


Abstract
DNA double strand breaks (DSBs) are one of the most deleterious lesions and if left unrepaired, they lead to cell death, genomic instability and carcinogenesis. Cells combat DSBs by two pathways: homologous recombination (HR) and non-homologous end-joining (NHEJ), wherein the two DNA ends are re-joined. Recently a back-up NHEJ pathway has been reported and is referred to as alternative NHEJ (aNHEJ), which joins ends but results in deletions and insertions. NHEJ requires processing enzymes including nucleases and polymerases, although the roles of these enzymes are poorly understood. Emerging evidence indicates that X family DNA polymerases lambda (Pol λ) and mu (Pol μ) promote DNA end-joining. Here, we show that DNA polymerase beta (Pol β), another member of the X family of DNA polymerases, plays a role in aNHEJ. In the absence of DNA Pol β, fewer small deletions are observed. In addition, depletion of Pol β results in cellular sensitivity to bleomycin and DNA protein kinase catalytic subunit inhibitors due to defective repair of DSBs. In summary, our results indicate that Pol β in functions in aNHEJ and provide mechanistic insight into its role in this process.
INTRODUCTION
DNA double strand breaks (DSBs) are one of the most deleterious types of cellular DNA damage. There are two major pathways for repairing DSBs. Homologous recombination (HR) is predominantly error-free repair and active predominantly during the S and G2 phases of the cell cycle (1,2). Non-homologous end-joining (NHEJ) can be either error-free or error-prone and it is active throughout the cell cycle (3–9). More recently, a number of non-canonical pathways have been grouped into a category termed alternative NHEJ (aNHEJ), which will be used here. This pathway is highly error-prone and mutagenic (10–16).
The classical or canonical NHEJ (cNHEJ) pathway repairs DNA by joining ends with minimal processing. The key factors in cNHEJ include the Ku 70 and 80 heterodimers, DNA Protein Kinase catalytic subunit (DNA-PKcs), X-Ray cross-complementing 4 (XRCC4), Ligase IV (Lig IV), XRCC like factor (XLF) and Artemis (17–19). The binding of the Ku 70/80 heterodimer to the broken DNA ends initiates the cNHEJ process. DNA-PKcs, XRCC4-Ligase IV, XLF, Artemis and DNA polymerases are then recruited to the site of damage. Upon binding to the Ku–DNA complex, DNA-PKcs is phosphorylated, facilitating end-processing and ligation of the broken DNA ends (17,20,21). Members of the DNA polymerase X family, namely, Pol λ and Pol μ have been implicated in cNHEJ (19,22–26). Previous work has also shown that Saccharomyces cerevisiae X family polymerase Pol 4, a Pol λ homolog, is required for gap filling during cNHEJ (27). It has been shown that aNHEJ requires poly-ADP-ribose polymerase 1 (PARP-1), XRCC1 and Lig I or III (28–30). The cNHEJ pathway has been described as error-prone but aNHEJ is suggested to be considerably more error prone as it tends to create larger deletions.
The mutational signature of aNHEJ includes small and large deletions with microhomology (μH) at the DNA junctions, and can result in chromosomal translocations (10,29–34). The aNHEJ machinery uses DNA resection to reveal single-stranded DNA with μH. Mre11 and CtIP have been shown to be involved in aNHEJ (35–37) and likely function in strand resection. Therefore, this process generally requires nucleolytic DNA end-processing. After DNA end-processing, DNA polymerases fill gaps generated as a result of the annealed μH in the DNA, and this is followed by DNA ligation (31,38). DNA Polymerase theta (DNA Pol θ) has recently been shown to participate aNHEJ (39,40), and for a review see (41). It is suggested that Pol θ participates in end-joining using long tracts of single-stranded DNA generated during strand resection. Pol θ is also likely to function in the generation of templated insertions at some sites of break joining (40,42,43).
V(D)J recombination is required for B and T cell development and involves somatic recombination that results in a diverse repertoire of antibodies and T-cell receptors. These programmed gene arrangements involve an end-joining mechanism (25,44,45). Our laboratory has recently provided evidence for a role for Pol β, another X family polymerase, in V(D)J recombination (46). Specifically, we showed that mice expressing the Y265C Pol β protein, which has a slow and inaccurate polymerase activity, have short CDR3 junctions in the immunoglobulin heavy chain, but not the light chain or in the junctions in T cells. This suggests that Pol β functions in NHEJ. Using a recently developed fluorescence based assay that monitors HR and aNHEJ, we provide evidence here that Pol β plays a role in the aNHEJ process.
MATERIALS AND METHODS
Cell lines and reagents
MCF7 cells are human breast adenocarcinoma cells derived from human mammary tissue (ATCC-HTB-22). These cells were maintained in RPMI medium (GIBCO) supplemented with 10% fetal bovine serum (FBS) (Gemini-Bio), 1% penicillin-streptomycin (GIBCO). U2OS cells are immortalized bone cancer cells (ATCC-HTB-96). These cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO) supplemented with 10% FBS and 1% penicillin-streptomycin. U2OS EJ-DR cells were maintained in DMEM, 10% Tetracyline-free FBS (Atlanta Biologicals) and 1% penicillin-streptomycin. All the cell lines were grown at 37°C in a 5% CO2 humidified incubator. DNA-PKcs inhibitors NU7441 and KU0060648 and triamcinolone (TA) ligand were kindly provided by Dr Ranjit Bindra, Yale University.
Transfection and expression analysis
siRNA for Pol β were obtained from Dharmacon (see Supplementary Table S1). siRNA was transfected in U2OS EJ-DR cells using Lipofectamine RNAiMAX (Life Technologies). Cells were incubated for 72 h post transfection and Pol β depletion was confirmed by western blotting. shRNA used for Pol β was a gift from Dr Robert Sobol (Addgene # 18663). shRNA was nucleofected into MCF7, U2OS and U2OS EJ-DR cells. Cells were incubated for 72 h after nucleofection and Pol β depletion was confirmed by western blotting.
For Pol β rescue experiments, a plasmid harboring siRNA resistant wild-type (WT) Pol β cDNA and Pol β-Y265C containing a C-terminal HA tag was generated. Cells were co-transfected with 1 μg of siRNA resistant plasmid and 60 pmol siRNA (si1) using nucleofection. Pol β knock down and expression of the siRNA resistant WT Pol β were confirmed by western blotting.
Western blot analysis
Western blotting was performed as described (47). Briefly, protein samples were loaded into polyacrylamide gels and subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis. To detect the protein of interest, the membranes were incubated with antibodies recognizing DNA Pol β (Abcam ab1831), β-Tubulin (2146S) and α-Tubulin (Cell signaling 2144S). The secondary antibodies used were anti-rabbit IgG, HRP-linked antibody (GE Healthcare Life Sciences NA9340) and anti-mouse IgG, HRP-linked antibody (GE Healthcare Life Sciences NA931).
Single cell gel electrophoresis assay (comet assay)
Equal numbers of cells (1 × 105) were plated into 12-well plates. Cells were treated 24 h after plating with 50 μg/ml bleomycin (BLM) for 1 h. After treatment, the cells were prepared and analyzed in neutral and alkaline comet assays at different recovery times, according to published procedures (48) using Comet slides (Trevigen Cat # 4250–200-03). Cells were also treated with 10 μM NU7441 for 24 h. After treatment, cells were prepared and analyzed in the neutral comet assay. Additionally, cells were also treated with 10 μM NU7441 for 24 h and then treated with 50 μg/ml BLM for 1 h. Following treatment, the cells were prepared and analyzed in the neutral comet assay at different recovery times. Image analysis of at least 50 cells was performed using CometScore software (TriTek, Sumerduck, VA, USA). Data are represented as mean ± SEM (n = 50).
DSB repair assays
The mutagenic end-joining assay was performed as described (49). Briefly, the aNHEJ assay consists of two separate constructs: a Tetracycline repressor gene (TetR) with the I-SceI endonuclease site incorporated in-frame in the TetR and a red fluorescent protein (RFP) gene under the control of cytomegalovirus (CMV) promoter. In between the CMV promoter and the RFP gene is the TetO binding site. (Figure 1A). A stably integrated inducible I-SceI expression vector is also incorporated in this cell line. In the I-SceI inducible system, the I-SceI gene is fused with a ligand-binding domain of rat glucocorticoid receptor (GR) at the C-terminus. The TA ligand for GR allows the I-SceI to be translocated into the nucleus. A destabilization domain (DD) is fused to the N-terminus of the I-SceI gene. This blocks the destabilizing effect, resulting in increased I-SceI levels. DSBs were induced by TA ligand and Shield1 (Clontech). Shield1 is a ligand that reversibly stabilizes and destabilizes the DD-tagged I-SceI in the U2OS EJ-DR cells. aNHEJ and HR repair activity were then assessed by quantification of the RFP+ and GFP+ cells, respectively, using a BD FACSCaliburH instrument. FL1 and FL2 channels were used to detect GFP and RFP, respectively, which were analyzed simultaneously to minimize spectral overlap. RFP+, GFP+ and parental cells were used as controls for optimization, and the data were analyzed using FlowJo 8.8.6 software. Experiments were performed three times and the data were graphed as mean ± SEM.
Figure 1.

(A) Schematic of the mutagenic end-joining. The aNHEJ construct consists of a TetR gene with the I-SceI endonuclease site incorporated in-frame so that Tet Repressor (TetR) is produced. It also consists the red expression protein (RFP) gene under the control of CMV promoter and the TetO binding site is in between the CMV promoter and the RFP gene. The purple circles represent the TetR that binds to the TetO and represses expression of RFP. Upon induction of I-SceI, if the TetR ORF is disrupted and if not joined correctly, results in loss of TetR binding to its recognition sequence TetO in the reporter gene plasmid, thus leading to the expression of RFP. Therefore, the loss of TetR is due to circumstances of mutagenic end-joining. (B) The GFP reporter is also shown. (C) Western blot showing depletion of Pol β using siRNA targeting of the POLB gene. WT is wild-type no treatment control, C denotes cells treated with siGENOME RISC-free negative control non-targeting siRNA, and si1, si3, and si4 denote cells treated with three different siRNAs targeting the POLB gene. α-tubulin is used as a loading control. Pol β/α-tubulin indicates the ratio of the intensity of the Pol β band over the α-tubulin band in each lane and is a reflection of the relative amount of Pol β that is depleted from the cells. (D) Western blot showing depletion of Pol β by an shRNA targeting the POLB gene. C denotes cells treated with a non-targeting shRNA, and sh-POLB denotes cells treated with an shRNA that targets the POLB gene. (E) aNHEJ is suppressed in cells treated with siRNA directed against POLB. Quantification of the flow cytometry plots for U2OS EJ-DR cells. WT, C, si1, si3 and si4 denote levels of RFP+ cells without ligands and WT+, C+, si1+, si3+ and si4+ denote RFP+ cells treated with TA and Shield1 to induce cutting at the I-SceI site. The percentage of RFP+ cells is plotted on the Y-axis. Data are graphed as mean ± SEM (n = 3) ** (P ≤ 0.0099), * (P ≤ 0.04) and P = ns (not significant). (F) HR is slightly suppressed in Pol β-depleted cells. Quantification of the flow cytometry plots for U2OS EJ-DRs cells. WT, C, si1, si3 and si4 denote levels of GFP+ cells without ligands. WT+, C+, si1+, si3+ and si4+ indicate GFP+ cells treated with TA and Shield1. C+ and si1+, si3+ and si4+. The percentage of GFP+ cells plotted on the Y-axis. Data are graphed as mean ± SEM (n = 3) ** (P = 0.003), * (P ≤ 0.04) and P = ns (not significant). (G) DNA end-joining is suppressed in Pol β-depleted cells. Pol β was depleted using an shRNA targeting the POLB gene. WT denotes mock-treated cells, C denotes cells treated with a non-targeting shRNA control and sh-POLB denotes cells treated with an shRNA targeted against the POLB gene. The percentage of RFP+ cells plotted on the Y-axis. Data are graphed as mean ± SEM (n = 3) ** (P = 0.001), * (P ≤ 0.004) and P = ns (not significant).
Cell cycle analysis
Cell cycle profiles of U2OS EJ-DR cells were assessed by propidium iodide (PI) staining followed by flow cytometry analysis, as described in (49).
Flow cytometry for γH2AX
MCF7 and MCF7-Pol β-depleted cells were treated with 50 μg/ml BLM for 1 h. Cells were then rinsed with phosphate-buffered saline (PBS) and replenished with fresh media. Cells were allowed to recover for 0, 2 and 4 h post treatment. Cells were harvested by trypsinization, washed once with PBS and then pelleted. The pellet was resuspended by 70% ice cold ethanol in a dropwise manner. Cells were fixed overnight at −20°C. The cells were incubated with primary γH2AX antibody (Cell Signaling 9718P) at 1:200 overnight at 4°C. Cells were then washed twice with PBS and incubated with anti-rabbit Alexa647 (Molecular probes, Invitrogen) at 1:100 for 1 h at room temperature. Finally, cells were washed twice with PBS and resuspended in 500 μl PI/RNase staining buffer (BD Pharmingen). Fluorescence was analyzed by flow cytometry using the BD FACSCalibur and analyzed using FlowJo 8.8.6 software.
Survival experiments
For the Cell TiterGlo assay (Promega), U2OS and U2OS-Pol β-depleted cells were dispensed into white-bottom 96-well plates and incubated for 24 h. Cells were then incubated with DNA damage-inducing agents for 24 h. After incubation, the cells were assayed for adenosine triphosphate luminescence as described in the manufacturer’s instructions using a microplate reader. For clonogenic survival experiments, MCF7 and MCF7-Pol β-depleted cells (1 × 105) were seeded into 12-well plates. Cells were treated with various concentrations of BLM (Enzo Life Science) and/or DNA-PKcs inhibitor (NU7441 and KU0060648) for 24 h followed by trypsinization, dilution and plating. Treated cells were allowed to form colonies for 10–14 days before staining with 0.25% crystal violet.
Breakpoint analyses and DNA sequencing
Genomic DNA was isolated from ∼100 000 control and Pol β knock down cells 96 h after induction of DSBs. Breakpoint analysis was performed using a highly reproducible and validated assay described previously (50). The I-SceI site and flanking sequence at the I-SceI and TetR locus was amplified from 300 ng of genomic DNA using the following primers: 5′-GCTAACCATGTTCATGCCTTC-3′ (forward) and 5′-ACCTAGCTTCTGGGCGAGTT-3′ (reverse). The polymerase chain reaction (PCR) amplicon was ∼291 base pairs (bp). For sequencing of the breakpoint junctions, PCR amplicons were gel-purified and Ion Torrent sequencing was performed at the Yale Keck facility.
PCR (30 cycles) was performed across the integrated I-SceI locus for maximum depth of coverage at the site of repair. Library preparation and sequencing were carried out according to the manufacturer’s protocol by the Keck DNA Sequencing Lab (New Haven, CT, USA) using an Ion Torrent Personal Genome Machine with accompanying Ion 316 chip (Life Technologies).
Sequencing data were filtered for quality and demultiplexed using the FASTX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) to ensure only high-quality, non-ambiguous read data remained for analysis. Reads were filtered such that only those containing 60% or more bases with a Phred+33 quality score of 30 or greater were considered for analysis to ensure only high quality reads remained. Remaining reads were aligned to known reference sequence using Bowtie-2 alignment software (50–52). A reconstruction experiment showed that we are able to detect events described in Tables 1–4 (Supplementary Figure S14).
Table 1. Types of mutations observed in the presence and absence of Pol β.
| Control | siPOLB | P-valued | |
|---|---|---|---|
| Total sequences | 260 485 | 129 193 | |
| Mutant sequences | 53 623 (20.6)a | 24 302 (18.8)a | 2.93 × 10−36 |
| Total deletions | 44 906 (17.2)b | 19 873 (15.4)b | 5.08 × 10−46 |
| Total insertions | 8717 (16.3)c | 4429 (18.2)c | 1 |
a([Total # mutant sequences/total sequences] × 100).
b([# of total deletions/total number of sequences] × 100).
c([# of total insertions/total number of sequences] × 100).
dChi-square test P-values adjusted with the Bonferroni correction.
Table 4. Types of insertions observed in control and Pol β-depleted cells.
| Type of insertion signature | Control # (%)5 | siPOLB # (%)5 | P-value6 |
|---|---|---|---|
| Templated insertions1 | 4297 (49.3) | 2487 (56.2) | 4.70 × 10−11 |
| Templated slippage2 | 55 (0.63) | 114 (2.6) | 2.18 × 10−18 |
| Misincorporation and synthesis across break3 | 108 (1.2) | 13 (0.29) | 5.46 × 10−5 |
| Complex insertions templating from the TetR4 | 7 (0.080) | 37 (0.8) | 1.723 × 10−9 |
1Templated insertions.
2Templated slippage.
3Misincorporation and synthesis across break.
4Complex insertions templating from TetR.
5([Number of insertion sequences in each type of insertion signature/Total # of insertion sequences] × 100).
6Chi-square test P-values adjusted with the Bonferroni correction.
RESULTS
DNA Pol β participates in aNHEJ
To determine whether Pol β functions in aNHEJ we used an aNHEJ assay developed by Bindra and Powell (49). The EJ-RFP cassette was previously integrated into U2OS cells harboring the DR-GFP reporter for HR enabling us to monitor both aNHEJ and HR at the same time as described in (49). A stably integrated inducible I-SceI expression system is also incorporated into this cell line, ensuring high levels of I-SceI expression and induction (49). Cleavage at the I-SceI site in the TetR plasmid can disrupt the open reading frame (ORF) resulting in loss of TetR binding to TetO in the reporter gene plasmid, with consequent expression of RFP. The aNHEJ and HR assays were performed as described (49); see ‘Materials and Methods’ section for additional details. aNHEJ and HR repair activity were assessed by quantification of the percentages of RFP+ and GFP+ cells, respectively. We point out that although precise rejoining of the ends of the break induced by I-SceI is possible in this assay, we are unable to distinguish between precise rejoining and uncut sequences.
Individual siRNAs targeting the POLB gene (see ‘Materials and Methods’ section for sequences) were transfected into the U2OS EJ-DR cells followed by 72 h incubation to ensure downregulation of Pol β. In parallel, cells were either mock-transfected or transfected with non-targeting siRNA as negative controls. Depletion of ∼95% of the Pol β protein by each siRNA targeting the POLB gene was confirmed by western blot (Figure 1C). Depletion of Pol β using shRNA against the 3′ UTR of the POLB transcript was confirmed by western blot (Figure 1D). Cells were then treated with Shield1 and TA for 24 h to induce DSBs at the I-SceI site within the reporter constructs. The percentages of RFP+, reflective of aNHEJ and GFP+ cells, reflective of HR, were quantified by flow cytometry as described (49). Representative flow cytometry plots for U2OS EJ-DR GFP+ and RFP+ cells are shown in Supplementary Figure S1.
Upon induction of I-SceI, we find that the percentage of RFP+ cells increases significantly compared to cells not induced to express I-Scel in mock (WT) and control (C) siRNA-transfected cells (Figure 1E). Importantly, upon depletion of Pol β by three different siRNAs, the percentage of RFP+ cells, representing cells that underwent aNHEJ, does not increase significantly over that of cells not induced to express I-SceI (Figure 1E). Comparison of the levels of RFP expressed in cells depleted of Pol β with the control (C) siRNA shows that significantly lower percentages of cells express RFP when they are depleted of Pol β. We did not find any significant differences in the percentage of GFP+ cells between the control and Pol β-depleted cells (Figure 1F), suggesting there was little effect on HR. We performed additional experiments in which we used shRNA against the 3′ UTR of the POLB transcript. Upon induction of I-SceI we observed a significant increase in RFP in control cells, but not in cells treated with the sh-POLB. Once again, the percentage of RFP+ cells was significantly lower than that observed in the control (C) cells when they were depleted of Pol β (Figure 1G). In combination, our results indicate that Pol β plays a role in aNHEJ.
The cell cycle is not perturbed by depletion of Pol β
It has been reported that aNHEJ is significantly repressed when cell growth is arrested (49). To determine if cells depleted of Pol β were growth arrested we analyzed the cell cycle profile of the U2OS EJ-DR cells in which Pol β was depleted either by siRNA or shRNA. Importantly, we did not observe cell cycle arrest when Pol β was depleted (Supplementary Figures S2 and 3). This demonstrates that the decreased aNHEJ in cells depleted of Pol β is not due to growth arrest or altered cell cycle kinetics.
Pol β is involved in aNHEJ
To determine if the presence of Pol β is important for aNHEJ, we asked whether overexpression of WT Pol β in Pol β-depleted cells rescues the decrease in aNHEJ. We constructed an siRNA-resistant POLB expression vector, POLB1 and co-transfected it together with the siRNA targeting the POLB gene. Similarly, we constructed an siRNA-resistant Y265C POLB expression vector, Y265C and co-transfected it together with the siRNA targeting the POLB gene. We show that we are able to express WT Pol β or Y265C-Pol β from their respective plasmids using western blotting, even in the presence of siRNA1 (Figure 2A and B, respectively). We note that the siRNA resistant POLB plasmids are only partially resistant to the siRNA but the levels of expression are similar to those of endogenous Pol β in control cells. Cells were treated with Shield1 and TA for 24 h to induce DSBs at the I-SceI site within the reporter construct. The percentage of RFP+ cells was monitored by flow cytometry. Upon induction of I-Scel, the percentage of RFP+ cells did not increase significantly when they were depleted of Pol β by siRNA targeting the POLB gene compared to the control. However, this phenotype was rescued by overexpression of siRNA-resistant Pol β (Figure 2C). Recent work from our laboratory indicates that expression of the Y265C Pol β protein in mice leads to short CDR3 junctions during V(D)J recombination, which is an end joining process (46). We have previously shown that the catalytic efficiency of the Y265C protein is significantly lower than that of WT Pol β and that Y265C is unable to support base excision repair (BER) (53,54). Importantly, overexpression of the Y265C protein does not complement the decrease in aNHEJ observed upon depletion of WT Pol β. This indicates that the catalytic activity of Pol β and not just its presence is important for aNHEJ (Figure 2C).
Figure 2.

Overexpression of Pol β rescues the deficiency of mutagenic end-joining in Pol β-depleted cells. (A) Western blot. Exogenous-Pol β (Exo-Pol β) is exogenously expressed Pol β from the siRNA-resistant plasmid harboring the POLB cDNA and endogenous-Pol β (Endo-Pol β) is endogenously expressed protein. C denotes negative non-targeting siRNA control; si1 denotes cells treated with si1 targeted against the POLB gene; POLB1 denotes overexpression of Pol β protein in cells by treating them with a plasmid harboring the cDNA of the POLB gene that is resistant to si1; POLBsi1 denotes cells treated with both siRNA1 targeting the POLB gene and a plasmid overexpressing Pol β. The exogenously expressed protein carries an HA epitope tag, resulting in its slower resolution, versus endogenous protein, in the gel. Quantification at the bottom of the image Exo-Pol β/tubulin. (B) Exogenous-Y265C-Pol β (Exo-Pol β-Y265C) is exogenously expressed Pol β from the siRNA-resistant plasmid harboring the Y265C POLB cDNA and endogenous-Pol β (Endo- Pol β) is endogenously expressed protein. Y265C denotes overexpression of Y265C-Pol β protein in cells by treating them with a plasmid harboring the cDNA of the Y265C POLB gene that is resistant to si1; Y265Csi1 denotes cells treated with both siRNA1 targeting theY265C POLB gene and a plasmid overexpressing Y265C-Pol β. The exogenously expressed protein carries an HA epitope tag, resulting in it's slower resolution, versus endogenous protein, in the gel. (C) Overexpression of Pol β rescues the aNHEJ deficiency in cells treated with siRNA targeting the POLB gene. But the overexpression of Y265C-Pol β does not rescue the aNHEJ deficiency. The percentage of RFP+ cells plotted on the Y-axis. Data are graphed as mean ± SEM (n = 3) **** (P < 0.0001) *** (P = 0.0009) and P = ns (not significant).
Next, we asked if inhibiting the ability of the cells to perform cNHEJ would increase aNHEJ in the presence and absence of Pol β. NU7441 is a highly potent and selective inhibitor of DNA-PKcs, which is essential for cNHEJ (55). We reasoned that treating cells with this drug would decrease cNHEJ and result in an increase in aNHEJ, the latter of which can be scored in our assay. Upon inhibition of cNHEJ with NU7441, we observe an increase in aNHEJ in the control cells (Figure 3). Therefore, inhibiting cNHEJ results in repair of more breaks by aNHEJ. In contrast, aNHEJ is still suppressed in cells treated with NU7441 and depleted of Pol β by si1 (Figure 3). This suggests that when the cNHEJ pathway is inhibited, the aNHEJ pathway relies, at least in part, on Pol β to repair DSBs. In combination, our results indicate a role for Pol β in aNHEJ.
Figure 3.
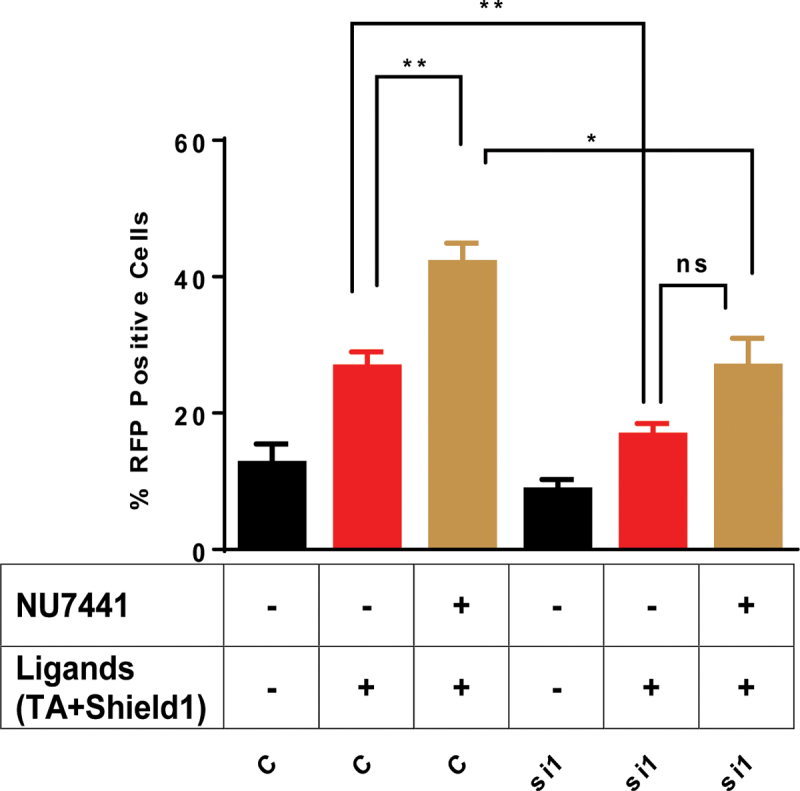
Mutagenic end-joining is suppressed in the presence of DNA-PKcs inhibitor upon depletion of Pol β. Quantification of the flow cytometry plots for U2OS EJ-DRs cells. Cells were treated with a non-targeting siRNA (C) or siRNA 1 that targets the POLB gene (si1). NU7741 is an inhibitor of DNA-PKcs and its addition to the cells is indicated by +. The addition of the ligands needed to induce expression of I-SceI is indicated by +. The percentage of RFP+ cells plotted on the Y-axis. Data are graphed as mean ± SEM (n = 3) ** (P ≤ 0.004), * (P = 0.02) and P = ns (not significant).
Pol β-depleted cells are sensitive to agents that induce double-strand breaks
Our finding that Pol β promotes aNHEJ suggests that this polymerase functions in DNA end-joining in cells. In addition, previous reports have suggested that expression of Pol β is linked to treatment of cells with agents that induce DSBs, predominantly BLM (56,57). To determine if Pol β functions in the repair of BLM induced breaks, we depleted Pol β in MCF7 and U2OS cells using an shRNA and treated them with BLM. Depletion of Pol β in these cell lines was confirmed by western blot (Supplementary Figure S4A and B). As shown in Figure 4A, depletion of Pol β results in greater cellular sensitivity to BLM compared to cells treated with a control shRNA that does not target Pol β. Similar results were observed for U2OS cells (Figure 4B).
Figure 4.

Pol β-depleted cells are sensitive to BLM and NU7441 and KU0060648. (A) MCF7-Pol β-depleted cells are sensitive to BLM. (B) U2OS-Pol β-depleted cells are sensitive to BLM. (C) MCF7-Pol β-depleted cells are sensitive to DNA-PKcs inhibitor NU7441. (D) U2OS-Pol β-depleted cells are sensitive to DNA-PKcs inhibitor NU7441. (E) MCF7-Pol β-depleted cells are sensitive to DNA-PKcs inhibitor KU0060648. (F) MCF7-Pol β-depleted cells are more sensitive to BLM when pre-treated with DNAPKcs inhibitor. Pol β-depleted MCF7 cells were pre-treated with 10 μM DNA-PKcs inhibitor for 24 h. Cells were then treated with a range of concentrations of 10, 50, 100 and 250 μg/ml BLM for 24 h. (G) U2OS-Pol β-depleted cells are more sensitive to BLM when pre-treated with DNA-PKcs inhibitor. Pol β-depleted U2OS cells were pre-treated with 10 μM DNA-PKcs inhibitor for 24 h. Cells were then treated with a range of concentrations of 10, 50, 100, 250, 500 and 1000 μg/ml BLM for 24 h. * (P = 0.05).
Inhibition of DNA-PKcs and therefore cNHEJ with NU7441 results in increased levels of aNHEJ for cells treated with control siRNA but not siRNA directed against POLB. This indicates that at least a subset of ends that are shunted to aNHEJ cannot be joined by this process upon depletion of Pol β. Therefore, we asked whether Pol β-depleted cells are sensitive NU7441. MCF7 and U2OS cells were nucleofected with shRNA that targets the 3′UTR of the POLB gene, treated with various concentrations of NU7441. We observe that MCF7 and U2OS cells depleted of Pol β have decreased cellular survival compared to mock-depleted cells in the presence of NU7441 (Figure 4C and D). Next, we asked whether Pol β-depleted cells are more sensitive to KU0060648, a different DNA-PKcs inhibitor, compared to mock-depleted cells. We found that MCF7 cells depleted of Pol β have significantly decreased cellular survival in the presence of KU0060648 (Figure 4E). We observe sensitivity of Pol β-depleted MCF7 and U2OS cells to BLM and NU7441 alone; however, significantly greater sensitivity to BLM is observed in both cell lines in the presence of NU7441 upon depletion of Pol β (Figure 4F and G). These results indicate that upon inhibition of cNHEJ by NU7441, the breaks induced by BLM are repaired by a Pol β-dependent pathway. Our data with the EJ-RFP reporter demonstrating that aNHEJ is utilized upon inhibition of cNHEJ by NU7441 suggests that this Pol β-dependent pathway is aNHEJ.
Double strand breaks accumulate in DNA Pol β-depleted cells
Given that Pol β-depleted cells are sensitive to BLM and that inhibition of cNHEJ by treatment with NU7441 increases sensitivity, we reasoned that depletion of Pol β would result in increased levels of DSBs in the presence of BLM. To test this, we monitored the levels of DSBs in the presence of BLM using γH2AX by flow cytometry. Upon treatment with BLM, we observe an increase in the levels of γH2AX in both control and Pol β-depleted MCF7 cells. After 4 h of recovery, γH2AX persisted in Pol β-depleted cells (Figure 5), but its levels decreased to those of baseline in MCF7 cells expressing Pol β.
Figure 5.

Persistence of γH2AX positive cells in Pol β-depleted cells even after 4 h recovery time. MCF7 and MCF7-sh-POLB cells were treated with 50 μg/ml BLM for 1 h and allowed to recover for 0, 2 and 4 h and analyzed by flow cytometry. Cells were stained with γH2AX antibody to assess the levels of DSBs. Data are plotted as the mean ± SEM (n = 3) *** (P < 0.0001).
We further corroborated the persistence of DSBs in Pol β-depleted cells using the neutral comet assay with WT and Pol β-depleted MCF7 and U2OS cells treated with BLM. We found that DSBs persisted in Pol β-depleted cells significantly longer and at greater levels compared to MCF7 and U2OS WT cells even after 4 h of recovery (Figure 6A and B). Representative images from each time point of recovery post BLM treatment for U2OS and U2OS Pol β-depleted cells are shown in Figure 6C. We also asked whether single strand break (SSBs) levels increased in these cells using the alkaline comet assay (Supplementary Figure S5). We observed that the levels of SSBs were low in Pol β-depleted and control MCF7 cells, suggesting that we are not interrogating base excision repair and/or single-strand break repair under our experimental conditions. We also found that DSBs persisted in Pol β-depleted cells at slightly, but statistically significant, greater levels compared to MCF7 and U2OS WT cells when treated with the DNA-PKcs inhibitor NU7441 for 24 h (Figure 6D and E). Additionally, we show that Pol β-depleted cells exhibit a significantly greater persistence of DSBs versus controls when they are treated with both BLM and NU7441, which blocks cNHEJ (Figure 6F and G). In combination, our results are consistent with the idea that Pol β functions in DSB repair through the aNHEJ pathway and that the deficiency of aNHEJ in the absence of Pol β sensitizes cells to BLM, a drug that predominantly forms DSBs.
Figure 6.

Pol β-depleted cells have increased levels of DSBs upon treatment with BLM or NU7441. (A) MCF7 and MCF7-sh-POLB cells were treated with 50 μg/ml BLM for 1 h and allowed to recover for 0, 2 and 4 h. DSBs were analyzed by the neutral comet assay. The tail moment is plotted on the Y-axis. ** (P = 0.0028); ****(P < 0.0001). (B) U2OS and U2OS-sh-POLB cells were treated with 50 μg/ml BLM for 1 h and allowed to recover for 0, 2 and 4 h. DSBs were analyzed by the neutral comet assay. The tail moment is plotted on the Y-axis. * (P = 0.03); ** (P = 0.005); ****(P < 0.0001). (C) Representative images from each time point of recovery post BLM treatment for U2OS and U2OS Pol β-depleted cells. (D) MCF7 and MCF7-sh-POLB cells were treated with 10 μM NU7441 for 24 h. DSBs were analyzed by the neutral comet assay. The tail moment is plotted on the Y-axis. ** (P = 0.0012). (E) WT U2OS and U2OS-sh-POLB cells were treated with 10 μM NU7441 for 24 h. DSBs were analyzed by the neutral comet assay. The percentage of tail DNA is plotted on the Y-axis. * (P = 0.020). (F) MCF7 and MCF7-sh-POLB cells were treated with 10 μM NU7441 for 24 h and then treated with 50 μg/ml BLM for 1 h and allowed to recover for 0, 2 and 4 h. DSBs were analyzed by the neutral comet assay. The tail moment is plotted on the Y-axis. ** (P = 0.003); *** (P = 0.0006); ****(P < 0.0001). (G) U2OS and U2OS-sh-POLB cells were treated with 10 μM NU7441 for 24 h and then treated with 50 μg/ml BLM for 1 h and allowed to recover for 0, 2 and 4 h. DSBs were analyzed by the neutral comet assay. The tail moment is plotted on the Y-axis. * (P = 0.03); ** (P = 0.009); ****(P < 0.0001).
Depletion of Pol β results in fewer small deletions
To ascertain the molecular events resulting from depletion of Pol β in the reporter assay, we characterized the DNA junctions resulting from end-joining after I-SceI cutting. Genomic DNA was isolated from control cells (non-targeting RISC-free siRNA) and cells depleted of Pol β using si1 after treatment with the TA ligand and Shield1 to induce I-SceI expression. Following PCR amplification of the break site using flanking primers within the TetR gene, ion torrent sequencing was performed, as described in the ‘Materials and Methods’ section. As shown in Table 1, depletion of Pol β results in significantly fewer mutant sequences than observed from cells treated with control, non-targeting siRNA. Importantly, Pol β-depleted cells have fewer deletions than control cells. These observations suggest that the end-joining differs in cells in the presence or absence of Pol β.
Pol β participates in gap filling during aNHEJ in the generation of deletions
There were too many total deletion events for us to analyze each one separately so we focused on events that occurred at significantly different frequencies in the control and Pol β-depleted cells. We binned identical deletion events and calculated the odds of observing these deletion events in the control cells and in the Pol β-depleted cells. We identified deletion events where the odds of these events occurring were statistically different in the control and Pol β-depleted cells based on chi-square tests, accounting for ∼15% of deletion events. These chi-square tests were adjusted for multiplicity with the Bonferroni correction and an alpha level of 0.001 was used to select these events. Events that occurred in less than 10 control cells and Pol β-depleted cells were further eliminated from analysis. The remaining deletion events among mutant sequences occurred just as frequently in the control and Pol β-depleted cells and were therefore not of interest in distinguishing differences between these two groups in the processing of I-SceI induced breaks.
Deletions of 20 bp or fewer, defined as small deletions, were the most common events observed among mutant sequences and were present in both control and Pol β-depleted cells. The small deletion events occurring at different frequencies in the control and Pol β-depleted cells are displayed in Supplementary Figure S6 (small deletion events more frequently observed in control cells), Supplementary Figure S7 (small deletion events more frequently observed in siPOLB cells), Supplementary Figure S9 (large deletion events more frequently observed in control cells), Supplementary Figure S10 (large deletion events more frequently observed in siPOLB cells). Next, we perused these sequences to identify potential deletion mechanisms. The rationale used to model deletion events was that if μH was present, even just one base, it would be used, and that fewer rather than greater numbers of bases would be removed by end-processing in order for joining to take place.
Modeling of the small deletions in this manner, displayed in Supplementary Figure S8A–F, produced six different deletion signatures as summarized in Table 2. These events include direct joins for which no μH could be identified (Supplementary Figure S8A), a Pol μ signature in which DNA synthesis is directed across strand breaks (Supplementary Figure S8B) (58) and four different signatures of μH-mediated end-joining involving either 3′ and 5′ end-processing activity and gap filling (Supplementary Figure S8C), just 3′ and 5′ end-processing activity (Supplementary Figure S8D), 3′ end-processing activity and gap filling (Supplementary Figure S8E) or just μH with only 3′ end-processing activity (Supplementary Figure S8F). Supplementary Tables S2 and 3 summarize the total counts for the deletion events in control and Pol β-depleted cells.
Table 2. Types of small deletions observed in control and Pol β-depleted cells.
| Type of deletion signature | Control # (%)7 | siPOLB # (%)7 | P-value8 |
|---|---|---|---|
| Direct end-joining1 | 176 (0.39) | 51 (0.26) | 1 |
| Pol μ signature2 | 7104 (15.8) | 3824 (19.2) | 3.42 × 10−24 |
| μH, 3′and 5′ end-processing + gap filling3 | 651 (1.4) | 125 (0.63) | 4.74 × 10−16 |
| μH, 3′ and 5′ end-processing4 | 788 (1.8) | 341 (1.7) | 1 |
| μH, 3′ end-processing+ gap filling5 | 1923 (4.3) | 607 (3.1) | 4.75 × 10−11 |
| μH, 3′ end-processing6 | 767 (1.7) | 476 (2.4) | 1.97 × 10−6 |
1Direct joins for which no μH could be identified;
2a Pol μ signature in which DNA synthesis is directed across strand breaks and four different signatures of
3μH-mediated end-joining involving 3′ and 5′ end-processing and gap filling;
43′ and 5′ end-processing;
53′ end-processing and gap filling; or
6μH plus 3′ end-processing. See text for more details.
7([Number of deletion sequences in each type of deletion signature/Total # of deletion sequences] X 100).
8Chi-square test p-values adjusted with the Bonferroni correction.
Signatures observed significantly more often in control cells include one that involves μH along with 3′ and 5′ end-processing and gap filling (Supplementary Figure S8C) and one that involves μH along with 3′ end-processing and gap filling (Supplementary Figure S8E). Signatures observed significantly more often in Pol β-depleted cells include the Pol μ signature (Supplementary Figure S8B), and one that involves μH and 3′ end-processing (Supplementary Figure S8F). Interestingly, all signatures of end-joining that employ gap filling are observed more often in control cells, suggesting that Pol β functions in gap filling during aNHEJ. Conversely, the Pol μ signature is observed more often in cells depleted of Pol β, suggesting that Pol μ is at least partially functionally redundant with Pol β during end-joining.
The large deletion signatures are summarized in Table 3. The large deletion signatures are displayed in Supplementary Figures S9 and 10. Supplementary Tables S4 and 5 summarize the total counts for the large deletion events in control and Pol β-depleted cells. Four deletion signatures were observed for the large deletions. The events significantly enriched in Pol β-depleted cells include direct end-joining for which no μH could be identified, and a potential Pol μ signature in which DNA synthesis is directed across strand breaks. In contrast, large deletions occurring more frequently in control cells involve μH along with 3′ and 5′ end-processing and gap filling. The signatures for large and small deletions are similar in regard to the Pol μ signature being more prevalent in Pol β-depleted cells, suggesting once again that Pol μ functions as a ‘backup’ polymerase for Pol β in some aspects of aNHEJ. In addition, joins resulting from end processing along with gap filling are observed more often in control cells also suggesting a role for Pol β gap filling in the prevention of large deletions. However, for large deletions, direct end joining occurs more often in Pol β-depleted cells, whereas for small deletions direct end joining is observed more often in control cells.
Table 3. Types of large deletions observed in control and Pol β-depleted cells.
| Type of Deletion Signature | Control # (%)5 | siPOLB # (%)5 | P-value6 |
|---|---|---|---|
| Direct end-joining1 | 2 (0.0045) | 44 (0.22) | 2.18 × 10−18 |
| Pol μ signature2 | 4 (0.008) | 24 (0.12) | 3.92 × 10−7 |
| μH, 3′ and 5′ end-processing+ gap filling3 | 167 (0.37) | 8 (0.040) | 4.76 × 10−11 |
| μH, 3′ and 5′ end-processing4 | 131 (0.29) | 35 (0.17) | 1 |
1Direct joins for which no μH could be identified;
2aPol μ signature in which DNA synthesis is directed across strand breaks;
3μH-mediated end-joining involving 3′ and 5′ end-processing and gap filling; and.
43′ and 5′ end-processing. See text for more details.
5([Number of deletion sequences in each type of deletion signature/Total # of deletion sequences] × 100).
6Chi-square test P-values adjusted with the Bonferroni correction.
Pol β prevents certain types of insertions
Insertions are observed as frequently in Pol β-depleted cells as in the control cells (Table 1). Specific insertion events where the odds of these events occurring were statistically different in the control and Pol β-depleted cells were identified in the same manner as for the deletion events. These insertion events occurring at different frequencies in the control and Pol β-depleted cells at an alpha level of 0.001 are displayed in Supplementary Figures S11 (control cells) and S12 (siPOLB cells). We perused these sequences to identify potential insertion mechanisms. Modeling of the insertions displayed in Supplementary Figure S13A–C, produced three different insertion signatures as summarized in Table 4. The most common insertion events are templated insertions involving one base of microhomology and gap filling synthesis (Supplementary Figure S13A). Although observed in both control and Pol β-depleted cells, templated insertions are more frequent in Pol β-depleted cells, suggesting that Pol β may prevent a subset of these events. DNA synthesis from a primer-template with one base of microhomology has been shown to be catalyzed by Pols μ and λ (59). A signature observed more often in control cells in which Pol β is present is misincorporation and synthesis across a break (Supplementary Figure S13C). In this case, after 3′end-processing, a non-templated base appears to be inserted before alignment and synthesis across the break. However, the base that is inserted may in fact be templated via slippage, as suggested in Supplementary Figure S13C. It is tempting to speculate that slippage may be an underlying mechanism for events modeled in Supplementary Figure S13C given that Pol β has long been known to function in slippage mediated DNA synthesis (for an excellent example see (60)), especially within or near to nucleotide repeats. Snap- back synthesis, perhaps catalyzed by Pol θ, in the case of one base pair is unlikely and either misincorporation or a more complex scheme may be an underlying mechanism of insertion mutagenesis. For insertion events modeled in Supplementary Figure S13C, we suggest that synthesis across breaks is catalyzed by Pol μ, and that one base gap filling on the ‘bottom’ strand may be catalyzed by Pol β. Signatures observed more often in Pol β-depleted cells include templated insertions, as described above, templated slippage and complex insertions involving templating from sequences far away (in our case from within the TetR gene) from the ends (43). Templated slippage (Supplementary Figure S13B) involves primer annealing, incorporation and then movement and annealing of the primer, sometimes in several cycles. This activity has been described for Pol θ as a result of its many contacts with the primer strand (61). Our results suggest that the presence of Pol β may protect cells from some of these types of insertion events and that in select cases, especially those with staggered ends such as those that result from cutting by I-SceI, Pol θ may be functionally redundant with Pol β.
DISCUSSION
Using a previously developed reporter assay (49), we found that aNHEJ decreases in Pol β-depleted cells (Figure 1E and G). DSB repair via end-joining is orchestrated by two pathways: cNHEJ and aNHEJ. Classical end-joining is a Ku- and DNA-PKcs-dependent pathway. Upon treatment of control cells with inhibitors of DNA-PKcs, aNHEJ increases, suggesting that the cells have greater reliance on aNHEJ when cNHEJ is inhibited. However, in Pol β-depleted cells, inhibition of cNHEJ does not result in increased aNHEJ, suggesting that Pol β functions in aNHEJ. To determine if Pol β functions in DSB repair in cells, we treated MCF7 and U2OS cells with BLM, NU7441 or a combination of both. Pol β-depleted cells are more sensitive than WT cells to BLM and to NU7441 alone, and they demonstrate additional sensitivity when the drugs are administered in combination. In combination, our results suggest that Pol β plays a role in aNHEJ.
Pol β-depleted cells are sensitive to DNA-PKcs inhibitors and to bleomycin
Depletion of Pol β in both MCF7 and U2OS cells sensitizes them to two different inhibitors of DNA-PKcs and this correlates with the presence of persistent DSBs in these cells, as assessed in the neutral comet assay. This suggests that endogenous DSBs that are not processed via cNHEJ due to inhibition of DNA-PKcs rely on aNHEJ that is at least in part catalyzed by Pol β. The same is true for breaks induced by BLM. BLM is a glycopeptide antibiotic with a unique mechanism of antitumor activity. Miller et al. showed that in both hamster and human cells, the sensitivity of BLM-induced repair synthesis to ddTTP inhibition was essentially identical as that observed for purified Pol β, indicating the repair process proceeded through a mechanism utilizing Pol β (62). Later it was found that the inhibitors of Pol β potentiated the action of the anticancer drug BLM in cultured A549 cells, without any influence on the expression of Pol β (63). Liu et al. showed that cell viability of Pol β-deficient mouse embryo fibroblasts (MEFs) was significantly lower than that of isogenic WT cells when treated with the same BLM dosage. BLM essentially induces two SSBs to create a DSB (for a review see (64)). Either staggered or blunt ends are produced, depending upon the sequence context. Along with the induction of SSBs, BLM can lead to the production of oxidized abasic sites or a base propenal and a gap with 3′-phosphoglycolate/5′-phosphate ends that are processed by apurinic/apyrimidic endonuclease 1 (APE1), followed by gap-filling by Pol β. Therefore, it is likely that survival of cells after treatment with BLM is dependent on both BER and double-strand break repair. Although we do observe greater levels of DSBs at 0, 2 and 4 h after treatment of cells with BLM or a combination of BLM and NU7441, we do not observe accumulation of SSBs within this timeframe. This indicates that there is either rapid repair of the SSBs or that conversion of the SSBs to DSBs occurs rapidly. Alternatively, in our hands the comets generated in alkaline buffer (Supplementary Figure S5) may not reflect SSBs, especially if they are closely spaced, as appears to be the case for BLM (64). Importantly, Pol β-depleted MCF7 and U2OS cells display greater sensitivity to a combination of BLM plus NU7441 compared to BLM alone. We suggest that the sensitivity of the cells to BLM in the absence of NU7441 reflects predominantly the BER role of Pol β, in filling gaps created by excision of oxidized bases. Furthermore, we suggest that the aNHEJ function of Pol β is revealed when cells are treated with both BLM and NU7441. Perhaps clustered damage consisting of oxidized bases along with breaks results in recruitment of Pol β via the BER machinery, localizing it such that it participates in gap filling during aNHEJ of breaks at these sites. Importantly, our results in combination with other studies suggest that the status of Pol β can impact treatment of tumors with BLM.
Pol β functions in gap filling DNA synthesis during aNHEJ
Comparison of events observed more frequently in control cells compared to Pol β-depleted cells permitted us to analyze potential mechanisms of aNHEJ that involve Pol β. Our results, at least for structures with 3′ overhangs generated by I-SceI, suggest that Pol β functions in gap filling during the generation of deletions. Pol β is known to be a gap filling polymerase and in fact exhibits higher catalytic activity and fidelity on a single nucleotide-gapped primer template (65). For insertions, it appears as if Pol β also functions in gap filling synthesis and that it may also be involved in misalignment-mediated DNA synthesis, and this has been known as a predominant signature of Pol β for quite some time (60). Importantly, upon depletion of Pol β, we observe the signatures of Pols μ and θ for insertions and deletions, respectively, suggesting that in some cases these enzymes are functionally redundant with Pol β. However, Pol β does not appear to play a role in the generation of complex insertions, which may result exclusively from the ability of Pol θ to utilize templates at a distance from the break during repair. We point out that previous in vitro studies of end joining suggested that Pol β did not function in this process (59). However, these studies were not conducted with 3′ overhangs similar to the structure that is generated upon digestion with I-SceI. We suggest that Pol β may function in aNHEJ at structures similar to the one used in our study and perhaps after some type of end-processing has already taken place.
In summary, we have demonstrated a role for Pol β in aNHEJ. Our results support our previous conclusion that Pol β functions in V(D)J recombination (46), which is an end joining process. During V(D)J recombination, Pol β likely functions during the joining of palindromic sequences by filling in gaps generated after the search for microhomology. Our results suggest that Pol β is important for aNHEJ especially in cellular processes in which genomic diversity is important, such as V(D)J recombination, and perhaps other processes that have yet to be revealed.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Abhijit Patel for very helpful comments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of Environmental Health Sciences (NIEHS) [5 R01 ES019179–07 to J.B.S.]. Funding for open access charge: NIEHS [5 R01 ES019179–07 to J.B.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Rothkamm K., Kruger I., Thompson L.H., Lobrich M.. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003; 23:5706–5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saleh-Gohari N., Helleday T.. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res. 2004; 32:3683–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jackson S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis. 2002; 23:687–696. [DOI] [PubMed] [Google Scholar]
- 4. Khanna K.K., Jackson S.P.. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 2001; 27:247–254. [DOI] [PubMed] [Google Scholar]
- 5. Raynard S., Niu H., Sung P.. DNA double-strand break processing: the beginning of the end. Genes Dev. 2008; 22:2903–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brandsma I., Gent D.C.. Pathway choice in DNA double strand break repair: observations of a balancing act. Genome Integr. 2012; 3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma Y., Lu H., Schwarz K., Lieber M.R.. Repair of double-strand DNA breaks by the human nonhomologous DNA end joining pathway: the iterative processing model. Cell Cycle. 2005; 4:1193–1200. [DOI] [PubMed] [Google Scholar]
- 8. Poplawski T., Stoczynska E., Blasiak J.. [Non-homologous DNA end joining–new proteins, new functions, new mechanisms]. Postepy Biochem. 2009; 55:36–45. [PubMed] [Google Scholar]
- 9. Badie S., Escandell J.M., Bouwman P., Carlos A.R., Thanasoula M., Gallardo M.M., Suram A., Jaco I., Benitez J., Herbig U. et al. . BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat. Struct. Mol. Biol. 2010; 17:1461–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuhfittig-Kulle S., Feldmann E., Odersky A., Kuliczkowska A., Goedecke W., Eggert A., Pfeiffer P.. The mutagenic potential of non-homologous end joining in the absence of the NHEJ core factors Ku70/80, DNA-PKcs and XRCC4-LigIV. Mutagenesis. 2007; 22:217–233. [DOI] [PubMed] [Google Scholar]
- 11. Decottignies A. Alternative end-joining mechanisms: a historical perspective. Front. Genet. 2013; 4:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Betermier M., Bertrand P., Lopez B.S.. Is non-homologous end-joining really an inherently error-prone process. PLoS Genet. 2014; 10:e1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang H., Perrault A.R., Takeda Y., Qin W., Wang H., Iliakis G.. Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res. 2003; 31:5377–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thode S., Schafer A., Pfeiffer P., Vielmetter W.. A novel pathway of DNA end-to-end joining. Cell. 1990; 60:921–928. [DOI] [PubMed] [Google Scholar]
- 15. Nussenzweig A., Nussenzweig M.C.. A backup DNA repair pathway moves to the forefront. Cell. 2007; 131:223–225. [DOI] [PubMed] [Google Scholar]
- 16. McVey M., Lee S.E.. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008; 24:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deriano L., Roth D.B.. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu. Rev. Genet. 2013; 47:433–455. [DOI] [PubMed] [Google Scholar]
- 18. Chang H.H., Watanabe G., Lieber M.R.. Unifying the DNA end-processing roles of the artemis nuclease: Ku-dependent artemis resection at blunt DNA ends. J. Biol. Chem. 2015; 290:24036–24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lieber M.R., Gu J., Lu H., Shimazaki N., Tsai A.G.. Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in humans. Subcell Biochem. 2010; 50:279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lieber M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010; 79:181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang W., Crowe J.L., Liu X., Nakajima S., Wang Y., Li C., Lee B.J., Dubois R.L., Liu C., Yu X. et al. . Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol. Cell. 2015; 58:172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lieber M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008; 283:1–5. [DOI] [PubMed] [Google Scholar]
- 23. Reid D.A., Keegan S., Leo-Macias A., Watanabe G., Strande N.T., Chang H.H., Oksuz B.A., Fenyo D., Lieber M.R., Ramsden D.A. et al. . Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E2575–E2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma Y., Lu H., Tippin B., Goodman M.F., Shimazaki N., Koiwai O., Hsieh C.L., Schwarz K., Lieber M.R.. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol. Cell. 2004; 16:701–713. [DOI] [PubMed] [Google Scholar]
- 25. Bertocci B., De Smet A., Weill J.C., Reynaud C.A.. Nonoverlapping functions of DNA polymerases mu, lambda, and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity. 2006; 25:31–41. [DOI] [PubMed] [Google Scholar]
- 26. Ramsden D.A., Asagoshi K.. DNA polymerases in nonhomologous end joining: are there any benefits to standing out from the crowd. Environ. Mol. Mutagen. 2012; 53:741–751. [DOI] [PubMed] [Google Scholar]
- 27. Wilson T.E., Grawunder U., Lieber M.R.. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nature. 1997; 388:495–498. [DOI] [PubMed] [Google Scholar]
- 28. Wang M., Wu W., Wu W., Rosidi B., Zhang L., Wang H., Iliakis G.. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006; 34:6170–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang H., Rosidi B., Perrault R., Wang M., Zhang L., Windhofer F., Iliakis G.. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005; 65:4020–4030. [DOI] [PubMed] [Google Scholar]
- 30. Audebert M., Salles B., Calsou P.. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004; 279:55117–55126. [DOI] [PubMed] [Google Scholar]
- 31. Dueva R., Iliakis G.. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer Res. 2013; 2:163–177. [Google Scholar]
- 32. Soni A., Siemann M., Grabos M., Murmann T., Pantelias G.E., Iliakis G.. Requirement for Parp-1 and DNA ligases 1 or 3 but not of Xrcc1 in chromosomal translocation formation by backup end joining. Nucleic Acids Res. 2014; 42:6380–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liang L., Deng L., Nguyen S.C., Zhao X., Maulion C.D., Shao C., Tischfield J.A.. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 2008; 36:3297–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robert I., Dantzer F., Reina-San-Martin B.. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J. Exp. Med. 2009; 206:1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rass E., Grabarz A., Plo I., Gautier J., Bertrand P., Lopez B.S.. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 2009; 16:819–824. [DOI] [PubMed] [Google Scholar]
- 36. Xie A., Kwok A., Scully R.. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 2009; 16:814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Y., Jasin M.. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat. Struct. Mol. Biol. 2011; 18:80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wilson T.E., Lieber M.R.. Efficient processing of DNA ends during yeast nonhomologous end joining. Evidence for a DNA polymerase beta (Pol4)-dependent pathway. J. Biol. Chem. 1999; 274:23599–23609. [DOI] [PubMed] [Google Scholar]
- 39. Mateos-Gomez P.A., Gong F., Nair N., Miller K.M., Lazzerini-Denchi E., Sfeir A.. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature. 2015; 518:254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wyatt D.W., Feng W., Conlin M.P., Yousefzadeh M.J., Roberts S.A., Mieczkowski P., Wood R.D., Gupta G.P., Ramsden D.A.. Essential roles for polymerase theta-mediated end joining in the repair of chromosome breaks. Mol. Cell. 2016; 63:662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wood R.D., Doublie S.. DNA polymerase theta (POLQ), double-strand break repair, and cancer. DNA Repair (Amst). 2016; 44:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kent T., Chandramouly G., McDevitt S.M., Ozdemir A.Y., Pomerantz R.T.. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015; 22:230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kent T., Mateos-Gomez P.A., Sfeir A., Pomerantz R.T.. Polymerase theta is a robust terminal transferase that oscillates between three different mechanisms during end-joining. Elife. 2016; 5:e13740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Raghavan S.C., Tong J., Lieber M.R.. Hybrid joint formation in human V(D)J recombination requires nonhomologous DNA end joining. DNA Repair (Amst). 2006; 5:278–285. [DOI] [PubMed] [Google Scholar]
- 45. Lieber M.R., Yu K., Raghavan S.C.. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair (Amst). 2006; 5:1234–1245. [DOI] [PubMed] [Google Scholar]
- 46. Senejani A.G., Liu Y., Kidane D., Maher S.E., Zeiss C.J., Park H.J., Kashgarian M., McNiff J.M., Zelterman D., Bothwell A.L. et al. . Mutation of POLB causes lupus in mice. Cell Rep. 2014; 6:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mahmood T., Yang P.C.. Western blot: technique, theory, and trouble shooting. N. Am. J. Med. Sci. 2012; 4:429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yamtich J., Nemec A.A., Keh A., Sweasy J.B.. A germline polymorphism of DNA polymerase beta induces genomic instability and cellular transformation. PLoS Genet. 2012; 8:e1003052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bindra R.S., Goglia A.G., Jasin M., Powell S.N.. Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res. 2013; 41:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Soong C.P., Breuer G.A., Hannon R.A., Kim S.D., Salem A.F., Wang G., Yu R., Carriero N.J., Bjornson R., Sundaram R.K. et al. . Development of a novel method to create double-strand break repair fingerprints using next-generation sequencing. DNA Repair (Amst). 2015; 26:44–53. [DOI] [PubMed] [Google Scholar]
- 51. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Soong C., Breuer G., Hannon R., Kim S., Salem A., Wang G., Yu R., Carriero N., Bjornson R., Sundaram R. et al. . Development of a novel method to create double-strand break repair fingerprints using next-generation sequencing. DNA Repair. 2015; 26:44–53. [DOI] [PubMed] [Google Scholar]
- 53. Senejani A.G., Dalal S., Liu Y., Nottoli T.P., McGrath J.M., Clairmont C.S., Sweasy J.B.. Y265C DNA polymerase beta knockin mice survive past birth and accumulate base excision repair intermediate substrates. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:6632–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Washington S.L., Yoon M.S., Chagovetz A.M., Li S.X., Clairmont C.A., Preston B.D., Eckert K.A., Sweasy J.B.. A genetic system to identify DNA polymerase beta mutator mutants. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:1321–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rassool F.V., Tomkinson A.E.. Targeting abnormal DNA double strand break repair in cancer. Cell Mol. Life Sci. 2010; 67:3699–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu S., Lai Y., Zhao W., Wu M., Zhang Z.. Links between DNA polymerase beta expression and sensitivity to bleomycin. Toxicology. 2011; 281:63–69. [DOI] [PubMed] [Google Scholar]
- 57. Muller W.E., Maidhof A., Arendes J., Geurtsen W., Zahn R.K., Schmidseder R.. Additive effects of bleomycin and neocarzinostatin on degradation of DNA, inhibition of DNA polymerase beta, and cell growth. Cancer Res. 1979; 39:3768–3773. [PubMed] [Google Scholar]
- 58. Pryor J.M., Waters C.A., Aza A., Asagoshi K., Strom C., Mieczkowski P.A., Blanco L., Ramsden D.A.. Essential role for polymerase specialization in cellular nonhomologous end joining. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E4537–E4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nick McElhinny S.A., Havener J.M., Garcia-Diaz M., Juarez R., Bebenek K., Kee B.L., Blanco L., Kunkel T.A., Ramsden D.A.. A gradient of template dependence defines distinct biological roles for family X polymerases in nonhomologous end joining. Mol. Cell. 2005; 19:357–366. [DOI] [PubMed] [Google Scholar]
- 60. Kunkel T.A. The mutational specificity of DNA polymerase-beta during in vitro DNA synthesis. Production of frameshift, base substitution, and deletion mutations. J. Biol. Chem. 1985; 260:5787–5796. [PubMed] [Google Scholar]
- 61. Zahn K.E., Averill A.M., Aller P., Wood R.D., Doublie S.. Human DNA polymerase theta grasps the primer terminus to mediate DNA repair. Nat. Struct. Mol. Biol. 2015; 22:304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Miller M.R., Chinault D.N.. The roles of DNA polymerases alpha, beta, and gamma in DNA repair synthesis induced in hamster and human cells by different DNA damaging agents. J. Biol. Chem. 1982; 257:10204–10209. [PubMed] [Google Scholar]
- 63. Gao Z., Maloney D.J., Dedkova L.M., Hecht S.M.. Inhibitors of DNA polymerase beta: activity and mechanism. Bioorg. Med. Chem. 2008; 16:4331–4340. [DOI] [PubMed] [Google Scholar]
- 64. Chen J., Stubbe J.. Bleomycins: towards better therapeutics. Nat. Rev. Cancer. 2005; 5:102–112. [DOI] [PubMed] [Google Scholar]
- 65. Chagovetz A.M., Sweasy J.B., Preston B.D.. Increased activity and fidelity of DNA polymerase beta on single-nucleotide gapped DNA. J. Biol. Chem. 1997; 272:27501–27504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.