

Key Points
FXIIIplasma, but not FXIIIplt, promotes RBC retention in thrombi and increases thrombus weight.
Partial FXIII reduction may reduce venous thrombosis.
Abstract
The transglutaminase factor XIII (FXIII) stabilizes clots against mechanical and biochemical disruption and is essential for hemostasis. In vitro and in vivo models of venous thrombosis demonstrate that FXIII mediates clot size by promoting red blood cell (RBC) retention. However, the key source of FXIII and whether FXIII activity can be reduced to suppress thrombosis without imposing deleterious hemostatic consequences are 2 critical unresolved questions. FXIII is present in multiple compartments, including plasma (FXIIIplasma) as a heterotetramer of A2 and B2 subunits and platelets (FXIIIplt) as an A2 homodimer. We determined the role of the FXIII compartment and level in clot contraction, composition, and size in vitro and using in vivo models of hemostasis and venous thrombosis. Reducing overall FXIII levels decreased whole blood clot weight but did not alter thrombin generation or contraction of platelet-rich plasma clots. In reconstituted platelet-rich plasma and whole blood clot contraction assays, FXIIIplasma, but not FXIIIplt, produced high-molecular-weight fibrin crosslinks, promoted RBC retention, and increased clot weights. Genetically imposed reduction of FXIII delayed FXIII activation and fibrin crosslinking, suggesting FXIII levels mediate the kinetics of FXIII activation and activity and that the timing of these processes is a critical determinant of RBC retention during clot formation and contraction. A 50% reduction in FXIIIplasma produced significantly smaller venous thrombi but did not increase bleeding in tail transection or saphenous vein puncture models in vivo. Collectively, these findings suggest that partial FXIII reduction may be a therapeutic strategy for reducing venous thrombosis.
Visual Abstract
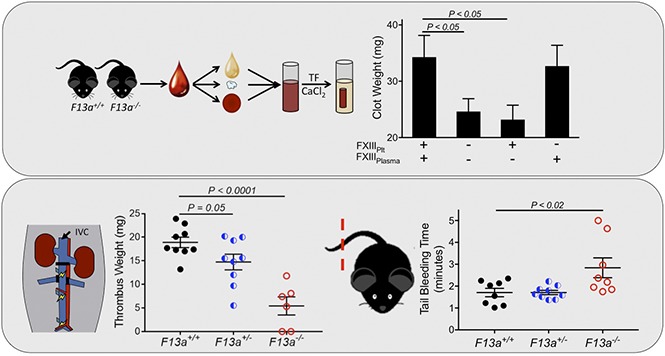
Introduction
Coagulation factor XIII (FXIII) is a protransglutaminase that plays an important role in clot stability. Activated FXIII (FXIIIa) introduces ε-N-(γ-glutamyl)-lysyl crosslinks between glutamine and lysine residues. Crosslinks between fibrin γ- and α-chains and between fibrin and other plasma proteins (eg, α2-antiplasmin, thrombin-activatable fibrinolysis inhibitor, and fibronectin) stabilize clots against mechanical disruption and fibrinolysis.1-4 FXIIIa also promotes red blood cell (RBC) retention in venous thrombi and genetic deletion or inhibition of FXIII zymogen or FXIIIa (FXIII[a]), respectively, reduces thrombus weight in vitro and in in vivo models of venous thrombosis.5
FXIII is present both in plasma and within cells, including platelets. Plasma FXIII (FXIIIplasma) is a 320-kDa noncovalent heterotetramer (FXIII-A2B2) consisting of 2 catalytic subunits (FXIII-A2) tightly associated (Kd ∼10−10 M)6 with 2 noncatalytic subunits (FXIII-B2). FXIIIplasma circulates at 14 to 28 µg/mL,7 nearly always in complex with fibrinogen. Assays in purified systems and platelet-poor plasma (PPP) show FXIIIplasma can efficiently crosslink fibrin and other plasma proteins to fibrin to collectively stabilize the fibrin network.2-4
Platelet FXIII (FXIIIplt) exists as a homodimer (FXIII-A2) in the platelet cytoplasm.8 During platelet activation, FXIIIplt is externalized on the platelet surface; exposure is maximized when platelets are activated by strong dual agonists (thrombin plus collagen).9 Although the concentration of FXIII in platelets is 150-fold higher than in plasma, its role during hemostasis and thrombosis is unclear. Three aspects of FXIIIplt function have been particularly controversial. First, it has been proposed that FXIIIplt is required for platelet contraction, as impaired contraction was observed in FXIII-deficient platelet-rich plasma (PRP) clots and in whole blood treated with transglutaminase inhibitors.10,11 However, other studies have reported little to no influence of FXIII on platelet contractile events.12-14 Thus, the contribution of FXIIIplt to platelet-mediated clot contraction remains unclear. Second, a previous report suggested that FXIII-A deletion induces a compensatory increase in platelet-derived tissue transglutaminase activity.15 Thus, this activity may have confounded earlier studies with F13a−/− mice. Third, although FXIIIplt can introduce crosslinks in fibrin and between fibrin and α2-antiplasmin,16-19 the effects of FXIIIplt on clot stability are only detected in vitro when FXIIIplasma levels are low (≤10%).9 Thus, the contributions of FXIIIplt to RBC retention in clots and during hemostasis and thrombosis in vivo are unknown. Determining the relative roles of FXIIIplasma and FXIIIplt in these processes is essential for understanding the biological importance of these compartments and the clinical significance of deficiencies in FXIIIplasma or FXIIIplt.
Understanding FXIII function in vivo is also necessary for optimizing therapeutic approaches to modify FXIII level or activity to mitigate bleeding and thrombosis. Notably, in spite of the established role of FXIIIa in clot stabilization, the FXIII level necessary for hemostasis is uncertain. Patients with <4% FXIIIa activity have a significantly increased risk of bleeding, including central nervous system and umbilical cord bleeding, hemarthroses, and hematomas, and experience recurrent miscarriage.20 However, patients with ∼4% to 30% FXIIIa activity exhibit high variability in bleeding severity,20 and individuals with ≥30% FXIIIa activity, including those with acquired FXIII deficiency, are usually asymptomatic.21,22 Interestingly, a recent analysis of hospitalized patients indicated that 21% of adults and 52% of children had FXIIIplasma levels <50 U dL−1, suggesting acquired FXIII deficiency is relatively common in patients after surgery and in the intensive care unit.23 Since hospitalized patients have increased risk for both bleeding and thrombosis, the role of FXIII in these situations is difficult to predict. Identifying a level of FXIII that reduces thrombus size without impairing hemostasis is critical for understanding the role of FXIII in clot function and developing novel antithrombotic therapies targeting FXIII(a).
Here, we used murine models of FXIII deficiency to determine the roles of plasma and platelet FXIII in platelet-mediated clot contraction, thrombin generation, FXIII activation, fibrin crosslinking, and RBC retention in contracted clots. We also determined the effect of FXIII reduction using in vivo models of hemostasis and thrombosis. Collectively, our findings suggest moderate reduction of FXIII may reduce venous thrombosis without significantly increasing bleeding.
Methods
Materials and methods
Sources of materials and methods for measuring thrombin generation, transglutaminase activity, FXIII activation, and fibrin crosslinking are detailed in supplemental Methods.
Murine blood draws and plasma preparation
Murine studies were approved by the University of North Carolina at Chapel Hill (UNC) and Cincinnati Children’s Hospital Medical Center Institution of Animal Care and Use Committees. F13a+/+, F13a+/−, and F13a−/− mice were backcrossed 6 generations on a C57BL/6J background.24 A separate line of F13a−/− mice (generous gift of CSL Behring) on a mixed 129Ola/CBACa background was maintained by homozygous breeding.25
Mice were anesthetized with 3% to 3.5% isoflurane in 2% oxygen, and blood was drawn from the inferior vena cava (IVC) into 3.2% citrate (10% vol/vol, final) in a terminal procedure. PRP was prepared by first centrifuging whole blood (125g, 5 minutes) and then centrifuging the platelet-enriched plasma fraction (100g, 5 minutes). PPP was prepared by centrifuging whole blood (5000g, 10 minutes).
Clot contraction assays
Whole blood was clotted with tissue factor (TF; Innovin, diluted 1:12 000 [1 pM TF], final) and CaCl2 (10 mM, final) in the absence or presence of the transglutaminase inhibitor T101 at the concentrations indicated. Clot formation and contraction proceeded in siliconized wells for 120 minutes at 37°C.
For PRP clot contraction, platelets in PRP were quantified (HV950FS Hemavet cell counter), and PRP was diluted with autologous PPP to obtain the final concentrations of platelets indicated. Clotting was triggered in siliconized aggregometer tubes at 37°C by adding TF and CaCl2 (1 pM and 10 mM, final, respectively). Photos of contracting clots were recorded every 5 minutes for the first hour, every 10 minutes for the second hour, and at 24 hours.
For assays with reconstituted whole blood, PRP was treated with prostaglandin-I2 (50 ng/mL, final), and platelets were pelleted by centrifugation (400g, 5 minutes). Platelet pellets were resuspended in Tyrode’s buffer. PPP was obtained by centrifuging whole blood (5000g, 10 minutes). RBCs were isolated and washed with citrated glucose saline buffer (1.3 mM sodium citrate, 3.3 mM glucose, 1.2 mM NaCl, pH 7.2) and packed by centrifugation. PPP and washed platelets (400 × 109/L, final) from C57BL/6J and F13a−/− mice were combined with RBCs (3.5 × 1012/L, final) from C57BL/6J mice to model reconstituted whole blood sufficient or deficient in FXIIIplasma and FXIIIplt. Clotting was triggered at 37°C in siliconized wells with TF and CaCl2 (1 pM and 10 mM, final, respectively) or collagen, thrombin, and calcium (20 mg/mL, 1 U/mL, and 10 mM final, respectively). Contracted clots were weighed at 2 hours.
Bleeding models
The tail transection model was performed on F13a+/+, F13a+/−, and F13a−/− mice on a C57BL/6J background, as described previously.26 Briefly, mice were anesthetized with ketamine/xylazine, 3 mm of the tail tip was excised using a scalpel blade (#11), and the tail was submerged in Tris-buffered saline (20 mM Tris, 150 mM NaCl, pH 7.5) containing 2 mM CaCl2 at 37°C. The time to blood flow cessation, defined as no flow for more than 15 s, was recorded as the bleeding time. The saphenous vein bleeding model was performed on F13a+/+, F13a+/−, and F13a−/− mice on a C57BL/6J background, as described previously.27 Briefly, the right saphenous vein was partially transected and opened further (longitudinally) with microscissors. Blood was gently wicked away under slow irrigation until hemostasis occurred. The clot was then disrupted using a 30-G needle, and blood was wicked away until hemostasis occurred again. Clot disruption was repeated after each incidence of hemostasis until 30 minutes after the initial injury. Two readouts were recorded: (1) the average time to hemostasis for each event in 30 minutes and (2) the total number of hemostatic events during the 30-minute period.
Venous thrombosis model
The IVC stasis model was performed as described previously28 on 8- to 12-week-old F13a+/+, F13a+/−, and F13a−/− mice on a C57BL/6J background. Briefly, anesthetized mice were subjected to sterile laparotomy, the IVC was exposed, side branches were ligated, and lumbar branches were cauterized. The IVC was separated from the aorta by blunt dissection and completely ligated. Mice recovered with analgesia (subcutaneous buprenorphine, 0.05 mg/kg) and were maintained on acetaminophen (6 mg/mL) in their drinking water. After 24 hours, mice were anesthetized and blood was drawn from the suprarenal IVC into 3.2% sodium citrate (10% vol/vol, final concentration). Thrombi were separated from the vein wall and weighed. Blood samples were centrifuged to isolate PPP and platelets, and FXIII-A2B2 was quantified by western blotting using standard curves with the appropriate species (human or mouse FXIII-A2B2).
Statistical methods
Descriptive statistics (mean and standard error of the mean [SEM]) were calculated for each experiment, and Lilliefors test was used to assess normality. Experiments with 2 groups and normally distributed data were compared by Student t test with equal or unequal variance, as appropriate. Experiments with more than 2 conditions were analyzed by analysis of variance (ANOVA) with Bonferroni or Dunnett’s post-hoc tests for between-group comparisons. P < .05 was considered significant.
Results
FXIII deficiency decreases RBC retention and clot weight without altering thrombin generation or platelet contraction
We previously showed that genetic deletion of FXIII-A (F13a−/−) reduces RBC retention in contracted clots and, consequently, clot weight.5 To now extend these findings to determine the dose relationship between FXIII level and clot weight, we first analyzed in vitro clot contraction of whole blood from F13a+/+, F13a+/−, and F13a−/− mice. Complete blood counts indicate all 3 genotypes have normal levels of leukocytes, RBCs, and platelets (supplemental Table 1). Compared with F13a+/+ mice, both a 50% reduction in FXIII-A as well as complete FXIII-A deficiency increased RBC extrusion from contracted clots (∼36% and ∼50% for F13a+/− and F13a−/− mice, respectively; Figure 1A) and decreased clot weight (∼32% and ∼71% for F13a+/− and F13a−/− mice, respectively; Figure 1B). Inhibition of FXIIIa activity using the irreversible transglutaminase inhibitor T101 (20 µM, final, a concentration that maximally inhibits FXIIIa activity and reduces clot weight; supplemental Figure 1A-B) increased RBC extrusion from contracted clots (∼69% and ∼28% for F13a+/+ and F13a+/− mice, respectively; Figure 1A) and reduced clot weight (∼67% and ∼70% for F13a+/+ and F13a+/− mice, respectively, Figure 1B). T101 had no effect on RBC retention or whole blood clot weight from F13a−/− mice, consistent with a specific role for FXIII(a)-dependent transglutaminase activity in determining clot composition and weight (Figure 1A-B).
Figure 1.

Inhibition or genetic deletion of FXIII(a) results in decreased clot weight. Whole blood from F13a+/+, F13a+/−, and F13a−/− mice was clotted with TF/CaCl2 in the absence or presence of the FXIII inhibitor T101 (20 µM, final) for 2 hours. (A-B) Serum RBC content (A) and contracted whole blood clot weight (B) were recorded. Each dot is a separate mouse; lines indicate mean ± SEM.
Thrombin generation did not differ between genotypes in PRP or PPP (Table 1), indicating that changes in RBC retention and clot weight were not due to altered plasma or platelet procoagulant activity. Because previous studies suggested FXIII is necessary for platelet-mediated contractile forces,10,11 we also analyzed the kinetics of clot contraction in F13a+/+, F13a+/−, and F13a−/− PRP. For all 3 genotypes, clot contraction occurred rapidly and was platelet concentration dependent. In PRP with 400 × 109 platelets/L, clots were 50% contracted within ∼20 minutes of triggering clotting and were fully contracted within ∼55 minutes. As the platelet concentration decreased, the onset time of clot contraction prolonged, and the rate and extent of maximum contraction decreased (Figure 2A-D; Table 2). However, there were no differences in contraction parameters between PRP clots from F13a+/+, F13a+/−, or F13a−/− mice (Figure 2A-D; Table 2), and inspection of contracted clots showed similar appearance in all 3 genotypes (Figure 2E). PRP clots from a separate line of F13a−/− mice on a 129Ola/CBACa background similarly showed 50% contraction with ∼17 minutes and complete clot contraction within ∼42 minutes (data not shown). Finally, because a previous study suggested F13a−/− mice may express a FXIII-independent platelet transglutaminase activity15 that could compensate for FXIII deficiency in these mice, we measured transglutaminase activity in platelets isolated from F13a+/+ and F13a−/− mice. These experiments showed little to no transglutaminase activity in platelets from F13a−/− mice on a C57BL/6J background or in platelets from F13a−/− mice on a 129Ola/CBACa background (Figure 2F). Collectively, these data show FXIII deficiency decreases RBC retention and contracted whole blood clot weight in a dose-dependent manner, without altering plasma or platelet procoagulant activity or platelet contraction.
Table 1.
Thrombin generation is similar in F13a+/+, F13a+/−, and F13a−/− mice
| PPP | PRP | |||||
|---|---|---|---|---|---|---|
| F13a+/+ | F13a+/− | F13a−/− | F13a+/+ | F13a+/− | F13a−/− | |
| Lag time, min | 1.6 ± 0.2 | 1.7 ± 0.1 | 1.6 ± 0.1 | 2.3 ± 0.2 | 2.3 ± 0.1 | 2.33 ± 0.2 |
| Time to peak, min | 3.6 ± 0.5 | 3.7 ± 0.1 | 3.5 ± 0.1 | 5.6 ± 0.3 | 6.0 ± 0.4 | 4.9 ± 0.2 |
| Velocity, nM/min | 35.8 ± 2.6 | 33.8 ± 1.2 | 34.1 ± 2.6 | 18.0 ± 2.7 | 13.8 ± 3.3 | 22.3 ± 5.3 |
| Peak thrombin, nM | 70.1 ± 2.9 | 66.7 ± 1.5 | 65.3 ± 3.9 | 55.2 ± 6.0 | 46.4 ± 6.6 | 52.7 ± 3.7 |
| Endogenous thrombin potential, nM*min | 387.7 ± 34.4 | 417.5 ± 68.7 | 284.6 ± 18.5 | 804.8 ± 164.6 | 621.0 ± 63.1 | 611.4 ± 50.0 |
Thrombin generation was measured by calibrated automated thrombography. Data show mean ± SEM and were analyzed by ANOVA with Dunnett’s post-hoc test (n = 4-6).
Figure 2.

FXIII is not required for platelet-mediated clot contraction. (A-D) PRP from F13a+/+, F13a+/−, and F13a−/− mice was clotted with TF/CaCl2 at 400 (A), 200 (B), 50 (C), and 10 × 109 platelets/L (D). Clot area over time was assessed as a percentage of initial clot area. Dots show mean ± SEM (n = 4); curves were fit to a nonlinear regression with a 1-phase decay equation. (E) Photographs of contracted clots at 24 hours. (F) Transglutaminase activity was measured in platelets from F13a+/+ and F13a−/− C57BL/6J mice and F13a−/− 129Ola/CBACa mice. Data show mean ± SEM (n = 3-5).
Table 2.
Kinetic parameters of PRP-mediated clot contraction in F13a+/+, F13a+/−, and F13a−/− mice
| Platelets (×109/L) | F13a+/+ | F13a+/− | F13a−/− |
|---|---|---|---|
| Onset time, min | |||
| 400 | 5.2 ± 0.9 | 7.1 ± 0.3 | 6.4 ± 1.5 |
| 200 | 8.0 ± 1.6 | 7.8 ± 1.7 | 10.3 ± 1.6 |
| 50 | 17.6 ± 1.6†† | 17.8 ± 4.4 | 19.8 ± 5.5 |
| 10 | 22.2 ± 3.2††† | 22.3 ± 4.5† | 28.5 ± 4.6†† |
| Rate, 10−3/min | |||
| 400 | 54.8 ± 1.4 | 55.7 ± 2.3 | 75.8 ± 5.1* |
| 200 | 33.6 ± 2.1††† | 37.1 ± 1.9† | 45.1 ± 1.1*† |
| 50 | 17.4 ± 3.2††† | 22.3 ± 0.2††† | 26.4 ± 3.4†† |
| 10 | 10.6 ± 1.1††† | 16.4 ± 6.1††† | 31.0 ± 13.0†† |
|
Maximum contraction (% of initial clot area) |
|||
| 400 | 6.4 ± 0.4 | 6.7 ± 0.1 | 6.1 ± 0.2 |
| 200 | 10.5 ± 1.4 | 8.8 ± 1.0 | 6.7 ± 0.6* |
| 50 | 24.0 ± 0.9†† | 26.9 ± 2.7†† | 18.1 ± 6.5 |
| 10 | 49.3 ± 5.6††† | 47.5 ± 6.0††† | 67.3 ± 7.3††† |
Clot contraction parameters were determined by a nonlinear regression curve fit with plateau followed by 1-phase decay. Data represent mean ± SEM and were analyzed by ANOVA with Dunnett’s post-hoc test (n = 4).
P < .05,
P < .005, and
P < .0001 between platelet count, compared to 400 × 109/L platelets.
P < .005 between genotypes compared to F13a+/+.
FXIIIplasma, but not FXIIIplt, mediates clot weight
Genetic reduction of FXIII-A in mice reduces FXIII-A antigen in both plasma and platelets (Figure 3A-B). Therefore, to determine the relative contributions of FXIIIplasma and FXIIIplt to RBC retention and clot weight, we isolated plasma and platelets from FXIII-sufficient and deficient (F13a−/−) mice and recombined these with RBCs to yield reconstituted whole blood with specific deficiencies in FXIIIplasma or FXIIIplt. We then triggered clotting with TF and CaCl2 and measured contracted clot weights. As expected, the absence of both FXIIIplasma and FXIIIplt significantly decreased clot weight (Figure 3C). Whereas reconstitution of FXIIIplt had no effect on RBC retention or clot weight, reconstitution of FXIIIplasma restored RBC retention and clot weight to levels seen in FXIII-replete reactions (Figure 3C). Because FXIIIplt is maximally externalized on the platelet surface by strong, dual agonists,9 we also analyzed effects of FXIIIplasma and FXIIIplt on the weight of contracted clots triggered by addition of thrombin and collagen. Similar to experiments with TF, these reactions showed FXIIIplasma, but not FXIIIplt, promoted RBC retention in clots and increased clot weight (Figure 3C).
Figure 3.

FXIIIplasma, but not FXIIIplt, mediates clot weight. (A-B) Representative western blot (A) and quantification (B) for FXIII-A in plasma and platelets from F13a+/+, F13a+/−, and F13a−/− mice (n = 3). (C) RBCs were reconstituted with C57BL6/J FXIII-sufficient (wild-type) and deficient (F13a−/−) plasma and platelets, and clotting was triggered with TF/CaCl2 (n = 9) or thrombin/collagen/CaCl2 (n = 6). Data show mean ± SEM. (D) Plasma and platelets from C57BL6/J FXIII-sufficient (wild-type) and deficient (F13a−/−) mice were recombined to make PRP sufficient or deficient in plasma or platelet FXIII. Clotting was triggered with TF/CaCl2, and reactions were quenched at the indicated time points and analyzed by SDS-PAGE with western blotting (n = 2). Fibrin crosslinking was detected using anti-fibrin(ogen) antibody, and identity of bands was confirmed by mass spectrometry.
To determine effects of these FXIII compartments on fibrin crosslinking, we triggered clotting in reconstituted PRP that was sufficient or deficient in FXIIIplasma or FXIIIplt and assessed fibrin crosslinking by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting. Only samples containing FXIIIplasma showed crosslinked high-molecular-weight (HMW) fibrin species (Figure 3D), indicating FXIIIplasma, but not FXIIIplt, generates these species. This finding unites the observation that FXIIIplasma, but not FXIIIplt, increases RBC retention and weight of contracted clots (Figure 3C) with previous findings that FXIII(a)-dependent retention of RBCs in contracted clots is associated with the formation of HMW crosslinked fibrin species.29 Together, these results indicate that FXIIIplasma, but not FXIIIplt, promotes RBC retention in contracted clots.
FXIII level mediates FXIII activation kinetics and, consequently, fibrin crosslinking
FXIIIplasma is activated by thrombin-mediated cleavage of 37-amino-acid (4-kDa) activation peptides on the N termini of the FXIII-A subunits and calcium-mediated dissociation of FXIII-B2.1 The observation that FXIII mediated clot weight in a gene-dose–dependent manner (Figure 1) suggested that the concentration of FXIII zymogen present in plasma determines clot weight by modulating FXIIIa generation and, consequently, fibrin crosslinking. To characterize this mechanism, we used SDS-PAGE and western blotting to compare FXIII activation and fibrin crosslinking kinetics during TF/CaCl2-initiated clotting of F13a+/+, F13a+/−, and F13a−/− PRP. As expected, F13a−/− clots showed no FXIII-A antigen present and, consequently, no fibrin crosslinking (data not shown). Compared with F13a+/+, F13a+/− clots exhibited an ∼3-minute delayed onset of FXIII activation, ∼2.7-fold decreased rate of FXIII activation, and ∼50% reduction in the total amount of FXIIIa generated at 15 minutes (Figure 4A-B). Moreover, compared with F13a+/+, F13a+/− clots demonstrated an ∼3-minute delayed onset and ∼2.5-fold decreased rate of γ-γ dimer formation (Figure 4C-E) and an ∼1.5-minute delayed onset and ∼1.4-fold decreased rate of HMW crosslinked fibrin formation (Figure 4C-D,F). Both F13a+/+ and F13a+/− clots showed complete crosslinking at ∼15 minutes. These data suggest reducing FXIIIplasma prolongs FXIII activation and delays, but does not prevent, maximal fibrin crosslinking.
Figure 4.

Compared with F13a+/+clots, F13a+/−clots have delayed FXIII activation and fibrin crosslinking. PRP was clotted with TF/CaCl2 and quenched at the indicated time points. FXIII activation (generation of FXIII-A′) and fibrin crosslinking were analyzed by SDS-PAGE with western blotting. (A) Representative western blots and (B) quantitation of FXIII-A′ over time for clot formation in F13a+/+ and F13a+/− PRP. Data show mean ± SEM (n = 3-5). (C-D) Representative western blots for fibrin crosslinking in F13a+/+and F13a+/− PRP clots. (E-F) Quantification of γ-γ (E) and HMW (F) crosslinked fibrin species formation. Data show mean ± SEMs (n = 3-5). A.F.U., arbitrary fluorescence units.
Partial reduction of FXIII does not prolong the time to hemostasis but significantly decreases thrombus size
FXIII is fundamentally different than other coagulation proteins, because FXIII activation and FXIIIa activity occur downstream of thrombin generation. Given our data indicating that FXIII influenced clot size without altering thrombin generation (Table 1) or platelet contractile events (Figure 2), we hypothesized that partial FXIII reduction would not impair hemostatic clot formation. To test this hypothesis, we subjected F13a+/+, F13a+/−, and F13a−/− mice to tail transection and saphenous vein puncture bleeding models. Both of these models are sensitive to coagulation and platelet defects and effects of conventional anticoagulants, including unfractionated and low-molecular-weight heparin.30-32 In the tail transection model, compared with F13a+/+ mice, F13a−/− mice had prolonged tail-bleeding times, but F13a+/− mice did not (Figure 5A). In the saphenous vein model, F13a+/+, F13a+/−, and F13a−/− mice had a similar number of hemostatic events in 30 minutes (data not shown) and similar average hemostasis times (Figure 5B).
Figure 5.

Partial FXIII deficiency does not increase the time to clot formation in murine models of hemostasis. (A-B) F13a+/+, F13a+/−, and F13a−/− mice were subjected to tail transection (A) and saphenous vein puncture (B). (A) Following 3-mm excision of the distal portion of the tail, bleeding was measured as described in “Methods.” Data show time to cessation of bleeding. (B) Following saphenous vein puncture, bleeding was measured after disruption of hemostatic clots as described in “Methods.” The data show average saphenous vein hemostasis times. Each dot represents a separate mouse; lines indicate mean ± SEM.
Finally, given our data demonstrating dose effects of FXIIIplasma on clot size in vitro (Figure 3), we tested the hypothesis that FXIIIplasma mediates venous thrombus weight in vivo. We first subjected F13a+/+, F13a+/−, and F13a−/− mice to the IVC ligation venous thrombosis model and harvested thrombi at 24 hours. We previously detected fully crosslinked fibrin in FXIII-sufficient mice and significantly smaller thrombi in F13a−/− mice at this time point (M. M. Aleman, University of North Carolina at Chapel Hill, unpublished data, 11 June 2011).5 We now demonstrated that this effect is gene-dose dependent during venous thrombosis in vivo (Figure 6A). To define the relationship between FXIIIplasma and thrombus weight, we used human plasma–derived FXIII-A2B2 to restore FXIIIplasma in F13a−/− mice to 10% to 100% of normal. Compared with mouse FXIII-A2B2, human FXIII-A2B2 has similar transglutaminase activity (data not shown), and similarly crosslinks mouse fibrin (supplemental Figure 2). We induced thrombus formation by IVC ligation, harvested thrombi and plasmas at 24 hours, and determined plasma and platelet FXIII-A levels by western blot. Thrombus weights in F13a+/+, F13a+/−, F13a−/−, and FXIII-A2B2-infused F13a−/− mice correlated positively and significantly with circulating FXIII-A antigen in plasma (R = 0.44, P < .005, Figure 6B). Importantly, since only a small amount of infused FXIII-A2B2 is endocytosed into platelets (Figure 6C),33 these experiments show FXIIIplasma, but not FXIIIplt, mediates RBC retention and thrombus weight in vivo.
Figure 6.

FXIIIplasmainfusion into F13a−/−mice rescues thrombus weight. F13a+/+, F13a+/−, and F13a−/− mice infused with vehicle and F13a−/− mice infused with FXIII-A2B2 were subjected to the IVC venous thrombosis model. (A) Thrombi were harvested after 24 hours and weighed. Each dot represents a separate mouse. (B) Correlation between FXIII-A level in plasma (measured by western blot and densitometry, shown as relative band intensity, arbitrary fluorescence units) and thrombus weights for F13a+/+, F13a+/−, F13a−/− (from panel A), and FXIII-A2B2-infused mice. Each dot represents a separate mouse; F13a+/+ (green closed circles), F13a+/− (blue half circles), F13a−/− (red open circles) mice, and FXIII-A2B2-infused F13a−/− mice targeting 100% (green triangles), 50% (blue triangles), 25% (pink triangles), and 10% (orange triangles) FXIII-A2B2. (C) Representative western blots showing FXIII-A in plasma immediately after infusion of 50% FXIII-A2B2 (plasma final, top), and in plasma (middle) and platelets (bottom) 24 hours after infusion and ligation. Note that infused human FXIII-A2B2 is more strongly detected than mouse.
Discussion
FXIII deficiency decreases RBC retention in thrombi and, consequently, decreases thrombus size.5 This finding identifies FXIII(a) as an intriguing candidate therapeutic target for preventing venous thrombosis, but it raises important questions. Herein, we sought to clarify the relative roles of FXIIIplasma and FXIIIplt and the effect of FXIII level on hemostasis and venous thrombosis. First, we showed that effects of FXIII reduction on clot weight are not due to reduced thrombin generation, decreased platelet-mediated clot contraction, or FXIII-independent transglutaminase activity. Second, we demonstrated that FXIIIplasma, but not FXIIIplt, promotes formation of HMW crosslinked fibrin species and RBC retention in clots and, therefore, mediates clot weight. Third, we characterized kinetic mechanisms relating FXIII levels with thrombus composition. Finally, we showed that partial reduction in FXIIIplasma reduces venous thrombus size but does not increase bleeding in vivo. These findings address previously published controversial observations and inform understanding of FXIII biological function. Collectively, these findings define a critical role for FXIIIplasma during coagulation in vivo and support the consideration of FXIII as a therapeutic target for anticoagulation.
In the course of this study, we investigated 3 observations that were previously, but controversially, associated with FXIII activity. First, although a subset of studies suggested FXIII contributes to platelet-mediated contractile events,10,11 our data indicate that PRP from FXIII-sufficient and deficient mice shows similar contraction kinetics. Possible discord between our results and the earlier findings may be explained by methodological differences, including the high centrifugal forces used for preparing PRP that may have resulted in lower platelet concentrations in the prior experiments10 or use of transglutaminase inhibitors (eg, cystamine)10,11 that can inhibit thrombin generation and activity.34 Our findings are consistent with several independent studies that showed no effect of FXIII on platelet contraction12-14 and no effect on cytoskeletal dynamics in Chinese hamster ovary cells transfected with FXIII.35 Second, although an earlier study detected transglutaminase-like activity in platelets from F13a−/− mice and suggested this activity compensates for FXIII deficiency in platelets from F13a−/− mice,15 we detected little to no transglutaminase activity in platelets from F13a−/− mice. Although tissue transglutaminase-2 (TG-2) antigen is present in mouse platelets, levels are 25-fold lower than FXIII.36 Moreover, human platelets do not express TG-2,9,37 and TG-2 does not substantively contribute to platelet thrombus formation.38 Thus, transglutaminase activity, be it from FXIII or other transglutaminase enzymes, appears dispensable for platelet contraction.
Third, although lysates from unstimulated platelets can crosslink fibrin,9 we were unable to demonstrate a functional effect of FXIIIplt on RBC retention in clots. This paradox may be explained by observations that FXIII exposed on the platelet surface is rapidly inactivated by an as yet unknown mechanism. Findings that FXIIIplasma has functions not reproduced by FXIIIplt are consistent with previous data9 but extend these observations to provide a biochemical explanation for these differences. Notably, although FXIIIplasma and FXIIIplt both have catalytic FXIII-A2 subunits, these compartments differ in subunit structure and activation kinetics. In FXIIIplasma, the FXIII-A2 subunit dimer is tightly bound to FXIII-B2,6 which mediates its association with the fibrin(ogen) γ-chain in plasma.39 Following activation of coagulation, FXIIIplasma is activated rapidly by thrombin and is immediately available to crosslink fibrin. In contrast, FXIIIplt lacks FXIII-B2 and resides in the platelet cytoplasm, where calcium-mediated activation and externalization occur more slowly.9 Indeed, presentation of platelet-derived FXIII activity to the plasma/thrombus milieu likely happens after fibrin formation, crosslinking, and initiation of platelet-mediated clot contraction occur. Thus, because FXIIIa activity is essential for RBC retention in clots while the clot is undergoing contraction,29 only FXIIIplasma can fulfill the immediate temporal requirement needed for this function. This premise is further supported by 2 previous observations. First, mice with normal FXIIIplt but delayed FXIII activation secondary to reduced binding of FXIIIplasma to fibrinogen (ie, Fibγ390-396A mice) phenocopy F13a−/− mice with small thrombi and reduced RBC content (loss of function).5 Second, the addition of plasma-derived FXIII-A2B2 to FXIII-deficient human plasma fully restores RBC retention in reconstituted, contracted whole blood clots (gain of function).5 Together, these experiments suggest FXIIIplasma, but not FXIIIplt, promotes RBC retention in thrombi and, consequently, influences venous thrombus size.
Given the prominent effect of FXIIIplasma on fibrin crosslinking, it is curious why FXIII is present in such high concentrations in platelets (3% of total protein).9 FXIIIplt has been reported to crosslink cytoskeletal proteins40-42 and regulate bidirectional platelet signaling via reorganization of the cytoskeleton and αIIbβ3 integrin.43 Although we did not detect platelet abnormalities in our assays, dysfunctions stemming from FXIII deficiency may be detectable in assays that assess platelet functions secondary to clot formation. In vitro studies and clinical observations suggest FXIIIplt may contribute to hemostasis in certain situations. For example, FXIII-B–deficient patients have reduced FXIIIplasma but normal FXIIIplt and a generally milder phenotype than FXIII-A–deficient patients.44 Although this difference has been attributed to only partial (vs total) loss of the FXIII-A2 catalytic subunits in plasma,44 the milder phenotype may also reflect residual hemostatic activity provided by FXIIIplt. Alternately, or perhaps in addition, because FXIII has nonhemostatic functions during wound healing and the immune response,45 FXIIIplt may contribute to these processes. In this regard, delayed exposure of FXIIIplt may be a critical aspect of its biological function.
The present study fills several critical knowledge gaps regarding FXIII biology and function, but it has potential limitations. First, although F13a+/− mice did not demonstrate bleeding in either hemostasis assay, studies of human patients suggest moderate FXIII deficiency is associated with increased bleeding risk in certain situations, including delayed bleeding after injury or surgery.20,23,46 Potential discord between mice and humans may reflect species differences, or it may indicate that murine hemostasis models are sensitive to initial clot formation, but not to clot stability, a parameter likely to be affected by FXIII deficiency. Indeed, even F13a−/− mice show only moderate (approximately twofold prolongation) in bleeding following tail transection assays (Figure 5A and Lauer et al25), and only slight, nonsignificant bleeding following digit amputation.24 Development of in vivo assays that assess clot stability are needed to fully elucidate the relationship between FXIII level and bleeding risk. Second, the IVC ligation model involves complete restriction of blood flow, which may minimize effects of platelets and, consequently, FXIIIplt on venous thrombosis. However, mice with reduced or delayed FXIII activation have smaller venous thrombi in both the IVC ligation (stasis) and stenosis (partial flow) models.5 Thus, the effects of FXIII(a) on RBC retention and thrombus weight are independent of local blood flow and mediated by FXIIIplasma in both models.
Identification of a role for FXIIIplasma in promoting RBC retention in clots and determining clot weight has implications for developing FXIII(a) inhibitors to reduce venous thrombosis. First, although FXIII is found in 2 separate compartments, potential inhibitors only need to inhibit FXIIIplasma to limit thrombosis severity and would not need to traverse the platelet membrane to access FXIIIplt. This premise simplifies the molecular design of potential drugs. Second, platelet proteins are somewhat “protected” from plasma inhibitors.47,48 Therefore, although FXIIIplasma and FXIIIplt share structural homology, having 2 separate FXIII compartments may conserve sufficient FXIII activity to preserve hemostasis. Thus, FXIII may have advantages not present in other therapeutic targets for thrombosis.
In summary, we have shown that FXIIIplasma mediates thrombus RBC retention and weight in a dose-dependent manner and that partial FXIII reduction reduces venous thrombus size without increasing bleeding in murine models of thrombosis and hemostasis. Collectively, these data define specific physiologic roles of FXIIIplasma and support the evaluation of FXIII(a) inhibition as a strategy to reduce venous thrombosis.
Supplementary Material
The full-text version of this article contains a data supplement
Acknowledgments
The authors thank Anna C. Silver and the UNC Michael Hooker Proteomics Core for excellent technical assistance.
This study was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute (T32HL69768, Integrative Biology Training Program) (UNC/S.K.), the American Heart Association (17PRE33660020) (S.K.), the National Science Foundation (graduate research fellowship DGE-1144081) (J.R.B.) (summer undergraduate research experience program 1559922) (UNC/S.M.M.), and National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL112603 (M.J.F.), F31HL139100 (S.K.), and R56HL094740 and R01HL126974 (A.S.W.).
Authorship
Contribution: S.K. designed and performed experiments, analyzed and interpreted the data, and wrote the manuscript; J.R.B., S.M.M., L.A.H., B.C.C., and M.J.F. performed experiments and analyzed data; A.S.W. designed the research, analyzed and interpreted the data, and wrote the manuscript; and all authors approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alisa S. Wolberg, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, 819 Brinkhous-Bullitt Building, CB #7525, Chapel Hill, NC 27599-7525; e-mail: alisa_wolberg@med.unc.edu.
References
- 1.Komáromi I, Bagoly Z, Muszbek L. Factor XIII: novel structural and functional aspects. J Thromb Haemost. 2011;9(1):9-20. [DOI] [PubMed] [Google Scholar]
- 2.Fraser SR, Booth NA, Mutch NJ. The antifibrinolytic function of factor XIII is exclusively expressed through α2-antiplasmin cross-linking. Blood. 2011;117(23):6371-6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen PH, Lorand L, Ebbesen P, Gliemann J. Type-2 plasminogen-activator inhibitor is a substrate for trophoblast transglutaminase and factor XIIIa. Transglutaminase-catalyzed cross-linking to cellular and extracellular structures. Eur J Biochem. 1993;214(1):141-146. [DOI] [PubMed] [Google Scholar]
- 4.Valnickova Z, Enghild JJ. Human procarboxypeptidase U, or thrombin-activable fibrinolysis inhibitor, is a substrate for transglutaminases. Evidence for transglutaminase-catalyzed cross-linking to fibrin. J Biol Chem. 1998;273(42):27220-27224. [DOI] [PubMed] [Google Scholar]
- 5.Aleman MM, Byrnes JR, Wang JG, et al. . Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest. 2014;124(8):3590-3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katona E, Pénzes K, Csapó A, et al. . Interaction of factor XIII subunits. Blood. 2014;123(11):1757-1763. [DOI] [PubMed] [Google Scholar]
- 7.Fickenscher K, Aab A, Stüber W. A photometric assay for blood coagulation factor XIII. Thromb Haemost. 1991;65(5):535-540. [PubMed] [Google Scholar]
- 8.Bagoly Z, Katona E, Muszbek L. Factor XIII and inflammatory cells. Thromb Res. 2012;129(Suppl 2):S77-S81. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell JL, Lionikiene AS, Fraser SR, Whyte CS, Booth NA, Mutch NJ. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood. 2014;124(26):3982-3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasahara K, Souri M, Kaneda M, Miki T, Yamamoto N, Ichinose A. Impaired clot retraction in factor XIII A subunit-deficient mice. Blood. 2010;115(6):1277-1279. [DOI] [PubMed] [Google Scholar]
- 11.Tutwiler V, Litvinov RI, Lozhkin AP, et al. . Kinetics and mechanics of clot contraction are governed by the molecular and cellular composition of the blood. Blood. 2016;127(1):149-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao KM, Newcomb TF. Clot retraction in a factor XIII free system. Scand J Haematol. 1980;24(2):142-148. [DOI] [PubMed] [Google Scholar]
- 13.Jelenska M, Kopeć M, Breddin K. On the retraction of collagen and fibrin induced by normal, defective and modified platelets. Haemostasis. 1985;15(3):169-175. [DOI] [PubMed] [Google Scholar]
- 14.Rijken DC, Uitte de Willige S. Inhibition of fibrinolysis by coagulation factor XIII. Biomed Res Int 2017;2017:1209676. [DOI] [PMC free article] [PubMed]
- 15.Jobe SM, Leo L, Eastvold JS, et al. . Role of FcRgamma and factor XIIIA in coated platelet formation. Blood. 2005;106(13):4146-4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Francis CW, Marder VJ. Rapid formation of large molecular weight alpha-polymers in cross-linked fibrin induced by high factor XIII concentrations. Role of platelet factor XIII. J Clin Invest. 1987;80(5):1459-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hevessy Z, Haramura G, Boda Z, Udvardy M, Muszbek L. Promotion of the crosslinking of fibrin and alpha 2-antiplasmin by platelets. Thromb Haemost. 1996;75(1):161-167. [PubMed] [Google Scholar]
- 18.Reed GL, Matsueda GR, Haber E. Fibrin-fibrin and alpha 2-antiplasmin-fibrin cross-linking by platelet factor XIII increases the resistance of platelet clots to fibrinolysis. Trans Assoc Am Physicians. 1991;104:21-28. [PubMed] [Google Scholar]
- 19.Reed GL, Matsueda GR, Haber E. Platelet factor XIII increases the fibrinolytic resistance of platelet-rich clots by accelerating the crosslinking of alpha 2-antiplasmin to fibrin. Thromb Haemost. 1992;68(3):315-320. [PubMed] [Google Scholar]
- 20.Menegatti M, Palla R, Bucciarelli P, Peyvandi F. Minimal factor XIII activity level to prevent major spontaneous bleeds [reply]. J Thromb Haemost. 2017;15(11):2280-2282. [DOI] [PubMed] [Google Scholar]
- 21.de Moerloose P, Schved JF, Nugent D. Rare coagulation disorders: fibrinogen, factor VII and factor XIII. Haemophilia. 2016;22(suppl 5):61-65. [DOI] [PubMed] [Google Scholar]
- 22.Biswas A, Ivaskevicius V, Thomas A, Oldenburg J. Coagulation factor XIII deficiency. Diagnosis, prevalence and management of inherited and acquired forms. Hamostaseologie. 2014;34(2):160-166. [DOI] [PubMed] [Google Scholar]
- 23.Lawrie AS, Green L, Mackie IJ, Liesner R, Machin SJ, Peyvandi F. Factor XIII--an under diagnosed deficiency—are we using the right assays? J Thromb Haemost. 2010;8(11):2478-2482. [DOI] [PubMed] [Google Scholar]
- 24.Souri M, Koseki-Kuno S, Takeda N, Degen JL, Ichinose A. Administration of factor XIII B subunit increased plasma factor XIII A subunit levels in factor XIII B subunit knock-out mice. Int J Hematol. 2008;87(1):60-68. [DOI] [PubMed] [Google Scholar]
- 25.Lauer P, Metzner HJ, Zettlmeissl G, et al. . Targeted inactivation of the mouse locus encoding coagulation factor XIII-A: hemostatic abnormalities in mutant mice and characterization of the coagulation deficit. Thromb Haemost. 2002;88(6):967-974. [PubMed] [Google Scholar]
- 26.Prasad JM, Gorkun OV, Raghu H, et al. . Mice expressing a mutant form of fibrinogen that cannot support fibrin formation exhibit compromised antimicrobial host defense. Blood. 2015;126(17):2047-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooley B, Funkhouser W, Monroe D, et al. . Prophylactic efficacy of BeneFIX vs Alprolix in hemophilia B mice. Blood. 2016;128(2):286-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aleman MM, Walton BL, Byrnes JR, et al. . Elevated prothrombin promotes venous, but not arterial, thrombosis in mice. Arterioscler Thromb Vasc Biol. 2013;33(8):1829-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Byrnes JR, Duval C, Wang Y, et al. . Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin α-chain crosslinking. Blood. 2015;126(16):1940-1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buyue Y, Whinna HC, Sheehan JP. The heparin-binding exosite of factor IXa is a critical regulator of plasma thrombin generation and venous thrombosis. Blood. 2008;112(8):3234-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastoft AE, Lykkesfeldt J, Ezban M, Tranholm M, Whinna HC, Lauritzen B. A sensitive venous bleeding model in haemophilia A mice: effects of two recombinant FVIII products (N8 and Advate(®)). Haemophilia. 2012;18(5):782-788. [DOI] [PubMed] [Google Scholar]
- 32.Vaezzadeh N, Ni R, Kim PY, Weitz JI, Gross PL. Comparison of the effect of coagulation and platelet function impairments on various mouse bleeding models. Thromb Haemost. 2014;112(2):412-418. [DOI] [PubMed] [Google Scholar]
- 33.Marx G, Korner G, Mou X, Gorodetsky R. Packaging zinc, fibrinogen, and factor XIII in platelet alpha-granules. J Cell Physiol. 1993;156(3):437-442. [DOI] [PubMed] [Google Scholar]
- 34.Aleman MM, Holle LA, Stember KG, Devette CI, Monroe DM, Wolberg AS. Cystamine preparations exhibit anticoagulant activity. PLoS One. 2015;10(4):e0124448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vijayan KV, Goldschmidt-Clermont PJ, Roos C, Bray PF. The Pl(A2) polymorphism of integrin beta(3) enhances outside-in signaling and adhesive functions. J Clin Invest. 2000;105(6):793-802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeiler M, Moser M, Mann M. Copy number analysis of the murine platelet proteome spanning the complete abundance range. Mol Cell Proteomics. 2014;13(12):3435-3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burkhart JM, Vaudel M, Gambaryan S, et al. . The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012;120(15):e73-e82. [DOI] [PubMed] [Google Scholar]
- 38.Mattheij NJ, Swieringa F, Mastenbroek TG, et al. . Coated platelets function in platelet-dependent fibrin formation via integrin αIIbβ3 and transglutaminase factor XIII. Haematologica. 2016;101(4):427-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Byrnes JR, Wilson C, Boutelle AM, et al. . The interaction between fibrinogen and zymogen FXIII-A2B2 is mediated by fibrinogen residues γ390-396 and the FXIII-B subunits. Blood. 2016;128(15):1969-1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Asijee GM, Muszbek L, Kappelmayer J, Polgár J, Horváth A, Sturk A. Platelet vinculin: a substrate of activated factor XIII. Biochim Biophys Acta. 1988;954(3):303-308. [DOI] [PubMed] [Google Scholar]
- 41.Cohen I, Glaser T, Veis A, Bruner-Lorand J. Ca2+-dependent cross-linking processes in human platelets. Biochim Biophys Acta. 1981;676(2):137-147. [DOI] [PubMed] [Google Scholar]
- 42.Serrano K, Devine DV. Intracellular factor XIII crosslinks platelet cytoskeletal elements upon platelet activation. Thromb Haemost. 2002;88(2):315-320. [PubMed] [Google Scholar]
- 43.Kasahara K, Kaneda M, Miki T, et al. . Clot retraction is mediated by factor XIII-dependent fibrin-αIIbβ3-myosin axis in platelet sphingomyelin-rich membrane rafts. Blood. 2013;122(19):3340-3348. [DOI] [PubMed] [Google Scholar]
- 44.Saito M, Asakura H, Yoshida T, et al. . A familial factor XIII subunit B deficiency. Br J Haematol. 1990;74(3):290-294. [DOI] [PubMed] [Google Scholar]
- 45.Dickneite G, Herwald H, Korte W, Allanore Y, Denton CP, Matucci Cerinic M. Coagulation factor XIII: a multifunctional transglutaminase with clinical potential in a range of conditions. Thromb Haemost. 2015;113(4):686-697. [DOI] [PubMed] [Google Scholar]
- 46.Duckert F, Jung E, Shmerling DH. A hitherto undescribed congenital haemorrhagic diathesis probably due to fibrin stabilizing factor deficiency. Thromb Diath Haemorrh. 1960;5:179-186. [PubMed] [Google Scholar]
- 47.Kuether EL, Schroeder JA, Fahs SA, et al. . Lentivirus-mediated platelet gene therapy of murine hemophilia A with pre-existing anti-factor VIII immunity. J Thromb Haemost. 2012;10(8):1570-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gewirtz J, Thornton MA, Rauova L, Poncz M. Platelet-delivered factor VIII provides limited resistance to anti-factor VIII inhibitors. J Thromb Haemost. 2008;6(7):1160-1166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.