exploitation of the gained knowledge for the general improvement of the catalytic system
Results
1. Investigations Toward the Isolation of the Catalytically Active Species
The possibility to isolate the catalytically active species through the reaction of a Lewis base and [PhS(SMe2)]SbCl6 was initially investigated. To avoid the disproportionation reaction caused by the lability of the Se–SPh bond when using a selenophosphoramide[29] the donor atom of the catalyst was replaced by a sulfur atom. Thiophosphoramides are comparably efficient and selective catalysts in sulfenoetherification reactions[21] and it is expected that disproportionation of the S–SPh bond in the active species would become thermodynamically less favorable in comparison to the selenophosphoramide analogue. However, when a thiophosphoramide Lewis base was combined with [PhS(SMe2)]SbCl6, an equilibrium between the reactants was established (Scheme 7). This equilibrium was evidenced by the 31P NMR chemical shift which, depending on the stoichiometry of the reactants employed, ranged between the 31P NMR chemical shift of the catalyst and the active species. When equimolar amounts of catalyst (±)-1a and [PhS(SMe2)]SbCl6 were used, a rather broad signal at 65.9 ppm in 31P NMR spectrum was observed. Employing an excess of the sulfenylating agent shifted the 31P NMR signal to 69.5 ppm (sharp signal) indicating the saturation of (±)-1a as (±)-2aa+ SbCl6−. In contrast, when an excess of catalyst (±)-1a was used, the 31P NMR signal of the equilibrium (78.6 ppm) was shifted toward the 31P NMR signal of (±)-1a.
Scheme 7.
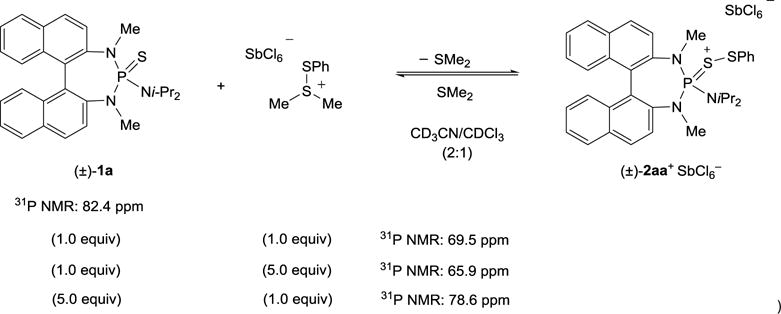
Equilibrium between catalyst (±)-1a and active species (±)-2aa+ SbCl6−.
Besides the complication arising from reversibility, solubility was a serious problem limiting the type of solvents that could be used for crystallization. At this point it was decided to change the strategy and move away from the use of [PhS(SMe2)]SbCl6. In order to counteract the reversible formation of the active species, thiophosphoramide (±)-1a was reacted with PhSCl and NaBArF24 furnishing (±)-2aa+ BArF24− as a pale yellow, air stable solid (Scheme 8). This approach not only enabled the irreversible generation of the active species (±)-2aa+, but also allowed for the introduction of the weakly coordinating BArF24− counteranion,[32] which greatly improved the solubility properties of complex (±)-2aa+ BArF24−. Also in this case, formation of (±)-2aa+ was initially confirmed by the 31P NMR chemical shift (65.8 ppm).
Scheme 8.

Synthesis of the catalytically active species (±)-2aa+ and its X-ray crystal structure. (Hydrogens and BArF24− counteranion omitted for clarity).
Ultimately, crystals suitable for X-ray analysis were obtained by slow evaporation of a saturated solution of (±)-2aa+ BArF24− in dichloromethane.[29] The molecular structure was confirmed by the X-ray crystal structure analysis of (±)-2aa+ (Scheme 8, (S)-enantiomer shown).[33] Several features of this structure are noteworthy. First, the diisopropylamino group is rotated such that the C(32)-N(3) bond occupies a nearly antiperiplanar orientation to the S(1)-P(1) bond while the C(29)-N(3) bond is synperiplanar to this. Torsion angles of 169.1 ° for S(1)-P(1)-N(3)-C(32) and −15.0 ° for S(1)-P(1)-N(3)-C(29), respectively, define this arrangement. The sum of the angles about the N(3) atom is >359.8 ° indicating near perfect planarity. This alignment is most likely a consequence of a generalized stereoelectronic effect in which the N(3) nonbonding electron pair is delocalized into the P(1)-N(1) and P(1)-N(2) σ* orbitals. Such stereoelectronic effects have been previously observed in similar structures.[34,35]
To avoid destabilizing steric interactions with the diisopropylamino group, the S–SPh subunit is oriented back toward the binaphthyl backbone as is evidenced by a 175.1° torsion angle of the N(3)-P(1)-S(1)-S(2) subunit. With a torsion of 96.0° of the P(1)-S(1)-S(2)-C(1) moiety, the P(1)-S(1) vector is almost perpendicular to the plane defined by the S(1)-S(2)-C(1) atoms. On the basis of the X-ray crystal structures of neutral 10-S-3 compounds[36,37] and by analogy to the well known 10-Se-3 T-shaped charge transfer complexes[30] the active sulfenylating agent was anticipated to be a bent ion pair complex (8-S-2) which is now verified crystallographically by the S(1)-S(2)-C(1) angle of 104.1°. The S(1)-S(2) bond (2.06 Å) is in the range of a sulfur–sulfur single bond length observed in disulfides.[38] The P(1)-S(1) bond (2.10 Å) of the active species is significantly lengthened compared to the 1.95 Å found for the P=S double bond in (Me2N)3P=S[39] and is closer to a typical P–S single bond.[40]
With a viable protocol for the irreversible generation of the catalytically active species from catalyst (±)-1a and PhSCl, the next objective was to determine if a similar species could be generated from the sterically demanding (2,6-diisopropylphenyl)sulfenyl chloride. As was the case previously (Scheme 8), the reaction of (±)-1a and (2,6-diisopropylphenyl)sulfenyl chloride yielded the active species (±)-2ab+ BArF24− as a pale yellow solid (upper, Scheme 9). The formation of (±)-2ab+ was evident from the 31P NMR chemical shift of 67.9 ppm. In addition, the reaction of a selenophosphoramide with the sterically demanding sulfenyl chloride was examined to furnish the corresponding active species 2bb+ BArF24− as a shiny yellow solid with a 31P NMR chemical shift of 61.3 ppm (upper, Scheme 9). Finally, an achiral version of the active species was accessed by reaction of HMPA(S) (1c) and PhSCl (lower, Scheme 9). All of the active species were surprisingly stable to air and moisture.
Scheme 9.

Preparation of various catalytically actives species.
2. Reaction of the Catalytically Active Species with Alkenes
The kinetic competency of the putative catalytically active species (±)-2aa+ BArF24− to sulfenylate alkene double bonds was demonstrated in a stoichiometric reaction with substrate 3a to afford thioetherification product (±)-4aa (Scheme 10). The reaction did not proceed to full conversion, most likely because, over time, the H+BArF24− generated effects a protonation of the nucleophilic OH group of the remaining substrate thus inhibiting product formation. However, more importantly, this control experiment substantiates the role of (±)-2aa+ as the active sulfenylating species.
Scheme 10.

Reaction of catalytically actives species (±)-2aa+ BArF24− with alkene 3a.
Given the accessibility of the catalytically active species, investigations on the fate of the Lewis base catalyst after transfer of the sulfenium ion to the alkene were carried out next. It has been proposed that the Lewis base remains coordinated to the thiiranium ion because in intermolecular sulfenofunctionalizations the site selectivity of the nucleophilic attack at the thiiranium intermediate is dependent on the catalyst. In the past, the existence of episulfuranes, i.e. three membered rings with a tetracoordinate sulfur atom, was a matter of debate.[41,42] In general, these reactive species occur only as transient/labile intermediates so that their existence is difficult to prove experimentally. It would be of interest to gain more insight into the nature of the interaction between Lewis base and thiiranium ion to enable the design of more efficient and selective catalysts. The challenge is to identify whether reactants, catalysts, and conditions can be adjusted such that Lewis base/thiiranium ion adducts can be observed by NMR spectroscopy or can even be isolated.
Preliminary investigations into the role of the Lewis base in stabilizing the thiiranium ion involved the combination of (±)-2aa+ BArF24− and (E)-4-octene (3b) in the absence of a nucleophile at room temperature. This experiment resulted in the formation of two new species that were initially assigned as diastereomeric, Lewis base-coordinated thiiranium ions 5ab+ BArF24− and epi-5ab+ BArF24− (right, Scheme 11). The ratio of these two species (85:15) reflects the enantiomeric ratio that would be obtained with enantiomerically enriched thiiranium ions upon capture with MeOH. Indeed, nucleophilic capture of 5ab+BArF24− and epi-5ab+BArF24− with MeOH resulted in the exclusive formation of the anti-configured product (in racemic form), which further supports the existence of the suggested intermediates.
Scheme 11.
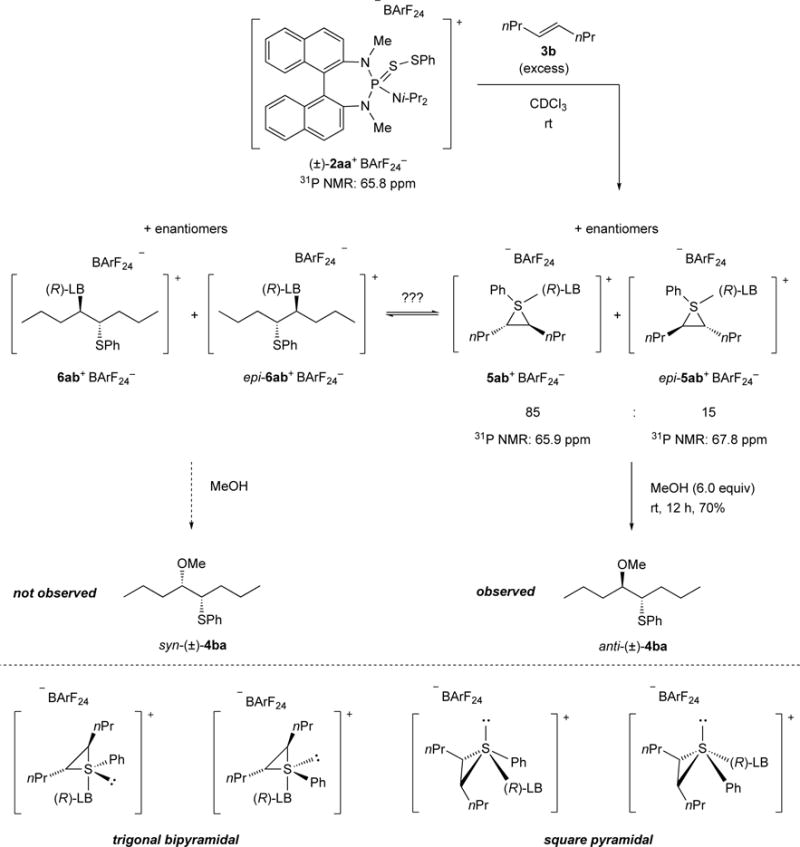
Reaction of the catalytically active species (±)-2aa+ BArF24− with (E)-4-octene (3b) in the absence of a nucleophile.
However, an alternative interpretation, that the observed intermediates represent structures 6ab+BArF24− and epi-6ab+BArF24− (left, Scheme 11) cannot be rigorously excluded. This kind of intermediate results from the nucleophilic opening of the thiiranium ions by the Lewis base itself.[41d,e,g] Since nucleophilic capture with MeOH produces exclusively anti-configured (±)-4ba, intermediates 6ab+ BArF24− and epi-6ab+ BArF24− would have to be unreactive and in equilibrium with 5a+ BArF24− and epi-5ab+BArF24−. Otherwise, direct nucleophilic displacement at 6ab+ BArF24− and epi-6ab+BArF24− would yield syn-(±)-4ba, which is not observed.
Furthermore, the presence of multiple signals in the 31P NMR spectrum could be a result of the stereogenicity at the sulfur atom. Taking the lone pair into account, the geometry at sulfur in these intermediates would be either trigonal bipyramidal[41d,f] or square pyramidal[41d] (lower, Scheme 11). However, it is unlikely that these rapidly interconverting species would be distinguishable at room temperature by 31P NMR spectroscopy.
Thus, neglecting stereogenicity at sulfur, the two observed 31P NMR signals represent diastereomeric intermediates arising from the use of a chiral Lewis base. Consequently, the same experiments with an achiral Lewis base should result in the formation of enantiomeric Lewis base-coordinated thiiranium ions and thus, only one 31P NMR signal should be observable. Accordingly, the achiral, catalytically active species 2ca+ BArF24− was employed in analogy to the previous reaction with (E)-4-octene (3b) (upper, Scheme 12). As predicted, only one 31P NMR signal was observed in this experiment at 66.1 ppm. Similarly, the reaction with cyclohexene resulted in the formation of only one intermediate that was captured with various nucleophiles to yield sulfenofunctionalized products with anti relative configuration (lower, Scheme 12). It is noteworthy that species 5cc+ BArF24− does not posses overall CS symmetry as might be expected from a meso-thiiranium ion. The 1H NMR spectrum clearly indicates the existence of an unsymmetrical intermediate. This observation is consistent with a Lewis base complexed thiiranium ion as neither of the four coordinate sulfur species possesses CS symmetry. However, a species of the type 6cc+ BArF24− cannot be unambiguously excluded.
Scheme 12.
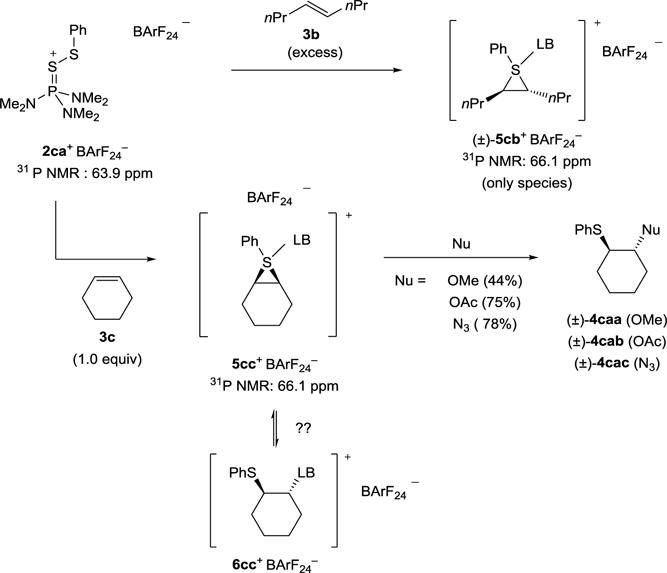
Reaction of the achiral, catalytically active 2ca+ BArF24− with (E)-4-octene (3b) and cyclohexene.
At this point, no direct experimental evidence to either confirm or rule out one of the conceivable intermediates could be provided. The most unambiguous proof would be an X-ray crystal structure. Interestingly, the observed intermediates are rather stable to air and moisture and even the nucleophilic capture is slow. In preliminary studies to obtain a suitable crystal of a stable intermediate, 1,4-dihydronaphthalene (3d) was investigated as the alkene (Scheme 13). Control experiments demonstrated the exclusive formation of the anti β-methoxy sulfide product 3da after nucleophilic capture of the putative episulfurane with MeOH. Unfortunately, crystallization attempts were unsuccessful.
Scheme 13.

Reaction of the achiral, catalytically active 2ca+ BArF24− with 1,4-dihydronaphthalene (3d) and crystallization attempts.
Interestingly, during the investigation into the use of new sulfenylating agents (vide infra), it was found that in the catalytic, intermolecular sulfenoetherification of a terminal alkene (1-octene (3e)), two phosphorus-containing species were present throughout the reaction. The major species observed at −20 °C has a chemical shift of 65.8 ppm, whereas the minor species has a chemical shift of 61.8 ppm in 31P NMR spectrum. This interesting observation was investigated in greater detail (Scheme 14). To this end, catalyst (R)-1b was combined with 1.0 equiv of sulfenylating agent 7b and 5.0 equiv of MsOH to form the active species (R)-2bb+ MsO− with a 31P NMR chemical shift of 61.8 ppm. Subsequently, 1-octene (3e) was added at −20 °C and the reaction was monitored by 31P NMR spectroscopy over a period of 12 h. During that time period, (R)-2bb+ MsO− (61.8 ppm) disappeared slowly and a new species at 65.8 ppm, potentially (R)-5be+ MsO−, grew in. Eventually, this species was treated with an excess of MeOH at room temperature to afford a 4:1 mixture of 4eb and 4eb′. The enantiomeric excess for compound 4eb was determined to be 99:1. The constitutional site selectivity as well as the enantioselectivity perfectly matches the results obtained from the catalytic sulfenoetherification process (vide infra, Table 2). These experiments provide additional support for the involvement of a Lewis base bound thiiranium ion intermediate.
Scheme 14.
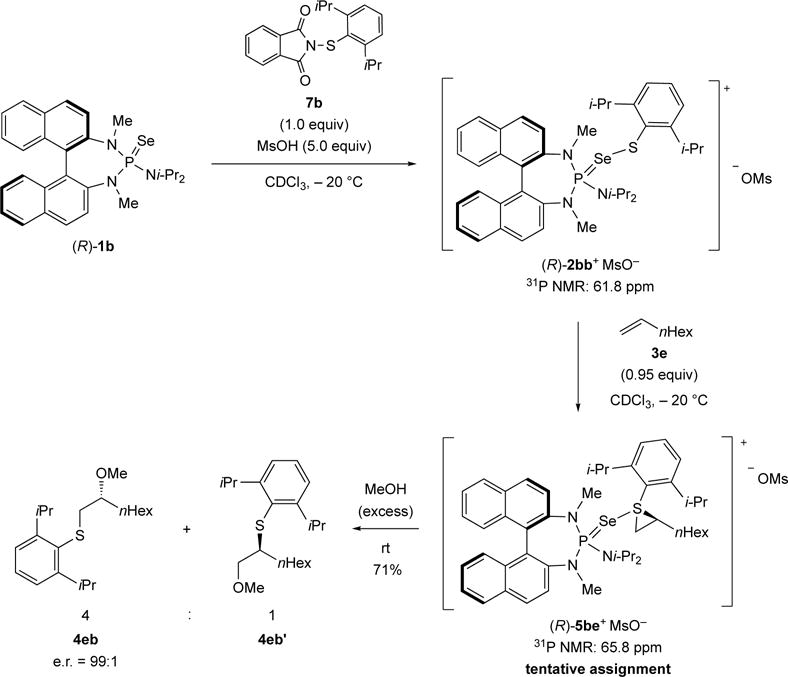
Stoichiometric generation of active species (R)-2bb+ MsO− and its reaction with 1-octene (3e)
Table 2.
Substrate scope with new sulfenylating agents.
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Alkene 3 | Sulfenylating agent 7 | Product 4 | Temp [°C] | Time [h] | Yield [%]e | e.r.i | |||
| 1 |

|
3a |

|
7a |

|
4aab | −20 | 36 | 78 | 95.6:4.4k |
| 2 |

|
3a |
|
7f |

|
4afa | −20 | 36 | 84 | 97.6:2.4k |
| 3 |

|
3a |

|
7h |

|
4aha | −20 | 36 | 87f | 98.4:1.6j,k |
| 4 |

|
3a |

|
7b |

|
4aba | 0 | 48 | 87 g | 99.3:0.7j,k |
| 5 |
|
3g |

|
7a |

|
4gab | 0 | 36 | 83 | 88.9:11.1k |
| 6 |
|
3g |

|
7b |

|
4gba | 0 | 36 | 94 | 98.6:1.4l |
| 7 |
|
3b |
|
7a |

|
4bab, c | −20 | 48 | 77 | 97.4:2.6l |
| 8 |
|
3b |

|
7b |

|
4bba, c | 0 | 36 | 85 | 99.2:0.8l |
| 9 |
|
3e |

|
7a |

|
4eab, c | 23 | 24 | 75 h | 87.8:12.2l |
| 10 |
|
3e |

|
7b |

|
4eba, c | 23 | 24 | 84h | 98.6:1.4l |
| 11 |
|
3h |

|
7a |

|
3haa, d | 0 | 48 | 85 | 83.7:16.3k |
| 12 |
|
3e |

|
7b |

|
3hba, d | 23 | 24 | 72 | 94.3:5.7k |
General reaction conditions: 3 (1.0 mmol), 7 (1.0 mmol), MsOH (1.0 mmol), (R)-1b (10 mol%), CH2Cl2 (0.2 M).
Reaction of literature described compounds21 conducted on 0.2 mmol scale.
MeOH used as external nucleophile.
0.5 equiv MsOH used.
Yield of isolated products.
Combined yield of a 97:3 mixture of tetrahydropyran/-furan.
Combined yield of a 93:7 mixture of tetrahydropyran/-furan.
Combined yield of a 4:1 mixture of 4e and its constitutional isomer (Supporting Information).
Absolute configuration assigned by comparison and in analogy to literature described compounds.
e.r. of the major isomer.
Determined by CSP-SFC analysis.
Determined by HPLC analysis.
Because putative intermediate (R)-5be+ MsO− was stable toward moisture and air, it was decided to generate this intermediate from the isolated active species (R)-2bb+ BArF24− (prepared as shown in Scheme 9) to avoid the presence of MsOH and phthalimide. Thus, (R)-2bb+ BArF24− was combined with an excess of 3e to form (R)-5be+ BArF24− as indicated by the 31P NMR chemical shift of 65.5 ppm (Scheme 15). After removal of excess alkene and CH2Cl2, intermediate (R)-5be+ BArF24− was obtained as a yellow solid that was stable to air and moisture. From an analysis of the 1H NMR spectrum of the intermediate, the relevant signals that belong to the episulfurane intermediate can be identified at 2.71 ppm (methine proton), 2.19 ppm and 1.65 ppm (diastereotopic methylene protons). However, from the coupling pattern and the coupling constants it is difficult to unambiguously assign whether these signals correspond to the proposed structure (R)-5be+ BArF24− or rather the Lewis base-opened structure (R)-6be+BArF24− (lower, Scheme 15). Attempts to crystallize the species were unsuccessful.
Scheme 15.

Stoichiometric reaction of (R)-5be+ BArF24− and 1-octene (3e).
3. Synthesis and Evaluation of New Lewis Base Catalysts
According to the proposed model for the stereochemical outcome of the sulfenofunctionalization,[24b] modification of the binaphthyl backbone was expected to have an impact on the stereochemical outcome. This assumption was corroborated by the insights gained from the X-ray crystal structure of (±)-2aa+ implicating a role for the biaryl backbone in the interaction with the approaching alkene during the stereodetermining thiiranium ion ring formation. Consequently, a Lewis base catalyst with substituents in the 6,6′-position of the binaphthyl backbone was synthesized first (Scheme 16). The synthesis involved site selective bromination of the enantiomerically enriched dicarbamate (S)-8 followed by a Suzuki coupling to yield the 6,6′-biphenyl-substituted dicarbamate (S)-9. After reduction of (S)-9 to the N,N-dimethylated diamine (S)-10 using LiAlH4, catalyst (S)-1d was obtained by deprotonation of (S)-10 with n-BuLi and reaction with (diisopropylamino)phosphorus dichloride followed by oxidation of the P(III)-intermediate with elemental selenium.
Scheme 16.
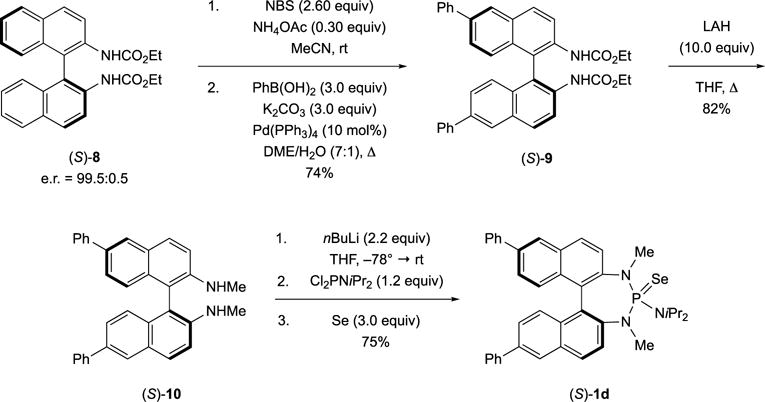
Synthesis of a 6,6′-diphenyl-substituted binaphthyl-derived Lewis base (S)-1d.
In addition to 6,6′-substitution, the introduction of substituents in the 3,3′-position was also investigated to achieve higher enantioselectivities. However, concomitant with the increase of steric demand a decrease in reaction rate has to be expected. Therefore, we decided to conduct the synthesis of the 3,3′-diphenyl-substituted binaphthyl-derived Lewis base first in racemic fashion and evaluate its general capability in catalyzing sulfenofunctionalizations.
A Pd-catalyzed, directed C–H arylation[43] was the key step for the introduction of phenyl groups ortho to the acetamido moieties ((±)-11 → (±)-12, upper, Scheme 17). After formation of the dicarbamate, its reduction using LAH yielded compound (±)-13. This reduction was slower than with (S)-9 and a higher boiling solvent (1,4-dioxane) was required to drive the reaction to completion. By analogy to the synthesis of (S)-1d Lewis base catalyst (±)-1e was obtained using the established procedures.
Scheme 17.

Synthesis of a 3,3′-diphenyl-substituted binaphthyl-derived Lewis base (±)-1e.
Further catalyst modification focused on increasing the p-surface area on the chiral backbone to have a beneficial effect on the enantiotopic face selection at the alkene. Therefore, a bipyrene-derived Lewis base catalyst was synthesized (Scheme 18). At the outset, reduction of pyrene (14)[44] was followed by a selective nitration[45] to give compound 15. Rearomatization gave access to 2-nitropyrene, which was reduced to 2-aminopyrene (16).[42] Subsequently, an oxidative coupling mediated by MnO2 yielded the desired bipyrenyl diamine.[46] Since the diamine was not particularly stable, it was immediately converted to the dicarbamate (±)-17 which was then reduced to (±)-18. Numerous attempts to resolve racemic (±)-18 by fractional crystallization with various chiral acids were unsuccessful. Finally, preparative HPLC was exploited for this separation.[47] The synthesis of the Lewis base (R)-1f was achieved by analogy to the aforementioned strategy, however, unreacted starting material (R)-18 (ca. 15%) was always isolated along with catalyst (R)-1f. Attempts to separate the starting material by column chromatography resulted in decomposition of the catalyst. Also, recrystallization did not provide satisfactory results. The catalyst/starting material mixture was used for the test reactions.
Scheme 18.

Synthesis of a bipyrene-derived Lewis base catalyst (R)-1f.
With the new Lewis base catalysts now in hand, their performance in sulfeno-functionalization reactions was tested (Scheme 19). For this purpose the well-investigated sulfenoetherification[22] was chosen as reference reaction. In addition, sulfenoaminations[25] were examined. The enantiomerically enriched 6,6′-diphenyl-substituted selenophosphoramide (S)-1d afforded good results with regard to conversion and enantioselectivity in both reactions. Despite the fact that no improvement was achieved with this particular catalyst, the high enantioselectivities give reason to hope for an improvement employing other, more bulky substituents in the 6,6′-position. Investigations toward 3,3′-substitution on the binaphthyl moiety revealed that reactions in the presence of catalyst (±)-1e are too slow, even at room temperature, so that the background reaction would erode any enantioselectivity using the enantiomerically enriched Lewis base. However, other substituents in this position have yet to be tested. The bipyrene-derived selenophosphoramide (R)-1f was successfully employed in sulfenofunctionalizations. Using the enantiomerically enriched catalyst an e.r of 94:6 was obtained in the sulfeno-etherification reaction. Despite the high enantioselectivity no improvement was achieved and, in the light of the synthetic effort required to access this catalyst, it would be reasonable to pursue the synthesis of other scaffolds, e.g. phenanthrene-derived selenophosphoramides.
Scheme 19.

Evaluation of new Lewis base catalysts in sulfenofunctionalization reactions.
To reduce the steric bulk at the 3,3′-position, the synthesis of a biphenathrene-based catalyst was investigated (Scheme 20). This substitution pattern should constitute a more rigid system that might combine high enantioselectivity with sufficiently high reactivity. The initial approach for the synthesis of the biphenanthrene backbone encompassed the oxidative coupling of 2-aminophenathrene (19) using a MnO2-mediated coupling[44] which resulted in the formation of the undesired carbazole 20 (first row, Scheme 20).[48] Next, Ullmann-type coupling reactions were examined (second and third row, Scheme 20). Unfortunately, these attempts resulted in debromination or no reaction at all. Finally, an attempted Suzuki-coupling was also unsuccessful (fourth row, Scheme 20).
Scheme 20.

Attempted synthesis of a biphenanthrenyldiamine.
The failure to access the biphenanthrenyldiamine backbone from precursors already bearing the nitrogen substituent directed efforts to the Bucherer reaction as a way to access the desired structure starting from 9-phenanthrol (26) and hydrazine.[49] Although this method is effective for the synthesis of BINAM, with 9-phenanthrol carbazole 20 was obtained (upper, Scheme 21).[43] Similarly, starting from the readily available biphenanthrol and MeNH2, the N-methylated carbazole was obtained as the major product (lower, Scheme 21). In view of the effort required for the synthesis of new Lewis base catalysts, at this point it was decided to pursue a different strategy to increase the enantioselectivity, namely changing the sulfenylating agent.
Scheme 21.

Attempted synthesis of a biphenanthrenyldiamine starting from 9-phenanthrol (26) a biphenanthrol (27), respectively.
4. Synthesis and Evaluation of New Sulfenylating Agents
To date, the most selective catalyst for the sulfenofunctionalization is the BINAM-derived selenophosphoramide 1b (vide infra, Table 1 and Table 2). Prior optimization studies identified the importance of a methyl substituent on the internal nitrogen atoms as well as the isopropyl groups on the external nitrogen.[50] As described in the previous section, attempts to further enhance the catalyst performance by modifying the biaryl backbone have not been successful. An alternative approach to improve the enantioselectivity of the process would be to change the sulfenylating agent (Scheme 22). Insights gained from the X-ray crystal structure of (±)-2aa+ BArF24− implicate a role for the arylsulfenyl group in interaction with the approaching alkene during the stereodetermining thiiranium ion ring formation. Thus, it appeared plausible that electronic and particularly steric properties of the arylsulfenyl group could impact the stereochemical outcome of the reaction. To test this hypothesis, a variety of electronically and sterically disparate N-arylsulfenylphthalimides were synthesized and tested.
Table 1.
Evaluation of new sulfenylating agents in the sulfenoetherification of 3a.

| |||||||
|---|---|---|---|---|---|---|---|
| Entrya | Sulfenylating agent 7 | Product 4 | Time [h] | Conversion [%] | e.r.b | ||
| 1 |

|
7a |

|
4aa | 24 | 93 | 95.3:4.7 |
| 2 |
|
7c |
|
4ac | 24 | 100 | 95.1:4.9 |
| 3 |

|
7d |

|
4ad | 24 | >95 | 95.6:4.4 |
| 4 |

|
7j |

|
4aj | 51 | 88 | 95.7:4.3 |
| 5 |

|
7e |
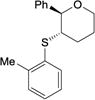
|
4ae | 21 | >95 | 97.2:2.8 |
| 6 |

|
7f |

|
4af | 21 | 100 | 97.1:2.9 |
| 7 |

|
7g |

|
4ag | 42 | >95 | 97.5:2.5 |
| 8 |

|
7h |

|
4ah | 24 | >95 | 98.0:2.0 |
| 9 |

|
7i |

|
4ai | 48 | 67 | 96.3:3.7 |
| 10 |

|
7b |

|
4ab | 65 | 70 | 99.1:0.9 |
| 11 |

|
7b |

|
4ab | 24 c | 80 | 99.0:1.0 |
| 12 |

|
7b |
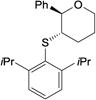
|
4ab | 12 d | 100 | 98.0:2.0 |
| 13 |

|
7k |

|
4ak | 24 | 80 | 98.0:2.0 |
| 14 |

|
7l |

|
4al | 48 | 80 | 94.0:6.0 |
| 15 |

|
7m |

|
4am | 21 | 95 | 92.6:7.4 |
| 16 |

|
7n |

|
4an | 48 | 90 | 91.8:8.2 |
General reaction conditions: 5 mm NMR tube, 4a (70.0 μmol), 7 (74.0 μmol), MsOH (70.0 μmol), (R)-1b (10 mol%), CDCl3 (0.11 M), −20 °C. Conversion was determined by assuming that, besides small quantities of the respective tetrahydrofuran (<5%), 3a was converted only to 4a, as no other significant product was detected by 1H NMR spectroscopy.
Determined by CSP-SFC analysis.
Reaction conducted at 0 °C.
Reaction conducted at room temperature
Scheme 22.
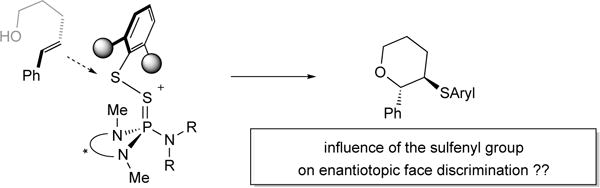
Influence of the sulfenylating agent on enantioselectivity.
4.1 Synthesis of Thiophenols
At the outset, thiophenols 30b, h, and l were synthesized by adapting a procedure used by Rundel[51] for the synthesis of sterically demanding thiophenols. Formation of the Grignard reagent from the corresponding aryl bromide was followed by capture with elemental sulfur and reduction with LiAlH4 (Scheme 23). Generally, moderate to good yields were obtained using this protocol. The 2,6-diisopropoxythiophenol (30k) was accessed by ortho-lithiation[52] and subsequent capture with elemental sulfur, however, the reaction did not proceed to full conversion and a mixture of 30k starting compound 31k was isolated. Attempts to separate the two compounds using chromatography were unsuccessful so that the mixture was used for the synthesis of the corresponding N-arylsulfenylphthtalimide. All other aryl thiophenols are commercially available.
Scheme 23.

Synthesis of thiophenols.
4.2 Synthesis of New Sulfenylating Agents
The synthesis of sulfenylating agents was accomplished using a slightly modified procedure from Reese in which the corresponding thiophenol was combined with bromine and phthalimide (Scheme 24).[53] Because conditions were not optimized for the individual thiophenols, the yields for the respective sulfenylating agent ranged from low (e.g. 18% for 7n) to high (e.g. 91% for 7c). Despite varying yields, this protocol provided access to the desired products. Only 4-nitrothiophenol was not compatible with these conditions. Thus, to obtain the desired sulfenylating agent (7j), 4-nitrophenylsulfenyl chloride was combined with phthalimide in the presence of Et3N. All of the sulfenylating agents were crystalline solids allowing for their purification by recrystallization.
Scheme 24.

Synthesis of New Sulfenylating Agents
4.3 Evaluation of New Sulfenylating Agents
To test the hypothesis that steric and electronic properties of the arylsulfenyl group could impact the stereochemical outcome of the reaction, N-arylsulfenylphthalimides 7a–7n were evaluated in the sulfenoetherification of hydroxyl substituted alkene 3a using selenophosphoramide (R)-1b as the catalyst (Table 1). The enantiomeric composition of the resulting tetrahydropyrans 4ab–4an were compared against the those obtained for the phenylsulfenyl substituted product 4aa (95.3:4.7 e.r., entry 1). Interestingly, almost no electronic effect on enantioselectivity was observed regardless of changes in the electron density of the phenyl ring (entries 2–4). More striking still was the lack of a significant electronic effect on reaction rate varying by only a factor of 2–3 between 7c (4-methoxy) and 7j (4-nitro).
To evaluate the influence of steric effects, different ortho-substituents were installed into the arylsulfenyl moiety. The presence of an ortho-methyl group increased the enantioselectivity of the reaction to 97.2:2.8 e.r. for tetrahydropyran 4ae (entry 5). Essentially the same ratio of enantiomers was obtained for product 4af containing two ortho-methyl substituents (entry 6). Surprisingly, no decrease in reactivity was encountered with these sulfenylating agents. On the contrary, the rate of formation of product 4af was faster as compared to that for 4aa and 4ae. Although a number of different, subtle factors could be contributing, this small increase could arise owing the electron donating effect of the methyl groups. Further increasing the steric bulk around the sulfur atom by incorporating ethyl substituents again led to a slight enhancement in enantioselectivity (entries 7, 8) Logically, the impact of ortho-isopropyl groups was next investigated (7i, 7b). Whereas monoisopropyl substituted product 4ai was obtained with somewhat diminished enantiomeric purity (96.3:3.7 e.r., entry 9), the sulfenoetherification with 7b bearing two isopropyl groups produced tetrahydropyran 4ab with a remarkably high enantiomeric ratio of >99:1 at −20 °C (entry 10). In this scenario, the reactivity gain from electron donation is obviously outweighed by the steric encumbrance leading to a slow conversion at −20 °C. Gratifyingly, at 0 °C the reaction proceeded at a reasonable rate (80% conv. after 24 h) accompanied by only a marginal erosion of the enantiomeric ratio (entry 11). Even at room temperature a high level of enantiocontrol was achieved (entry 12). For the sake of completeness, other N-arylsulfenylphthalimides 7k–7n were inspected (entries 13–16), all being inferior to sulfenylating agent 7b.[29]
Apart from the improvement of the enantioselectivity, another intriguing observation was made, namely the marginal difference in reaction rate when using electronically disparate sulfenylating agents. Even more surprising is the fact that electron-donating sulfenylating agents seem to accelerate the reaction. This is counterintuitive because it was hypothesized that electron-withdrawing groups on the aryl ring increase the electrophilicity of the sulfur atom rendering it even more reactive toward the nucleophilic attack by the olefin during the rate-determining thiiranium ion formation. Since this was not the case, it was interesting to obtain more quantitative results so as to confirm the initial observations. To that end, a kinetic study using NMR spectroscopic analysis with the new sulfenylating agents was conducted whereby product formation and starting material consumption was measured against an internal standard.
In the series of para-substituted phenylsulfenyl-derived reagents, the electron rich 4-methoxyphenyl (7c) was the fastest (t1/2 ≈ 4 h), whereas the parent reagent 7a and 4-trifluoromethylphenyl substituted 7d reacted at comparable rates (t1/2 ≈ 9 h, Scheme 25). 4-Nitrophenyl derivative 7j was not part of the study due to solubility problems of the sulfenylating agent.
Scheme 25.
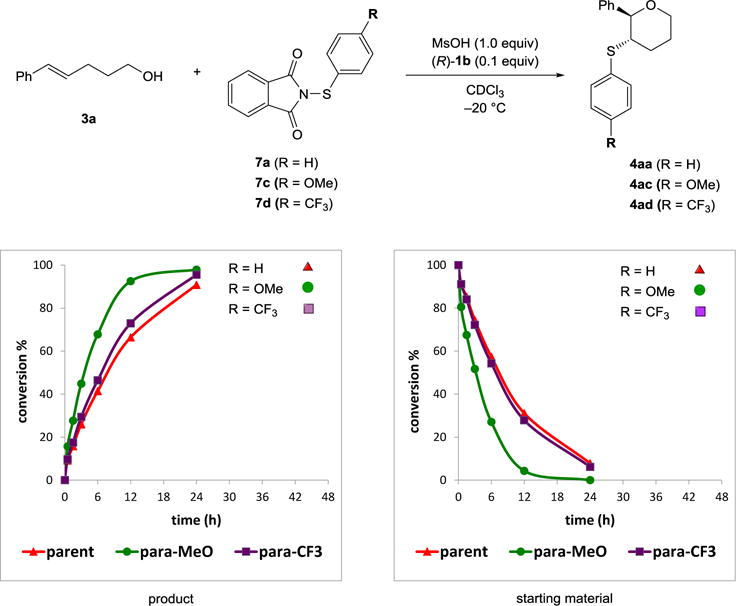
Comparison of reaction rates of para-substituted phenylsulfenylphthalimides.
Comparing ortho alkyl-substituted arylsulfenyl reagents, the methyl-substituted 7e is slightly faster (t1/2 ≈ 6 h) than the parent sulfenylating agent (Scheme 26). The ethyl-substituted 7g, on the other hand, is slower (t1/2 ≈ 12 h), which might be explained by the increased steric bulk of the substituent. As expected, the isopropyl-substituted reagent 7i led to a significantly decreased reaction rate (t1/2 ≈ 30 h).
Scheme 26.

Comparison of reaction rates of ortho-monosubstituted phenylsulfenylphthalimides.
Introduction of a second methyl substituent in the 2-position of the phenyl group (7f) results yet again in an increase of the reaction rate (t1/2 ≈ 3 h, Scheme 27). Also, with a second ethyl group (7h, t1/2 ≈ 6 h), the reaction rate is enhanced as compared to the parent reagent (7a, t1/2 ≈ 9 h) as well as to the mono ethyl-substituted reagent (t1/2 ≈ 12 h, Scheme 27). As expected from previous results, incorporation of a second isopropyl group leads to a very slow reaction. The apparent non-dependency of reaction rate on the alkene concentration (indicated by a straight line for 7b, Scheme 27) suggests a change in the mechanism. Under these conditions, opening of the thiiranium ion might become rate-determining, which would display 0th order kinetics. However, more accurate measurements are needed to confirm this proposition.
Scheme 27.

Comparison of reaction rates of ortho-disubstituted phenylsulfenylphthalimides.
Because the reaction catalyzed by 7b was so slow at −20 °C, the reaction rates at 0 °C and at 23 °C (room temperature) were recorded (Scheme 28. A significant increase in reaction rate was observed at 0 °C (t1/2 ≈ 12 h). Changing from 0 °C to 23 °C, the reaction rate increased again by a factor of 4 (t1/2 ≈ 3 h). As mentioned above, increasing the temperature led to only a marginal erosion of enantiomeric purity (c.f. Table 1, entries 10–12).
Scheme 28.

Comparison of reaction rates of 7b at different temperatures.
For the sake of completeness, N-alkylsulfenylphthalimides have also been examined in the course of this study (Scheme 29). However, these sulfenylating agents afforded generally slower conversion or no reaction at all (as compared to N-arylsulfenylphthalimides). For instance, methyl-substituted 7o did not proceed to full conversion within 48 h at −20 °C. Isobutyl-substituted 7p and benzyl-substituted 7q did not react at −20 °C at all, and even at room temperature the reaction was relatively slow. Isopropyl reagent 7r did not react at all, not even at room temperature.
Scheme 29.

Reaction of 4a with N-alkylsulfenylphthalimides.
Since the presence of the catalytically active species was confirmed by 31 P NMR spectroscopy for all of the sulfenylating agents shown in Scheme 30, it can be ruled out that the formation is problematic. Consequently, the differences in reaction rates probably arise from a decreased rate of the thiiranium ion formation. Although the lack of reactivity for 7r could be rationalized by bulkiness of the isopropyl group, the slower reaction of 7o in comparison to 7a (Table 1, entry 1) is difficult to explain by steric effects. Thus, a profound difference with regard to the electronic distribution/delocalization (on the sulfur) can be expected between arylsulfenyl-derived active species on the one hand, and alkylsulfenyl-derived active species on the other.
Scheme 30.
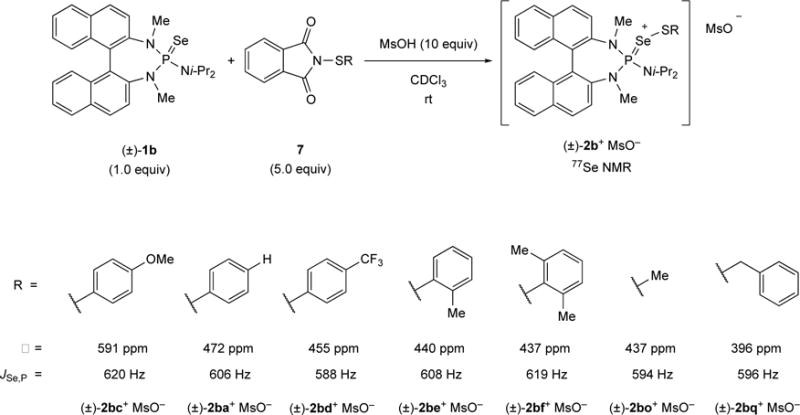
Comparison of the 77Se NMR chemical shifts of different active species.
To obtain additional insights into the electronic perturbation in the proximity of the reactive sulfur center, the 77Se NMR chemical shift of the active species was considered as a potential experimental indicator. Generation of the active species in stoichiometric quantities was achieved using the conditions shown in Scheme 30 and the 77Se NMR chemical shifts were recorded at room temperature. The 4-MeO-substituted species (±)-2bc+ displayed the most downfield shifted signal at 591 ppm. On the other hand, the alkyl-derived (±)-2bo+ and (±)-2bq + display the highest upfield shifts at 437 ppm and 396 ppm, respectively. If the ortho-substituted arylsulfenyl derived active species (±)-2be + and (±)-2bf + are ignored in this series, a trend can be established, namely that an increased down field shift of the selenium atom accounts for higher reactivity. However, when (±)-2be + and (±)-2bf + are considered, there is no obvious correlation between the chemical shift of the active species and its reactivity. The increased reactivity of (±)-2be + and (±)-2bf + compared to (±)-2ba+ might be a result of electron donation (by analogy to (±)-2bc+), but it could also arise from a better accessibility of the sulfur atom in the active species. Assuming that additional substituents in the ortho-position of the aryl group can push the reactive Se=S–Aryl moiety away from the binaphthyl backbone and render the reactive sulfur center more accessible, an increase in reaction rate is conceivable. At this point, it is not possible to rationalize the reaction rate differences by either steric or electronic effects, most likely it is combination of both.
4.4 Substrate Scope
The general applicability of sterically demanding N-arylsulfenylimides as efficient reagents for the highly enantioselective sulfenoetherification of other substrate classes was next evaluated (Table 2). At first, reaction of 3a was conducted on a preparative scale with 7a, 7b, 7f, and 7h to confirm the results from the initial survey (entries 1–4). Subsequently, representative alkenes were subjected to the sulfenoetherification using sulfenylating agent 7b. Because a different selenophosphoramide had been used in the originally reported conditions,[21] reactions with the parent sulfenylating agent 7a and catalyst (R)-1b were run in parallel to provide a valid comparison with 7b. A significant improvement of enantioselectivity was seen in the reaction of terminal alkene 3g and 7b as compared to 7a (entry 5 vs. 6). Despite the already high enantiopurity of product 4ba obtained from an intermolecular sulfenoetherification of 4-octene (3b), 7a and MeOH, the enantiopurity of the corresponding product 4bb was further increased by using 7b (entry 7 vs. 8). The suitability of a terminal double bond was also showcased in an intermolecular functionalization with 1-octene (3e) and MeOH as the nucleophile (entry 9 vs. 10). Product 4eb was accessed with excellent enantioselectivity (98.6:1.4), whereas 4ea gave only a moderate selectivity which is in line with stereochemical outcome previously observed with terminal alkene 4bb. Apart from the sulfenoetherification, we were able to demonstrate the benefit of 7b in a sulfenoamination reaction[23] using alkenyl sulfonamide 3h. Again an enhanced enantioselectivity was noted with 7b forming product 4hb in 94.3:5.7 e.r. (entry 11 vs. 12).[29]
Although enantiotopic face differentiation was improved for terminal and E-1,2-disubstituted alkenes, 1,1-disubstituted and trisubstituted double bonds did not respond to this modification affording comparable results with 7a and 7b. For instance, reaction of the 1,1-disubstituted alkene 3i afforded tetrahydropyran 4ib with low enantioselectivity (e.r. = 65:35) which is in the same range as with sulfenylating agent 3ia[21] (upper, Scheme 31). The trisubstituted alkene 3j and 7b afforded a moderate e.r. of 85:15 for the corresponding tetrahydropyran 4jb (lower, Scheme 31) providing no improvement compared to 7a. [22] Because the reactions were conducted at 0 °C (instead of −20 °C), both of the substrates 3i and 3j suffered proton-initiated cyclization which resulted in the formation of 4ib′ and 4jb′, respectively.
Scheme 31.

Evaluation of other alkene classes.
Discussion
1. Isolation of the Catalytically Active Species and Its Reactivity
To understand the factors that have an impact on the reactivity and selectivity of the reaction, a refinement of the mechanistic picture of the Lewis base catalyzed sulfenofunctionalization was desired. A major part of the work was dedicated to the isolation and crystallographic characterization of the Lewis base/sulfenylating agent adduct (±)-2aa+BArF24−. Because it had already been shown that selenophosphoramides undergo a disproportionation when reacted with [PhS(SMe2)]SbCl6,[29] the first modification involved the use of thiophosphoramides to avoid this undesired reaction. However, with sulfur as donor atom, an equilibrium between the free Lewis base and the sulfenylated Lewis base was observed. This result suggests that Me2S is not innocent in the process allowing for the establishment of the equilibrium. Therefore, a method for the irreversible formation of (±)-2aa+X− was investigated using PhSCl and NaBArF24. This approach allowed for the use of a weakly coordinating anion[30] to crystallinity. These anions display very low nucleophilicity as well as basicity because the negative charge is delocalized over many atoms, they are extremely resistant to oxidation and strong electrophiles, and they improve the solubility properties of ionic complexes. With BArF24− as the counteranion, suitable crystals of [PhS–LB]BArF24 for X-ray analysis were obtained by crystallization from CH2Cl2. As postulated and predicted by computational studies,[29] the SPh-moiety points back toward one of the naphthyl rings, so that the alkene must approach along the trajectory of the σ* orbital of the S–S-bond. This important result guided the search for a more selective Lewis base. In addition, with the information gained from the geometries of the active sulfenylating species, more quantitative computational studies of the transition state of the transformation were conducted. These studies allowed for a thorough analysis of the origin of the enantioselectivity of the Lewis base catalyzed sulfenofunctionalization.[29]
An important result that substantiates the role of (±)-2aa+ BArF24− as the active sulfenylating agent was obtained by its stoichiometric reaction with substrate 3a to form the thioetherification product 4aa (Scheme 10). Furthermore, the accessibility of the catalytically active species enabled an initial investigation toward the fate of the Lewis base after transfer of the sulfenium ion to the alkene. It has been postulated that the Lewis base resides in the periphery of the thiiranium ion because for intermolecular sulfenofunctionalizations, the site selectivity of the nucleophilic attack at the thiiranium intermediate is dependent on the chosen catalyst. Indeed, the findings presented in Results Section 2 are consistent with the formation of an episulfurane intermediate[42] upon combination of an alkene and (±)-2aa+ BArF24− in the absence of a nucleophile. Although spectroscopic analysis does not allow for an unambiguous conclusion, the fact that nucleophilic capture of the observed intermediates results in the formation of products with the expected anti-relative configuration is compelling. The stereochemical outcome was confirmed by comparison to known compounds or authentic samples.
The stoichiometric reaction between (R)-2bb and 1-octene shown in Scheme 15 enabled the observation of successively formed active species, including the reaction with the alkene and nucleophilic capture of the newly formed intermediates. The constitutional isomer composition as well as the enantiomeric ratio of the product from this experiment matched exactly the results from the catalytic thiofunctionalization reaction. Therefore, it is likely that same pathways are taken in the stoichiometric and the catalytic reaction, and this has to involve the formation a thiiranium ion. Moreover, the 31P NMR chemical shift of the putative episulfurane intermediate (R)-5be+ BArF24− (65.5 ppm) is in accordance with the major species (31P NMR: 65.8 ppm) that is observed during the catalytic reaction (at −20 °C). The catalytically active species (R)-2bb+, on the other hand, is the minor component observed during the catalytic reactions. This fact indicates that particularly for the intermolecular sulfenofunctionalization of a terminal alkene the turnover-limiting step is likely the opening of the thiiranium ion. Overall, quantitative kinetics have to be acquired to confirm this hypothesis.
Despite all these observations, we cannot rule out that the observed signal in the 31P NMR represents a species that is formed by nucleophilic opening of the thiiranium ion by the Lewis base itself.[41e] This kind of intermediates have been described previously.[41d,e,g] If so, this species would need to be in equilibrium with the episulfurane intermediate since its nucleophilic capture furnishes the anti-configured products exclusively. The 1H NMR spectrum of the observed intermediates is ambiguous because the geometry at sulfur in the episulfurane must be either trigonal bipyramidal[41d,f] or square pyramidal[41d] thus rendering this species unsymmetrical. It is also difficult to assign the structure on the basis of the observed couplings in the 1H NMR spectra. A possible way to confirm the formation of an Lewis base opened intermediate such as (R)-6be+ BArF24− (lower, Scheme 15) would be a 1H,77Se-2D experiment that can establish a correlation between the methylene protons on the terminal carbon and the selenium atom. Also, we have confirmed that the observed intermediate is rather stable to air and moisture and even its nucleophilic capture is relatively slow. These observations offer grounds for hope that its crystallization for X-ray structure determination is achievable with an appropriate alkene.
2. Improvement of Enantioselectivity
As an ongoing part of our investigations toward the improvement of the enantioselectivity in the Lewis base-catalyzed sulfenofunctionalization of alkenes, the synthesis of new catalyst scaffolds was conducted. Important conclusions can be drawn from these studies (upper, Scheme 19). Substitution of the binaphthyl backbone at 6,6′-position as well extension of the p-surface in the remote part of the catalyst appear to have little or no impact at all on the enantiotopic face discrimination. On the other hand, 3,3′-substituents decrease the reaction rate to an extent such that the racemic background reaction would override any potentially intrinsically high enantioselection by the chiral Lewis base. Hence, a proper balance between steric demand at this position and the efficacy of the catalyst with respect to reaction rate has to be found. Our idea was to reduce the steric bulk at the 3,3′-position by incorporating the substituent into a cyclic framework as it is the case in the biphenanthrene-based catalyst 32 (Scheme 32). Unfortunately, the synthesis of the biphenanthryl diamine backbone 36 has not yet been successful.
Scheme 32.

New Lewis base catalysts.
A route that could enable the synthesis of this motif is based on the Buchwald-Hartwig reaction (middle, Scheme 32). The known biphenanthrol (27) could be converted into a bismesylate or bistosylate (34) that in turn could be reacted with benzylmethylamine to give 35. Transition metal catalyzed coupling reactions using benzylmethylamine are reported. After reductive removal of the benzyl group, the immediate precursor (36) for the synthesis of the catalyst 32 could be accessed.
Moreover, the introduction of aryl groups at the 4,4′-position can be considered 33, upper, Scheme 32). Regarding the steric demand at the active site of the catalyst, this scaffold should be an intermediate between the 3,3′-disubstituted and the bipyrene-derived Lewis base. Its potential to differentiate the enantiotopic faces of the alkene substrate is certainly of interest.
Direct functionalization of the parent binaphthyldiamine would be an attractive approach for the synthesis of 33. To achieve this, the meta-selective Cu(II)-catalyzed C–H-bond arylation developed in the Gaunt laboratories can be exploited (lower, Scheme 32).[54] Under these conditions the aryl group is introduced selectively meta to the directing amido-substituent. As exemplified, biphenyls (37) serve as appropriate substrates for this reaction. Application of the method to pivalate- or carbamate-protected binaphthyldiamine (8) seems to be worth investigating. This kind of functionalization would provide a straightforward access to the desired scaffold (39) and could be carried out on already enantiomerically enriched precursors.
Apart from efforts to increase the enantioselectivity of the Lewis base-catalyzed sulfenofunctionalization by tuning the catalyst itself, the impact of different phthalimide-derived sulfenylating agents was investigated (Results Section 4). Electronic modifications in the para-position had essentially no effect on the stereochemical outcome suggesting that discrimination of the enantiotopic faces of the alkene is predominantly governed by steric effects. This hypothesis was verified by the incorporation of ortho-substituents. With increased steric bulk of the substituents the enantiomeric ratio of the corresponding products increased. Obviously, with this modification, the system reacts more sensitive to how the alkene is approaching. The generality of this modification was demonstrated with other substrate classes (Table 2). However, although enantiotopic face differentiation was improved for terminal and E-1,2-disubstituted alkenes, 1,1-disubstituted and trisubstituted double bonds did not respond to this modification affording comparable results with 7a and 7b. With the current mechanistic understanding, it is not surprising that no improvement in enantioselectivity is observed for these alkene classes. The reason for this is the topology of the given catalyst and the active species, respectively. Having diagonally symmetric quadrants of occupied and unoccupied space, the catalytically active species is suited for differentiating the enantiotopic faces of E-alkenes, but not other alkenes. Hence, for 1,1-disubstituted and trisubstituted alkenes as well as for Z-configured 1,2-disubstituted substrates new catalyst structures have to be tested or developed.
Although not the major focus of the study, interesting trends were observed with regard to reactivity of the new sulfenylating agents. For instance, when the electronic nature of the sulfenylating agents was significantly changed through incorporation of para-substituents the reaction rates changed only slightly or moderately. Contrary to our understanding, electron-donating groups such as MeO increased the reaction rate. This is counterintuitive because we assumed that electron-withdrawing groups on the aryl ring would increase the electrophilicity of the sulfur atom rendering it even more reactive toward the nucleophilic attack by the alkene. Maybe the fact that the sulfur atom has to contribute a lone pair to the formation of the thiiranium ring as well cannot be neglected. Considering this, it appears plausible that electron donation accounts for a higher readiness of the sulfur atom to share its lone pair. This would mean that electron-withdrawing substituents should decrease the reaction rate. However, this is not the case with a CF3-group that displays results comparable to the parent sulfenylating agent. Overall, more subtle effects might influence the reaction rate and the observed differences are not large enough to allow for an obvious interpretation. Especially the increased reaction rates with ortho-substituted derivatives could be a result of two concurrent effects: In accordance to the para-OMe derivative, an increased electronic density might be one of the reasons. The other reason might involve steric repulsion between the arylsulfenyl group and one of the naphthyl rings rendering the reactive sulfur atom more accessible.
Interestingly, alkyl-derived sulfenylating afforded slower reaction rates or no reaction at all (Scheme 29). Since the catalytically active species derived from these sulfenylating agents was confirmed by 31P NMR spectroscopy the failure to generate this species can be ruled out. Consequently, the differences in reaction rates probably arise from a decreased rate of the thiiranium ion formation. Thus, a profound difference with regard to the electronic distribution/delocalization (on the sulfur) can be expected between arylsulfenyl-derived active species on the one hand, and alkylsulfenyl-derived active species on the other hand. Attempts to quantify the expected differences by 77Se NMR were not successful since no obvious trends could be established.
Another explanation is provided by the ability of the Lewis base to act as a leaving group from different active species (Scheme 33). Assuming that the approaching alkene displaces the Lewis base through the attack at the sulfur atom, the displacement is expected to occur more readily at a “benzylic” sulfur atom (middle, Scheme 33) as compared to an unactivated α-alkyl sulfur atom (left, Scheme 33). This picture contradicts the assumption that the Lewis base resides on the sulfur during the thiiranium ion formation. However, if the tendency to form a tetracoordinate episulfurane type intermediate correlates with leaving group properties of the Lewis base in the differently substituted active species, this suggestion could still be valid. If this assumption is correct, acylated derivatives should undergo a rapid thiiranium ring formation because the sulfur atom is located at an α-carbonyl position (right, Scheme 33) which is known to easily undergo SN2 displacements. Therefore, N-acylsulfenylphthalimides should be tested for the sulfenofunctionalization reaction. The steric bulk and thus a potential influence on the enantioselectivity can be adjusted by judicious choice of the R′ group.
Scheme 33.

Differently substituted active species.
Conclusions
In conclusion, spectroscopic, crystallographic and preparative investigations have shed light on the mechanistic pathway of the Lewis base catalyzed sulfenofunctionalization. Initially, the catalytically active species was identified by studying the kinetic parameters[29] of the reaction. Its isolation and crystallographic characterization, provided crucial insights into the factors that govern the stereochemical course of the reaction. These insights supported our empirical efforts in synthesizing new Lewis base catalysts with modified biaryl backbones. Although no improvement was achieved with this approach, important conclusion can be drawn with regard to the design of new catalysts, particularly in combination computational docking studies. Gratifyingly, using the information gained from the solid-state structure of the catalytically active species, the enantioselectivity of the process was improved by optimizing the sulfenylating agent. These insights are guiding the design of more selective Lewis base catalysts.
Experimental Section
Typical procedure for Lewis base catalyzed, enantioselective sulfenofunctionalization of alkenes
Following the literature procedure,[22] an oven-dried Schlenk flask was charged with sulfenylating agent 7b (351 mg, 1.03 mmol, 1.03 equiv), catalyst (R)-1b (51.0 mg, 0.098 mmol, 0.098 equiv), substrate 3a (162 mg, 1.00 mmol) and CH2Cl2 (5.0 mL). The flask was capped with a septum and placed into an i-PrOH bath, which was cooled to 0 °C using a Cryocool unit. The temperature of the mixture was monitored via a thermocouple digital temperature probe. After the temperature stabilized, MsOH (65 μL, 1.0 mmol, 1.0 equiv) was added via syringe and the mixture was allowed to stir for 48 h at 0 °C. Upon complete reaction (TLC monitoring), the mixture was quenched while cold by the addition of Et3N (0.20 mL). The mixture was poured into aqueous HCl (1.0 M, 20 mL) in a separatory funnel, CH2Cl2 (30 mL) was added and the layers were thoroughly mixed. The organic layer was poured into aqueous NaOH (1.0 M, 20 mL), and the layers were thoroughly mixed and then separated. The acidic layer was back-extracted with CH2Cl2 (30 mL) which was poured into the basic layer and used to extract that layer as well. Both organic portions were combined, dried over MgSO4, filtered through glass wool and concentrated in vacuo (20–23 °C, 20 mmHg). Purification by flash column chromatography (SiO2, 65 g, 35 mm Ø, hexane/MTBE, 60:1) afforded 310 mg (87%) of a 93:7 mixture of 4ab and the corresponding tetrahydrofuran (4ab′) as a pale yellow oil. Partial separation of isomers was accomplished by flash column chromatography using high porosity silica gel (SiO2, 65 g, 35 mm Ø, hexane/MTBE, 80:1→5:1) yielding 271 mg of a 97:3 mixture of 4ab and 4ab′ and 30 mg of a 60:40 mixture of 4ab and 4ab′. Rf = 0.44 4ab and 4ab′ and (hexanes/EtOAc, 15:1). [α]D24 = −9.7 (c = 0.27, CHCl3). IR (NaCl plate): 3054, 3026, 2960, 2864, 2718, 2360, 2333, 1941, 1866, 1801, 1573, 1493, 1454, 1420, 1381, 1360, 1325, 1307, 1260, 1226, 1178, 1106, 1079, 1053, 1028, 969, 943, 905, 883, 820, 801, 773, 754. 1H-NMR (500 MHz, CDCl3): 1.09 (br d, J = 6.5 Hz, 6H, CH3), 1.12 (d, J = 6.9 Hz, 6H, CH3), 1.60–1.72 (m, 3H, CH2 and CH), 1.85–1.95 (m, 1H, CH), 2.91–3.00 (m, 1H, CH), 3.45–3.71 (m, 3H, CH2 and CH), 4.04–4.10 (m, 1H, CH), 4.25 (d, J = 10.1 Hz, 1H, CH), 7.09 (d, J = 7.6 Hz, 2H, CH), 7.26 (t, J = 7.4 Hz, 1H, CH), 7.32–7.36 (m, 1H, CH), 7.38 (t, J = 8.0 Hz, 2H, CH), 7.45 (d, J = 7.9 Hz, 2H, CH). 13C-NMR (125 MHz, CDCl3): 23.8, 24.6, 27.0, 31.3, 31.8, 51.9, 68.6, 84.3, 123.4, 127.4, 128.1, 128.3, 129.0, 129.1, 140.1, 153.8. HR-MS: 355.2096 ([M + H]+, C23H31OS+; calc. 355.2097); SFC: (2R,3S)-4ab, tR 10.06 min (99.3%); (2S,3R)-4ab, tR 5.06 min (0.7%) (Chiralpak OD, 10% MeOH in CO2, 2.0 mL/min, 220 nm, 40 °C).
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (GM R01-085235) for generous financial support. E. H. thanks the Deutscher Akademischer Austausch Dienst for a postdoctoral fellowship.
Footnotes
Supplementary Material
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/MS-number.
Author Contribution Statement
E. H. planned and carried out all of the experimental work. S.E.D. initiated and directed the project. E. H. wrote the manuscript with the assistance of S.E.D.
References
- 1.Metzner P, Thuillier A. Sulfur Reagents in Organic Synthesis. Academic Press; San Diego, CA: 1994. [Google Scholar]
- 2.Page P, editor. Organosulfur Chemistry: Synthetic Aspects. Academic Press; London: 1995. [Google Scholar]
- 3.Prinsep MR. Sulfur-containing natural products from marine invertebrates. Studies in Natural Products Chemistry. 2003;28:617–751. [Google Scholar]
- 4.a) Rayner CM. In: Organosulfur Chemistry: Synthetic Aspects. Page P, editor. Academic Press; London: 1995. pp. 89–131. [Google Scholar]; b) Mueller WH. Thiiranium Ions as Reaction Intermediates. Angew Chem. 1969;8:482–492. [Google Scholar]; c) Smid VA, Zefirov V, Bodrikov IV, Krimer MZ. Episulfonium Ions: Myth and Reality. Acc Chem Res. 1979;12:282–288. [Google Scholar]
- 5.de la Mare PBD, Bolton R. In: Electrophilic Additions to Unsaturated Systems. 2nd. de la Mare PBD, Bolton R, editors. Vol. 9. Elsevier; Amsterdam: 1982. pp. 198–246. [Google Scholar]
- 6.Marigo M, Wabnitz TC, Fielenbach D, Jørgensen KA. Enantioselective Organocatalyzed α-Sulfenylation of Aldehydes. Angew Chem Int Ed. 2005;44:794–797. doi: 10.1002/anie.200462101. [DOI] [PubMed] [Google Scholar]
- 7.Sobhani S, Fielenbach D, Marigo M, Wabnitz TC, Jørgensen KA. Direct Organocatalytic Asymmetric α-Sulfenylation of Activated C–H Bonds in Lactones, Lactams, and β-Dicarbonyl Compounds. Chem Eur J. 2005;11:5689–5694. doi: 10.1002/chem.200500512. [DOI] [PubMed] [Google Scholar]
- 8.Han Z, Chen W, Dong S, Yang C, Liu H, Pan Y, Yan L, Jiang Z. Highly Enantioselective Organocatalytic Sulfenylation of 3-Aryloxindoles. Org Lett. 2012;14:4670–4673. doi: 10.1021/ol3021176. [DOI] [PubMed] [Google Scholar]
- 9.Guan H, Wang H, Huang D, Shi Y. Enantioselective Oxysulfenalytion and Oxyselenylation of Olefins Catalyzed by Chiral Brønsted Acids. Tetrahedron. 2012;68:2728–2735. [Google Scholar]
- 10.Hamilton GL, Kanai T, Toste FD. Chiral Anion-Mediated Asymmetric Ring Opening of meso-Aziridinium and Episulfonium Ions. J Am Chem Soc. 2008;130:14984–14986. doi: 10.1021/ja806431d. [DOI] [PubMed] [Google Scholar]
- 11.Lin S, Jacobsen EN. Thiourea-Catalysed Ring Opening of Episulfonium Ions with Indole Derivatives by Means of Stabilizing non-Covalent Interactions. Nature Chem. 2012;4:817–824. doi: 10.1038/nchem.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasquato L, Modena G. The Complete and Irreversible Conversion of a cis Carbon-Substituted Thiiranium Ion into the trans Isomer. Chem Commun. 1999:1469–1470. [Google Scholar]
- 13.Owsley DC, Helmkamp GK, Spurlock SN. Episulfonium Salts. I. Carbon vs. Sulfur Attack in the Reaction of Cyclooctene-S-Methylepisulfonium Ion with Nucleophiles. J Am Chem Soc. 1969;91:3606–3609. [Google Scholar]
- 14.Denmark SE, Collins WR, Cullen MD. Observation of direct sulfenium and selenenium group transfer from thiiranium and seleniranium ions to alkenes. J Am Chem Soc. 2009;131:3490–3492. doi: 10.1021/ja900187y. [DOI] [PubMed] [Google Scholar]
- 15.Archer NJ, Rayner CM, Bell D, Miller D. Synthetic Routes to Novel Homochiral Sulfenyl Sulfonium Salts and Their Use as Potential Enantioselective Sulfenylating Agents. Asymmetric Synthesis via Homochiral Thiiranium ions. Synlett. 1994:617–619. [Google Scholar]
- 16.Lucchini V, Modena G, Pasquato L. Enantiopure Thiosutfonium Salts in Asymmetric Synthesis. Face Selectivity in Electrophilic Additions to Unfunctionalised Olefins. J Chem Soc, Chem Commun. 1994:1565–1566. [Google Scholar]
- 17.Denmark SE, Beutner GL. Lewis Base Catalysis in Organic Synthesis. Angew Chem Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 18.Vedejs E, Denmark SE. Lewis Base Catalysis in Organic Synthesis. Wiley-VCH; Weinheim: 2016. pp. 1–3. [Google Scholar]
- 19.Denmark SE, Collins WR. Lewis Base Activation of Lewis Acids: Development of a Lewis Base Catalyzed Selenolactonization. Org Lett. 2007;9:3801–3804. doi: 10.1021/ol701617d. [DOI] [PubMed] [Google Scholar]
- 20.Denmark SE, Kalyani D, Collins WR. Preparative and Mechanistic Studies toward the Rational Development of Catalytic, Enantioselective Selenoetherification Reactions. J Am Chem Soc. 2010;132:15752–15765. doi: 10.1021/ja106837b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denmark SE, Vogler T. Synthesis and Reactivity of Enantiomerically Enriched Thiiranium Ions. Chem Eur J. 2009;15:11737–11745. doi: 10.1002/chem.200901377. [DOI] [PubMed] [Google Scholar]
- 22.Denmark SE, Kornfilt DJP, Vogler T. Catalytic Asymmetric Thiofunctionalization of Unactivated Alkenes. J Am Chem Soc. 2011;133:15308–15311. doi: 10.1021/ja2064395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denmark SE, Chi HM. Catalytic, Enantioselective, Intramolecular Carbosulfenylation of Olefins. Mechanistic Aspects: a Remarkable Case of Negative Catalysis. J Am Chem Soc. 2014;136:3655–3663. doi: 10.1021/ja413270h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Denmark SE, Jaunet A. Catalytic, Enantioselective, Intramolecular Carbosulfenylation of Olefins. A J Am Chem Soc. 2013;135:6419–6422. doi: 10.1021/ja401867b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Denmark SE, Jaunet A. Catalytic, Enantioselective, Intramolecular Carbosulfenylation of Olefins. Preparative and Stereochemical Aspects. J Org Chem. 2014;79:140–171. doi: 10.1021/jo4023765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denmark SE, Chi HM. Lewis Base Catalyzed, Enantioselective, Intramolecular Sulfenoamination of Olefins. J Am Chem Soc. 2014;136:8915–8918. doi: 10.1021/ja5046296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Denmark SE, Kornfilt DJ-P. Catalytic, Enantioselective, Intramolecular Sulfenofunctionalization of Alkenes with Phenols. J Org Chem. 2017;82:3192–3222. doi: 10.1021/acs.joc.7b00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denmark SE, Chi HM. Catalytic, Enantioselective, Intramolecular Sulfenoamination of Alkenes with Anilines. J Org Chem. 2017;82:3826–3843. doi: 10.1021/acs.joc.7b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denmark SE, Rossi S, Webster MP, Wang H. Catalytic, Enantioselective Sulfenylation of Ketone-Derived Enoxysilanes. J Am Chem Soc. 2014;136:13016–13028. doi: 10.1021/ja506133z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Denmark SE, Hartmann E, Kornfilt DJP, Wang H. Mechanistic, Crystallographic, and Computational Studies on the Catalytic, Enantioselective Sulfenofunctionalization of Alkenes. Nature: Chemistry. 2014;6:1056–1064. doi: 10.1038/nchem.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Godfrey SM, Ollerenshaw RTA, Pritchard RG, Richards CL. Structural Isomerism in R3PSe(Ph)I Compounds: the Ionic Structure of [(Me2N)3PSePh]I. J Chem Soc, Dalton Trans. 2001:508–509. [Google Scholar]
- 31.Kal’yan YB, Krimer MZ, Cherepanova EG, Bogdanov VS, Smit VA. Arylthiosulfonium Salts as Transfer Agents of the S-aryl Group to Double Bonds. Russ Chem Bull. 1982;31:342–349. [Google Scholar]
- 32.Krossing I, Raabe I. Noncoordinating Anions – Fact or Fiction? A Survey of Likely Candidates. Angew Chem Int Ed. 2004;43:2066–2090. doi: 10.1002/anie.200300620. [DOI] [PubMed] [Google Scholar]
- 33.The crystallographic coordinates for (±)-2aa+ BArF24– have been deposited in the CCDC (1006831). These data can be obtained free of charge from the Cambridge Crystallographic Data Center (www.ccdc.cam.ac.uk).
- 34.Hartmann F, Dahlems T, Mootz DZ. Crystal Structure of Hexamethylphosphoric Triamide (C2H3N)3PO. Z Kristallogr-New Cryst Struct. 1998;213:639–640. [Google Scholar]
- 35.Denmark SE, Eklov BM. Neutral and Cationic Phosphoramide Adducts of Silicon Tetrachloride: Synthesis and Characterization of Their Solution and Solid-State Structures. Chem Eur J. 2008;14:234–239. doi: 10.1002/chem.200701466. [DOI] [PubMed] [Google Scholar]
- 36.Arduengo AJ, Burgess EM. Tricoordinate Hypervalent Sulfur-Compounds. J Am Chem Soc. 1977;99:2376–2378. [Google Scholar]
- 37.Kuhn N, Bohnen H, Fahl J, Bläser D, Boese R. Koordination oder Reduktion? Zur Reaktion von 1,3-Diisopropyl-4,5-dimethylimidazol-2-yliden mit Schwefelhalogeniden und Schwefeloxidhalogeniden. Chem Ber. 1996;129:1579–1586. [Google Scholar]
- 38.Sacerdoti M, Gilli G, Domiano P. Comparison of Two Independent Structure Determinations of Diphenyl Disulphide. Acta Cryst. 1975;B31:327–329. [Google Scholar]
- 39.Rudd MD, Lindeman SV, Husebye S. Structural Characteristics of Three-Coordinate Arylhalide Tellurium(II) Complexes with Chalcogen Ligands. Synthesis, Spectroscopic Characterization and X-ray Structural Studies of Bromo[N-methylbenzothiazole-2(3H)selone]phenyltellurium(II), bromophenyl[tris-(dimethylamino)phosphaneselenide]tellurium(II) and Tris(dimethylamino)phosphanesulfide. Acta Chem Scand. 1996;50:759–774. [Google Scholar]
- 40.Acheson RM, Lines CT, Bryce MR, Dauter Z, Reynolds CD, Schmidpeter A. Synthesis and X-Ray Crystal Structures of 2,3-Dihydro-2-mercapto-2,1,3-benzophosphadiazine-4(1H)-thione 2-Sulphide Derivatives. J Chem Soc Perkin Trans. 1985;2:1913–1917. [Google Scholar]
- 41.a) Pettitt DJ, Helmkamp GK. Stable Oxonium Salts and Alkylation of Episulfides and Disulfides. J Org Chem. 1963;28:2932–2936. [Google Scholar]; b) Pettitt DJ, Helmkamp GK. Episulfonium Ion Intermediates in an Addition Reaction of Cyclooctene. J Org Chem. 1964;29:2702–2706. [Google Scholar]; c) Owsley DC, Helmkamp GK, Spurlock SN. Episulfonium Salts. I. Carbon vs. Sulfur Attack in the Reaction of Cyclooctene-S-Methylepisulfonium Ion with Nucleophiles. J Am Chem Soc. 1969;91:3606–3609. [Google Scholar]; d) Owsley DC, Helmkamp GK, Rettig MF, F M. Episulfonium Salts II. Detection of an Unusual Intermediate alt with Chloride Ion. J Am Chem Soc. 1969;91:5239–5242. [Google Scholar]; e) Csizmadia VM, Schmid GH, Mezey PG, Csizmadia IG. Ab initio SCF-MO Study of the Reaction Intermediates Formed by Addition of Thiohypochlorous Acid to Ethylene. J Chem Soc, Perkin Trans. 1977;2:1019–1024. [Google Scholar]; f) Carretero JC, García Ruano JL, Rodríguez JH. Stable Episulphuranes. Tetrahedron Lett. 1987;28:4593–4596. [Google Scholar]; g) Glass RS, Jouikov VV, Bojkova NV. Electrochemical Activation of Dimethyl Disulfide for Electrophilic Aromatic Substitution. J Org Chem. 2001;66:4440–4443. doi: 10.1021/jo010156x. [DOI] [PubMed] [Google Scholar]
- 42.Fachini M, Lucchini V, Modena G, Pasi M, Pasquato L. Nucleophilic Reactions at the Sulfur of Thiiranium and Thiirenium Ions. New Insight in the Electrophilic Additions to Alkenes and Alkynes. Evidence for an Episulfurane Intermediate. J Am Chem Soc. 1999;121:3944–3950. [Google Scholar]
- 43.Scarborough CC, McDoonald RI, Hartmann C, Sazama GT, Bergant A, Stahl SS. Steric Modulation of Chiral Biaryl Diamines via Pd-Catalyzed Directed C–H Arylation. J Org Chem. 2009;74:2613–2615. doi: 10.1021/jo802632v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Šoustek P, Michl M, Almonasy N, Machalický O, Dvořák M, Lyčka A. The Synthesis and Fluorescence of N-Substituted 1- and 2-Aminopyrenes. Deys & Pigments. 2008;78:139–147. [Google Scholar]
- 45.Yang T, Huang Y, Cho BP. Synthesis and Characterization of Enantiomeric anti-2-Fluorobenzo[a]pyrene-7,8-dihydrodiol-9,10-epoxides and Their 2′-Deoxyguanosine and Oligodeoxynucleotide Adducts. Chem Res Toxicol. 2006;19:242–254. doi: 10.1021/tx050308+. [DOI] [PubMed] [Google Scholar]
- 46.Wang K, Hu Y, Li Z, Wu M, Liu Z, Su B, Yu A, Liu Y, Wang Q. A Simple and Efficient Oxidative Coupling of Aromatic Nuclei Mediated by Manganese Dioxide. Synthesis. 2010:1083–1090. [Google Scholar]
- 47.We are grateful to Dr. Christina Kraml (Lotus Separations, Princeton University) for carrying out this preparative separation.
- 48.Vykočil Š, Smerčina M, Lorenc M, Tišlerová I, D: Brooks R, Kulagowski JJ, Langer V, Farrugia LJ, Kočovský P. Copper(II)-Mediated Oxidative Coupling of 2-Aminonaphthalene Homologues. Competition between the Straight Dimerization and the Formation of Carbazoles. J Org Chem. 2001;66:1359–1365. doi: 10.1021/jo005691w. [DOI] [PubMed] [Google Scholar]
- 49.By analogy to the reaction of 2-naphthol:; Brown KJ, Berry MS, Murdoch JR. Synthesis of Optically Active 2,2′-Dihalo-1,1′-binaphthyls via Stable Diazonium Salts. J Org Chem. 1985;50:4345–4349. [Google Scholar]
- 50.Vogler T. Postdoctoral Research Report. Univeristy of Illinois; Urbana-Champaign: 2009. [Google Scholar]
- 51.Rundel W. Notiz Über die Darstellung tert.-butylierter Thiophenole, Diphenyldisulfide und Thianthrene. Chem Ber. 1968;101:2956–2962. [Google Scholar]
- 52.Milne JE, Buchwald SL. An Extremely Active Catalyst for the Negishi Cross-Coupling Reaction. J Am Chem Soc. 2004;126:13028–13023. doi: 10.1021/ja0474493. [DOI] [PubMed] [Google Scholar]
- 53.Klose J, Reese CB, Song Q. Preparation of 2-(2-Cyanoethyl)sulfanyl-1H-isoindole-1,3-(2H)-dione and Related Sulfur-Transfer Agents. Tetrahedron. 1997;53:14411–14416. [Google Scholar]
- 54.Phipps RJ, Gaunt M. A meta-Selective Copper-Catalyzed C-H Bond Arylation. Science. 2009;323:1593–1597. doi: 10.1126/science.1169975. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.