

Abstract
This letter demonstrates a simple method to achieve high-yields of 1H semiconducting MoS2 monolayers in concentrated, colloidally-stable aqueous suspension. The method is based on oxidation suppression during the hydrothermal processing step used for metal-to-semiconductor phase reversion. Accompanying DFT calculations on elementary steps in the MoS2 wet oxidation reaction suggest that a two-site corrosion mechanism is responsible for the observed high reactivity and low stability of 1T metallic MoS2.
Transition metal dichalcogenide (TMD) nanosheets are an emerging class of two-dimensional (2D) materials with significant application potential. Among the TMDs is molybdenum disulfide (MoS2), which shows a transition from an indirect to a direct band gap upon full exfoliation to the monolayer form.1 This indirect-to-direct transition is accompanied by the emergence of photoluminescence leading to applications in electronics and optoelectronics.2–5 Photoluminescent MoS2 monolayers can be prepared by chemical vapor deposition (CVD)6–8 or by mechanical exfoliation,3,9,10 but low throughput (for CVD) or low yield (for mechanical exfoliation) limits the development of some applications. Solvent- or surfactant-assisted liquid exfoliation is a scalable method, but typically most of the raw product is in multilayer form,11,12 and the solvents or surfactants require removal by additional processing steps.13,14
A potential high-yield route to monolayer MoS2 without surface contamination involves chemical exfoliation using n-butyl-lithium intercalation to weaken interlayer interactions.15 Forced hydration of the Li-intercalated MoS2 generates aqueous suspensions of monolayer MoS2, which are colloidally stable due to negative charges transferred during the Li interaction.16 Unfortunately, chemical exfoliation converts a large fraction of the nanosheets from trigonal prismatic (semiconducting 1H/2H phase) to an octahedral configuration (metallic 1T phase),17 with corresponding loss of the intrinsic semiconducting properties and photoluminescence.2 For this reason, significant efforts have been made to restore the semiconducting 1H/2H phase by processing ce-MoS2 monolayers.2,18,19 Eda et al.2 restored the 2H phase by annealing ce-MoS2 films, but the drying process leads to nanosheet restacking within the film, which limits further processability and also suppresses photoluminescence. It would be desirable to carry out the phase reversion of 1T monolayers in their original aqueous suspension, but this has proven problematic. The temperature required for the metal-to-semiconductor phase reversion exceeds the normal boiling point of water.2,19 Also the 1H/2H form is reported to be hydrophobic,20 leading to instability, flocculation, and restacking in aqueous solutions. Guardia et al.21 successfully maintained colloidal stability by using very dilute nanosheet suspensions, but dilute processing is a disadvantage for high-volume production.
Phase reversion in aqueous systems is also restricted by potential oxidation and degradation of the MoS2 nanosheets at the elevated temperatures required.19 Oxidation by O2 has been reported at elevated temperature for MoS2 nanosheets22 and for bulk MoS2 and its friction films.23,24 Over longer time scales, oxidative degradation of MoS2 nanosheets has even been observed at room temperature in humid air,25 and in aqueous suspensions.26 A recent study observed much faster oxidation kinetics for chemically exfoliated MoS2 (mixed 1T/1H phase) than surfactant-assisted liquid exfoliated MoS2 (2H phase), but did not propose a mechanism to explain the origin of the difference.26 To overcome the various limitations in fabricating semiconducting monolayer nanosheets (denoted as 1H-MoS2), Chou et al.19 functionalized chemically exfoliated MoS2 surfaces to allow dispersion in a high-boiling point organic solvent followed by dispersion, annealing and phase reversion to 1H in that solvent.
We became interested in the possibility of a simple technique that would directly convert 1T MoS2 nanosheets into monolayer 1H nanosheets in their original aqueous suspensions, and without the requirement of organic solvents, nanosheet functionalization, or surfactants or other dispersion aids that could complicate downstream processing. Given the experimentally-measured oxidative instability of chemically-exfoliated MoS2, we hypothesized that oxidation suppression would be the key, and therefore coupled chemical exfoliation with hydrothermal phase reversion under carefully controlled low-O2 conditions. This communication documents the resulting improved synthesis method, which not only avoids oxidative degradation of the nanosheets and yield loss, but surprisingly also solves the previously observed problem of colloidal instability.21,27
The chemical exfoliation method using n-butyllithium intercalation produced highly water-dispersible MoS2 sheets predominantly in monolayer form (see previous work15). The colloidal stability has been attributed to negative surface charge on the 1T-phase nanosheets imparted by the electron transfer during Li-intercalation.16 Note that previous studies indicate that a distorted 1T phase (denoted as 1T′) is also present in these chemically exfoliated MoS2 (ce-MoS2) samples (see modelling section below).19,28 For reversion to the 1H phase we used hydrothermal treatment at ∼200 °C, which is the minimal temperature where annealing can effectively convert dry multilayer ce-MoS2 films.2 Without exclusion of O2, we found that the reversion was accompanied by aggregation and flocculation (Fig. 1) at high MoS2 concentration (∼200 mg L−1 of Mo – see Fig. S1†), consistent with prior observations by Guardia et al.21 Although the use of dilute MoS2 (40 mg L−1 of Mo) suspensions is useful for preventing flocculation,21 ICP-OES measurements of soluble Mo species show that a large fraction of the MoS2 nanosheets are destroyed by oxidative dissolution (∼50% at initial loading 40 mg L−1 of Mo) under these dilute conditions (Fig. S1†). We hypothesized further that this unwanted oxidation may also be responsible, indirectly, for the flocculation phenomenon. MoS2 wet oxidation has been reported to generate protons,26 which accumulate and counteract the negative surface charge through surface protonation and/or electrical double-layer charge screening (Fig. 1). Indeed the pH of remaining suspension (starting concentration ∼200 mg L−1 of Mo) dropped from ∼5 to ∼2 after 2 hours of hydrothermal treatment, during which 33% of Mo was oxidized and released as soluble species measured by ICP-OES (Fig. S1†). The magnitude of this pH change is consistent with the reaction stoichiometry shown in Fig. 1. We further tested the hypothesis by decreasing either the temperature or the initial MoS2 concentration, and both changes could eventually avoid aggregation (Fig. S1†). Temperature reduction is not viable as a synthesis approach, however, as we find >170 °C to be necessary to achieve the metal-to-semiconductor phase reversion (Fig. 2). Any combination of slow kinetics (low temperature) or low MoS2 concentration failed to generate enough protons to destabilize the remaining nanosheets. In these experiments pH values were measured to be >2.5, which is consistent with previous observations that the MoS2 nanosheet aggregation begins to occur near pH 2.26 Overall, hydrothermal treatment in simple air-exposed aqueous suspensions is not effective for metal-to-semiconductor phase reversion because of oxidative dissolution that consumes a large portion of the nanosheet product and can give rise to proton-induced aggregation and restacking of the remaining MoS2 nanosheets.
Fig. 1.
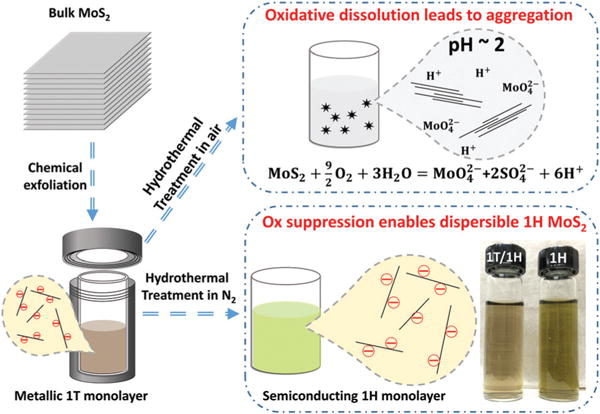
Overview of 1H MoS2 nanosheet fabrication routes with oxidation control (lower branch) and without (upper branch). The common starting step is chemical exfoliation of bulk MoS2, which produces primarily 1T monolayer MoS2 that is colloidally stable in aqueous media. When hydrothermal phase reversion is conducted in air-exposed suspension (upper branch) a large fraction of the 2D material is oxidatively degraded and/or the remaining nanosheets flocculate and restack. In O2-free suspensions (lower branch), high nanosheet yield is obtained and the monolayer 1H material retains colloidal stability.
Fig. 2.
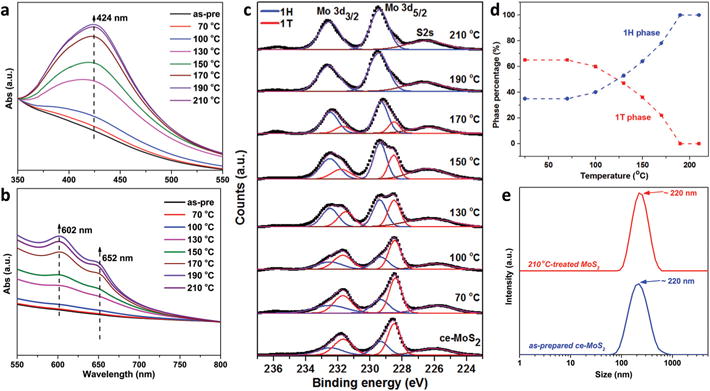
Monitoring of phase composition and nanosheet size during hydrothermal treatment with oxidation suppression. (a, b) UV-vis absorption spectra; (c) XPS spectra (Mo 3d core level peak region) for ce-MoS2 nanosheets treated hydrothermally for 2 h. In order to show relative intensity increases, the spectra in (a) and (b) are normalized using peak intensities at 350 and 800 nm, respectively. Mo 3d peaks were deconvoluted into 1T/1T′ (red) and 1H (blue) components, and the relative fractions extracted and shown in (d). (e) The hydrodynamic sizes of as-prepared ce-MoS2 and treated sample at 210 °C measured by dynamic light scattering indicate no aggregation and thus colloidal stability.
To minimize oxidative dissolution, the starting MoS2 suspensions were deoxygenated in a N2 glove box (O2 concentration <1 ppm) prior to hydrothermal reaction. With this step, no aggregation was observed in any of the hydrothermal products up to 400 mg L−1 nanosheet loading and temperatures up to 210 °C, (Fig. 1 inset) and the suspensions retained colloidal stability over 6-month storage. ICP-OES measurement of soluble Mo revealed <5% dissolution at all temperatures, and no significant pH changes were observed. Fig. 2 tracks the phase reversion by UV-visible spectroscopy. The initially featureless spectrum of ce-MoS2 slowly develops peaks at ∼424, 602 and 652 nm as hydrothermal temperature increases. Peaks at ∼652 and 602 nm represent characteristic direct-gap transitions A and B at the K-point of semiconducting 2H MoS2, respectively, while the broad peak at ∼424 nm is the convolution result of C and D excitonic peaks.2,29 We used XPS (Fig. 2c) to estimate 1T/1H ratio (Fig. 2d). The ce-MoS2 sample consists of two polymorphs, of which 1T/1T′ phase is the primary component with peaks at 228.5 and 231.7 eV, corresponding to Mo4+ 3d5/2 and Mo4+ 3d3/2, respectively. The positions of the 1H component peaks are at higher binding energies, ∼0.8 eV, as reported previously.2 The phase ratios estimated from XPS data show ∼70% 1T/1T′ phase initially with the metal-to-semiconductor reversion starting at ∼100 °C and being complete above ∼190 °C. This temperature range is similar to that observed previously under non-hydrothermal conditions.2,19 In the S 2p spectrum, the convoluted two-component peak shifts to higher binding energies during the metal-to-semiconductor transition (Fig. S2†). Dynamic light scattering data shows no change in hydrodynamic size of the MoS2 nanosheets during hydrothermal reversion (Fig. 2e), which provides additional evidence that this method produces stable nanosheet colloids that avoid the aggregation and flocculation seen without O2 control.
Fig. 3 provides additional characterization of the nanosheet products made by this new method. The typical HRTEM image of 1H-MoS2 is given in Fig. 3a. Its electron diffraction pattern indicates the singlet component (Fig. S3†), distinct from that of ce-MoS2, where the presence of additional spots implies the mixture of 1H and 1T phases.30 AFM shows thicknesses typical of monolayers on substrates (1.1–1.2 nm) and lateral sizes from 100–500 (see full distribution in Fig. S4†). The lateral dimensions and thicknesses are similar to the parent (un-converted) ce-MoS2, which supports the conclusion of no significant aggregation or morphology change during hydrothermal treatment. In addition, both the parent and converted MoS2 monolayers in this work and in literature reports2,21 show apparently higher thickness values than those reported for mechanically exfoliated monolayers (0.65–0.7 nm),8,29 which could be due to the presence of a residual distorted phase and/or molecular adsorbates.2 Fig. 3c shows, remarkably, that the 1H monolayers retain significant negative charge, which is the origin of their colloidal stability. The zeta potential/pH relationship is similar to that of the precursor 1T/1H mixed phase fabricated by chemical exfoliation (see Wang et al.26), implying that the negative charge is preserved by our phase reversion protocol. Previous reports that phase-reverted 2H MoS2 nanosheets were unstable in aqueous suspension21 must be the consequence of the unintended role of dissolved O2 in removing the surface charge or electrostatically shielding them by proton generation. Using the O2-free protocol we observed no change in the dispersibility of the nanosheets after hydrothermal treatment.
Fig. 3.
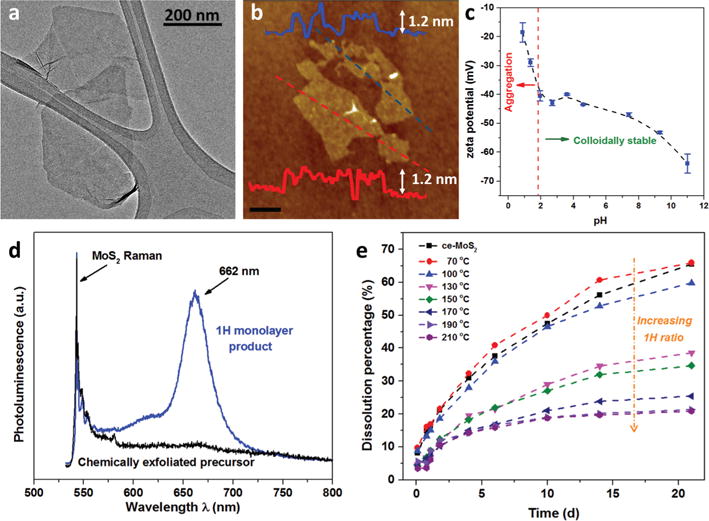
Morphology and properties of 1H monolayer nanosheet product made by the proposed O2-free hydrothermal method. (a) Lateral dimension by TEM and (b) thickness by tapping-mode AFM of 1H-MoS2 nanosheets after hydrothermal treatment at 210 °C for 2 h (scale bar 200 nm). (c) pH-Dependent zeta potential of fully phase reverted 1H MoS2 in aqueous suspensions; the pH of the 1H-MoS2 solution was adjusted by addition of HCl or NaOH. The dashed line shows the colloidal stability/instability occurs at ∼pH 2 based on our experimental observations. (d) Photoluminescence and Raman spectra of ce-MoS2 and converted 1H-MoS2 nanosheets. (e) Oxidative dissolution rates for ce-MoS2, partially and fully phase-reverted samples in HEPES buffer (pH 7) at 25 °C. The gradual phase reversion to 1H is accompanied by systematic decreases in the sub-sequent oxidation reactivity at 25 °C.
Monolayer MoS2 has been reported to be a direct band gap semiconductor with the lowest energy interband transition occurring at the K point of the Brillouin zone, emitting the photons with energy of 1.9 eV when relaxed.29 Fig. 3d shows Raman and photoluminescence (PL) spectra of ce-MoS2 and the converted 1H-MoS2 nanosheet product. As expected, metallic ce-MoS2 nanosheets show no photoluminescence, while 1H-MoS2 nanosheets exhibit clear photoluminescence with a two-peak spectrum (major peak 660 nm – see deconvolution analysis Fig. S5†). The PL spectrum as well as the UV-vis spectrum shown in Fig. 2 are in good agreement with those of mechanically exfoliated monolayer samples,29 indicating they arise from the intrinsic direct band-gap emission. The ratio of MoS2 photoluminescence and Raman intensity is considered as a criterion to evaluate the intrinsic luminescence property by ruling out external effects.29 The ratio for our 1H-MoS2 sample is comparable with that for the monolayers and bilayers exfoliated mechanically.29 The restoration of strong photoluminescence of MoS2 clearly demonstrates the metal-to-semiconductor conversion and the preservation of the monolayer nature of MoS2 nanosheets during the oxygen-suppressed hydrothermal treatment. We anticipate that this approach can be extended to phase reversion in some other transition metal dichalcogenides (see preliminary data on WS2 and MoSe2, Fig. S6†).
Finally, Fig. 3e shows the effect of hydrothermal phase reversion on oxidation reactivity. Our previous study showed much faster oxidation rates for ce-MoS2 nanosheets (mixed 1T/1H) relative to the pure 2H material fabricated by ultrasonic exfoliation.26 The present results (Fig. 3e) show that oxidation reactivity decreases steadily and systematically as the hydrothermal phase reversion proceeds. Since the hydrothermal treatment does not significantly change morphology or colloidal state, this result provides further evidence that the different oxidation reactivities are intrinsic properties of the 1T and 1H phases.
The fundamental origin of the higher oxidation reactivity of the 1T (or 1T′) phase relative to the 1H phase is currently unknown. Understanding these oxidative degradation pathways under ambient conditions is critical for technological applications that require device stability, and also for understanding their environmental and biological behaviors that govern persistence and risk.31,32 We therefore carried out electronic structure calculations in density functional theory to search for possible phase-dependent steps in the oxidation mechanism. Initial work studied the O2/MoS2 system for both 1T/1T′ and 1H monolayer nanosheet models, and oxygen binding energies were found to be similar for the two phases (1T/1T′ and 1H). The lack of phase-dependence led to an alternative hypothesis: that the faster oxidation of the 1T/1T′ phase is the result of a two-site corrosion-type mechanism. In this mechanism, the MoS2 surface serves as both an anode and a cathode; electrons are generated at one site through an anodic reaction and conduct to the second site where a cathodic reaction occurs with the surrounding medium (see eqn (1) and (2)). The rapid oxidation of the 1T/1T′ phase is hypothesized to be the direct result of its electrical conductivity,33–35 which allows the required internal electron transfer, while the insulating 1H phase does not. The driving force for the corrosion-type mechanisms can be calculated using the computational hydrogen electrode (CHE) technique36,37 applied to these two initiation reactions:
| (1) |
| (2) |
where “*” denotes a site on the MoS2 nanosheet.
We examined the Mo-edge sites of MoS2 with monolayer S coverage (Fig. 4), which is considered to be the most stable structure in aqueous solutions without a sulfur source.38 In this context, the first step of the anodic reaction is the oxidation of an H2O molecule releasing a proton–electron pair (to the solution and surface, respectively) and leaving an OH adsorbate on the S atom. Using the CHE approach, we calculated the ΔG of this step to be −0.16 eV when the electron is referenced to 0 VRHE. On a cathodic site, O2 is reduced by a proton–electron pair to form an OOH adsorbate, similar to oxygen reduction processes on other cathodes.36,39 We calculated its ΔG to be −1.78 eV when the electron is at the same reference potential. To show the driving force, we illustrate several other choices of internal electron potential in Fig. 4b, along with the calculated equilibrium potentials for each half reaction. We would expect the internal potential of the reacting MoS2 to equilibrate at an intermediate potential as such elementary reactions proceed. In net, the total driving force is −1.94 eV, making this paired redox reaction highly favorable.
Fig. 4.
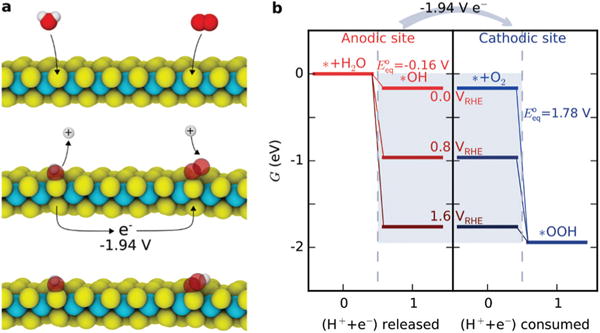
DFT calculations and hypothesized mechanism for enhanced oxidation rates of 1T vs. 1H-phase MoS2 nanosheets. (a) Initial elementary steps of corrosion-type oxidation on 1T′ MoS2. Yellow: S, cyan: Mo, red: O, white: H. (b) The free energy diagram of the anodic and cathodic redox reactions. The energy levels in the gray box are assumed potentials of the electron in the computational hydrogen electrode model, which reveals a 1.94 eV net driving force for the coupled reaction. The values represent the equilibrium potentials of the anodic and cathodic reactions at 25 °C.
After the initial steps, additional OH and/or OOH species must adsorb on the S atoms to release sulfate, and to further oxidize the Mo atom after an S vacancy is formed. The overall reaction 2MoS2 + 6H2O + 9O2 → 2H2MoO4 + 4H2SO4 can be calculated to have a driving force of −14.4 eV per Mo atom (note that E° = 0.8 V and the MoS2 oxidation half reaction involves 18 electrons). We proposed a possible set of sub-sequent steps in the ESI (Table S1†) and recommend a more exhaustive study to understand the complete reaction mechanism.
Conclusions
In summary, oxidation by environmental O2 has been shown to be an important limitation for the aqueous processing and storage of MoS2 nanosheets. The undesired effects are particularly significant at long times, or at elevated temperatures such as those used in hydrothermal phase reversion. The effects are particularly significant for the 1T/1T′ phase, whose rapid oxidation may be the result of a corrosion-type mechanism enabled by its metallic nature. An improved synthesis protocol is developed and proposed here to produce monolayer 1H nanosheets that combines many desired features: processing in the absence of organic solvents or functionalization; pure 1H crystal structure in the product; monolayer thickness with its corresponding photoluminescence; and hydrophilicity due to retention of negative surface charge, and long-term colloidal stability in the aqueous phase. We also propose a new hypothesis, based on DFT calculations, that oxidation of MoS2 nanosheets in the metallic 1T/1T′ phase proceeds by a two-site corrosion mechanism, which is the origin of their high reactivity relative to the 1H phase. In the future, the roles and mechanisms of oxidative degradation during synthesis and processing deserve more attention for other transition metal dichalcogenides.40
Supplementary Material
Acknowledgments
Financial support for this work was provided by the National Science Foundation (Grant: INSPIRE Track 1 CBET-1344097) the Superfund Research Program of the National Institute of Environmental Health Sciences (Grant P42 ES013660). Electronic structure calculations were conducted at Brown University’s Center for Computation and Visualization. We acknowledge Wenhao Li and Prof. Rashid Zia for the photoluminescence and Raman measurements and analysis, as well as Thomas Valentin for assistance with the XPS characterization.
Footnotes
Electronic supplementary information (ESI) available: Experimental details and data. DFT modelling. See DOI: 10.1039/c7nr01193h
References
- 1.Mak KF, Lee C, Hone J, Shan J, Heinz TF. Phys Rev Lett. 2010;105:136805. doi: 10.1103/PhysRevLett.105.136805. [DOI] [PubMed] [Google Scholar]
- 2.Eda G, Yamaguchi H, Voiry D, Fujita T, Chen M, Chhowalla M. Nano Lett. 2011;11:5111–5116. doi: 10.1021/nl201874w. [DOI] [PubMed] [Google Scholar]
- 3.Radisavljevic B, Radenovic A, Brivio J, Giacometti V, Kis A. Nat Nanotechnol. 2011;6:147–150. doi: 10.1038/nnano.2010.279. [DOI] [PubMed] [Google Scholar]
- 4.Mouri S, Miyauchi Y, Matsuda K. Nano Lett. 2013;13:5944–5948. doi: 10.1021/nl403036h. [DOI] [PubMed] [Google Scholar]
- 5.Huang X, Zeng Z, Zhang H. Chem Soc Rev. 2013;42:1934–1946. doi: 10.1039/c2cs35387c. [DOI] [PubMed] [Google Scholar]
- 6.Lee YH, Zhang XQ, Zhang W, Chang MT, Lin CT, Chang KD, Yu YC, Wang JTW, Chang CS, Li LJ, Lin TW. Adv Mater. 2012;24:2320–2325. doi: 10.1002/adma.201104798. [DOI] [PubMed] [Google Scholar]
- 7.Zhan Y, Liu Z, Najmaei S, Ajayan PM, Lou J. Small. 2012;8:966–971. doi: 10.1002/smll.201102654. [DOI] [PubMed] [Google Scholar]
- 8.Najmaei S, Liu Z, Zhou W, Zou X, Shi G, Lei S, Yakobson BI, Idrobo JC, Ajayan PM, Lou J. Nat Mater. 2013;12:754–759. doi: 10.1038/nmat3673. [DOI] [PubMed] [Google Scholar]
- 9.Yin Z, Li H, Li H, Jiang L, Shi Y, Sun Y, Lu G, Zhang Q, Chen X, Zhang H. ACS Nano. 2012;6:74–80. doi: 10.1021/nn2024557. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Wu J, Yin Z, Zhang H. Acc Chem Res. 2014;47:1067–1075. doi: 10.1021/ar4002312. [DOI] [PubMed] [Google Scholar]
- 11.Lee K, Kim HY, Lotya M, Coleman JN, Kim GT, Duesberg GS. Adv Mater. 2011;23:4178–4182. doi: 10.1002/adma.201101013. [DOI] [PubMed] [Google Scholar]
- 12.Coleman JN, Lotya M, O’Neill A, Bergin SD, King PJ, Khan U, Young K, Gaucher A, De S, Smith RJ. Science. 2011;331:568–571. doi: 10.1126/science.1194975. [DOI] [PubMed] [Google Scholar]
- 13.Akesson B, Paulsson K. Occup Environ Med. 1997;54:236–240. doi: 10.1136/oem.54.4.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halim U, Zheng CR, Chen Y, Lin Z, Jiang S, Cheng R, Huang Y, Duan X. Nat Commun. 2013;4:2213. doi: 10.1038/ncomms3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joensen P, Frindt R, Morrison SR. Mater Res Bull. 1986;21:457–461. [Google Scholar]
- 16.Heising J, Kanatzidis MG. J Am Chem Soc. 1999;121:11720–11732. [Google Scholar]
- 17.Py M, Haering R. Can J Phys. 1983;61:76–84. [Google Scholar]
- 18.Fan X, Xu P, Zhou D, Sun Y, Li YC, Nguyen MAT, Terrones M, Mallouk TE. Nano Lett. 2015;15:5956–5960. doi: 10.1021/acs.nanolett.5b02091. [DOI] [PubMed] [Google Scholar]
- 19.Chou SS, Huang YK, Kim J, Kaehr B, Foley BM, Lu P, Dykstra C, Hopkins PE, Brinker CJ, Huang J, Dravid VP. J Am Chem Soc. 2015;137:1742–1745. doi: 10.1021/ja5107145. [DOI] [PubMed] [Google Scholar]
- 20.Ozcan O. Int J Miner Process. 1992;34:191–204. [Google Scholar]
- 21.Guardia L, Paredes JI, Munuera JM, Villar-Rodil S, Ayán-Varela M, Martínez-Alonso A, Tascón JMD. ACS Appl Mater Interfaces. 2014;6:21702–21710. doi: 10.1021/am506922q. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto M, Einstein TL, Fuhrer MS, Cullen WG. J Phys Chem C. 2013;117:25643–25649. [Google Scholar]
- 23.Windom BC, Sawyer W, Hahn DW. Tribol Lett. 2011;42:301–310. [Google Scholar]
- 24.Khare H, Burris D. Tribol Lett. 2014;53:329–336. [Google Scholar]
- 25.Gao J, Li B, Tan J, Chow P, Lu TM, Koratkar N. ACS Nano. 2016;10:2628–2635. doi: 10.1021/acsnano.5b07677. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, von dem Bussche A, Qiu Y, Valentin TM, Gion K, Kane AB, Hurt RH. Environ Sci Technol. 2016;50:7208–7217. doi: 10.1021/acs.est.6b01881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou SS, De M, Kim J, Byun S, Dykstra C, Yu J, Huang J, Dravid VP. J Am Chem Soc. 2013;135:4584–4587. doi: 10.1021/ja310929s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eda G, Fujita T, Yamaguchi H, Voiry D, Chen M, Chhowalla M. ACS Nano. 2012;6:7311–7317. doi: 10.1021/nn302422x. [DOI] [PubMed] [Google Scholar]
- 29.Splendiani A, Sun L, Zhang Y, Li T, Kim J, Chim CY, Galli G, Wang F. Nano Lett. 2010;10:1271–1275. doi: 10.1021/nl903868w. [DOI] [PubMed] [Google Scholar]
- 30.Maitra U, Gupta U, De M, Datta R, Govindaraj A, Rao C. Angew Chem, Int Ed. 2013;52:13057–13061. doi: 10.1002/anie.201306918. [DOI] [PubMed] [Google Scholar]
- 31.Lowry GV, Gregory KB, Apte SC, Lead JR. Environ Sci Technol. 2012;46:6893–6899. doi: 10.1021/es300839e. [DOI] [PubMed] [Google Scholar]
- 32.Gondikas AP, Morris A, Reinsch BC, Marinakos SM, Lowry GV, Hsu-Kim H. Environ Sci Technol. 2012;46:7037–7045. doi: 10.1021/es3001757. [DOI] [PubMed] [Google Scholar]
- 33.Fan XL, Yang Y, Xiao P, Lau WM. J Mater Chem A. 2014;2:20545–20551. [Google Scholar]
- 34.Lin YC, Dumcenco DO, Huang YS, Suenaga K. Nat Nanotechnol. 2014;9:391–396. doi: 10.1038/nnano.2014.64. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Lu Z, Xu S, Kong D, Cha JJ, Zheng G, Hsu PC, Yan K, Bradshaw D, Prinz FB. Proc Natl Acad Sci U S A. 2013;110:19701–19706. doi: 10.1073/pnas.1316792110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nørskov JK, Rossmeisl J, Logadottir A, Lindqvist L, Kitchin JR, Bligaard T, Jonsson H. J Phys Chem B. 2004;108:17886–17892. [Google Scholar]
- 37.Peterson AA, Abild-Pedersen F, Studt F, Rossmeisl J, Nørskov JK. Energy Environ Sci. 2010;3:1311–1315. [Google Scholar]
- 38.Huang Y, Nielsen RJ, Goddard WA, III, Soriaga MP. J Am Chem Soc. 2015;137:6692–6698. doi: 10.1021/jacs.5b03329. [DOI] [PubMed] [Google Scholar]
- 39.Suntivich J, Gasteiger HA, Yabuuchi N, Nakanishi H, Goodenough JB, Shao-Horn Y. Nat Chem. 2011;3:546–550. doi: 10.1038/nchem.1069. [DOI] [PubMed] [Google Scholar]
- 40.Wang Z, Zhu W, Qiu Y, Yi X, von dem Bussche A, Kane A, Gao H, Koski K, Hurt R. Chem Soc Rev. 2016;45:1750–1780. doi: 10.1039/c5cs00914f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.