

Abstract
Promyelocytic leukemia (PML) proteins have been implicated in antiviral responses but PML and associated proteins are also suggested to support virus replication. One isoform, PML-II, is required for efficient transcription of interferon and interferon-responsive genes. We therefore investigated the PML-II contribution to human adenovirus 5 (Ad5) infection, using shRNA-mediated knockdown. HelaΔII cells showed a 2–3-fold elevation in Ad5 yield, reflecting an increase in late gene expression. This increase was found to be due in part to the reduced innate immune response consequent upon PML-II depletion. However, the effect was minor because the viral E4 Orf3 protein targets and inactivates this PML-II function. The major benefit to Ad5 in HelaΔII cells was exerted via an increase in HSP70; depletion of HSP70 completely reversed this replicative advantage. Increased Ad5 late gene expression was not due either to the previously described inhibition of inflammatory responses by HSP70 or to effects of HSP70 on major late promoter or L4 promoter activity, but might be linked to an observed increase in E1B 55K, as this protein is known to be required for efficient late gene expression. The induction of HSP70 by PML-II removal was specific for the HSPA1B gene among the HSP70 gene family and thus was not the consequence of a general stress response. Taken together, these data show that PML-II, through its various actions, has an overall negative effect on the Ad5 lifecycle.
Keywords: adenovirus,; PML protein,; HSP70
INTRODUCTION
The promyelocytic leukemia (PML) gene encodes a series of protein isoforms via alternative splicing (Jensen et al., 2001). Most of these contribute to the formation of PML nuclear bodies (PML-NB) that also transiently or permanently include many other proteins (Van Damme et al., 2010). PML proteins and/or PML-NB are implicated in a wide range of cellular functions, including innate and intrinsic immune responses (Bernardi & Pandolfi, 2007; Geoffroy & Chelbi-Alix, 2011). The PML gene itself is an interferon-stimulated gene (ISG) (Chelbi-Alix et al., 1995; Stadler et al., 1995), which suggests PML might be an effector of IFN responses. PML isoform II (PML-II) in particular is also required for the effective induction of IFNβ and ISG expression, which it achieves by promoting the assembly of functional transcription complexes at target promoters (Chen et al., 2015; Kim & Ahn, 2015). Thus this PML isoform can act upstream of IFN production to create a feed-forward loop that potentiates type I IFN responses.
PML-NBs are intimately associated with the replication cycles of nucleus-replicating DNA viruses. Incoming viral genomes are found located in close proximity to PML-NBs (Ishov & Maul, 1996) and, for herpes simplex virus type 1 (HSV-1), PML-NBs have been shown to disassemble and reform close to the site of virus entry into the nucleus (Everett & Murray, 2005), suggestive of an early response by the cell to infection. During infection, PML-NBs are then targeted by proteins encoded by a wide variety of viruses (Leppard & Dimmock, 2006; Leppard & Wright, 2012). These findings fit a model in which PML-NBs or their components are broadly antiviral and hence viruses have evolved functions to disrupt these activities in order to favour virus replication. Alternatively, and not mutually exclusively, the interaction of viruses with PML-NB may have been selected to favour the virus, with disruption of PML-NBs liberating proteins that act to increase virus production (Berscheminski et al., 2014).
One well-characterized example of PML-NB disruption is the action of HSV1 ICP0 protein (Maul & Everett, 1994). Supporting an involvement of PML in IFN responses, HSV1 ICP0 mutants have a significant growth defect and attenuated pathogenicity in mice, and both of these properties are substantially recovered in animals that are deficient in IFN responses (Halford et al., 2006; Leib et al., 1999); the ability of IFN to inhibit growth of ICP0 mutants in cell culture is also greatly reduced when PML-null or knockdown cells are used (Chee et al., 2003; Everett et al., 2008b). However, PML and other PML-NB components are also directly inhibitory to HSV1 independent of IFN (Everett et al., 2008a, b), with PML-I and PML-II being particularly implicated (Cuchet et al., 2011). More recently, PML-II was also found to be the most potent inhibitor, among the six nuclear PML isoforms, of transduction by a recombinant parvovirus AAV-2 vector (Mitchell et al., 2014).
Human adenovirus type 5 (HAdV-C5, Ad5) infection also targets PML, rearranging it from PML-NB into track-like structures (Carvalho et al., 1995; Doucas et al., 1996); other PML-NB components are also redistributed, including some into virus replication centres (Berscheminski et al., 2014; Doucas et al., 1996). The Ad5 E4 Orf3 protein, which forms nuclear tracks by self-association (Ou et al., 2012; Patsalo et al., 2012), acts directly on PML-II, binding to its unique C-terminal domain to cause the redistribution of all PML isoforms (Hoppe et al., 2006; Leppard et al., 2009). Functionally, E4 Orf3 is necessary for Ad5 to replicate in the face of a pre-established IFN response (Ullman et al., 2007) and E4 Orf3 also disrupts the intrinsic antiviral effects of PML and another PML-NB component, Daxx (Ullman & Hearing, 2008). Taken together, these observations support the idea that the E4 Orf3 interaction with PML-II opposes antiviral responses so as to favour productive viral infection, which fits well with the more recent finding that PML-II is needed for a robust type 1 IFN response (Chen et al., 2015). However, it has also been reported that PML-II serves a positive function during Ad5 infection (Berscheminski et al., 2013).
In light of these findings, we investigated the circumstances under which PML-II could provide a positive or negative influence on Ad5 infection, and the mechanisms underlying these influences. Viral gene expression and replication were increased by the removal of PML-II within a background of ongoing expression of other PML isoforms, leading to an increase in virus yield. One factor in this increase was the reduction in the interferon type I response in PML-II depleted cells. The other more significant factor was the increased level of HSP70 protein in PML-II depleted cells, which was found to support elevated Ad5 gene expression.
RESULTS
Stable knockdown of PML-II in Hela cells
To investigate the effect of PML-II on the well-characterized Ad5 productive infection of Hela cells, we first generated PML-II knockdown Hela cells (HelaΔII) by lentiviral shRNA transduction, along with matched empty vector cells (HelaEV). HelaΔII cells were fully viable in long-term culture, showed similar morphology to both parental Hela cells and HelaEV cells (Fig. 1a) and grew at only a slightly slower rate than HelaEV cells under puromycin selection. HelaΔII cells showed significant reductions in PML-II mRNA (Fig. 1b) and protein (Fig. 1c). These cells also displayed functional knockdown of PML-II based on their reduced ability to express IL-6 and ISG56 (Fig. 1d), which was shown previously to depend on the presence of PML-II (Chen et al., 2015).
Fig. 1.
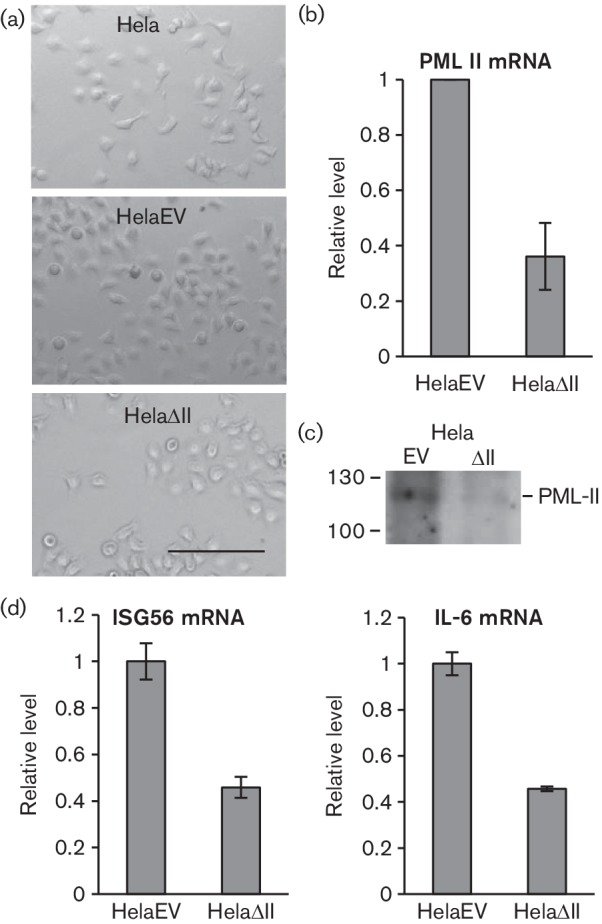
HelaΔII cells show physical and functional knockdown of PML-II. (a) Phase-contrast microscopic images of control Hela and shRNA Hela cells; bar, 100 µm. (b, c, d) HelaEV and HelaΔII cells were plated for 24 h, then RNA or protein samples were harvested. (b) PML-II mRNA was detected by RT-qPCR; results are normalized to the level detected in HelaEV cells and are the means and standard deviation of three technical replicates. (c) PML-II protein was detected by Western blotting. (d) IL-6 and ISG56 mRNAs were analysed as in (b).
Depletion of PML-II increases the productivity of Ad5 infection
To establish the effect of PML-II on Ad5 infection, HelaΔII and HelaEV cells were infected in parallel with wild-type (wt) Ad5. Looking at protein expression over a time course, there was a strikingly higher level of late protein expression in HelaΔII cells (Fig. 2a); with an exposure selected to avoid grossly overexposing the HelaΔII lane, late proteins in the HelaEV cells were barely detectable. In contrast, expression of the early protein E2A 72K DNA binding protein (DBP) was much less affected by the removal of PML-II though the E1B 55K protein was, by the late stage of infection (24 h post-infection), significantly increased (Fig. 2a). The expression of late proteins in HelaEV cells was similar to that in untransduced standard Hela cells (Fig. 2b), confirming that the difference between HelaΔII and HelaEV infections was not due to any unexpected negative effect of introducing the retroviral vector alone in HelaEV cells. The effect of PML-II depletion on viral gene expression was confirmed and quantified by flow cytometry (Fig. 2c); both the proportion of cells positive for late proteins and their mean fluorescence intensity were increased in HelaΔII cells. The increased late protein expression in HelaΔII cells was reflected in a 2–3-fold higher virus yield/cell as compared with HelaEV cells, measured at 24 h and 48 h post-infection (Fig. 2d).
Fig. 2.

Removal of PML-II increases Ad5 protein expression and virus yield. (a) HelaEV and HelaΔII cells were infected with wild-type Ad5 at m.o.i. of 5, and total protein extracts at various times post-infection were analysed by Western blotting. Upper panel: Ad5 late protein; middle panels: Ad5 E1B 55K and E2A DNA binding protein (DBP); lower panel: GAPDH. The 24 h samples were loaded at a 1 : 100 dilution compared to the earlier time points. (b) Total protein extracts of HelaEV and standard Hela cells, infected for 20 h as in (a), were analysed for hexon expression. (c) Adenovirus gene expression by FACS analysis. Upper panel: late gene expression; lower panel: DBP expression; grey curves are the background (mock-infected cells) while the black curves represent infected cells; the % of fluorescence-positive cells and their mean fluorescence intensity (MFI) are indicated on each panel. (d) Total virus in infected culture lysates was determined by fluorescent focus assay. Error bars show the standard deviation of replicates within an experiment; the experiment shown is representative of multiple experiments.
Lack of IFN response partially explains the beneficial effect of PML-II depletion
Infection by Ad5 is intrinsically an IFN response-inducing event, with both virus entry itself and later gene expression events triggering interferon and inflammatory signalling (Hartman et al., 2007; Hendrickx et al., 2014; Zhu et al., 2007). Since PML-II plays an important role in the activation of an IFN response (Chen et al., 2015), we considered the possibility that, even though Ad5 encodes functions that inhibit the IFN response in various ways, the beneficial effect of PML-II removal on Ad5 infection might nonetheless arise because of the consequent further defect in the IFN response. To test this, we directly disabled the IFN response by knockdown of IRF3, which is a key transcription factor in the induction of type 1 IFN responses (Au et al., 1995). Physical and functional depletion of IRF3 from HelaEV cells (Fig. 3a, b) increased Ad5 wt300 late gene expression by a modest amount (Fig. 3c), indicating that the Ad5 functions deployed to inhibit IFN responses are not 100 % effective. However, hexon expression under IRF3 knockdown in HelaEV cells was still very substantially lower than seen in HelaΔII cells, in which IRF3 knockdown had little additional effect. Thus, while some part of the benefit to Ad5 of PML-II removal reflects the loss of the IFN response, there is a significant additional component to be accounted for; this is considered further below.
Fig. 3.
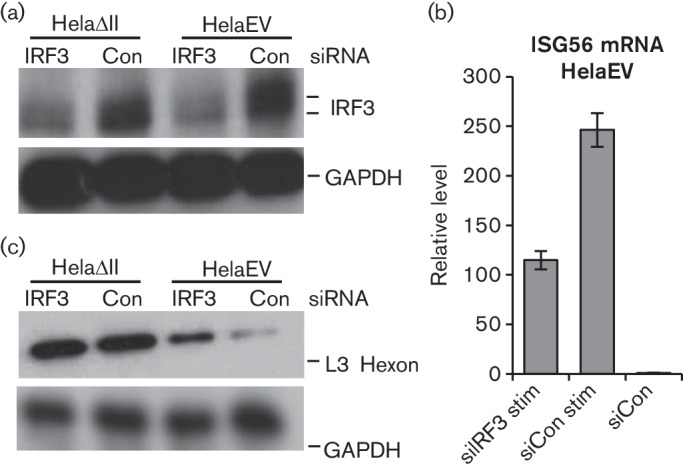
PML-II inhibits Ad5 infection by both IFN-dependent and -independent mechanisms. HelaEV and HelaΔII cells were plated for 24 h, transfected with 62.5 pmol ml−1 siRNA as indicated for 48 h and then either stimulated with poly I : C for 16 h (a, b) or infected with Ad5 wt300 at m.o.i. of 5 for 20 h (c), after which samples were prepared for analysis. (a) IRF3 protein or GAPDH (loading control) was detected by Western blot. (b) RT-qPCR analysis detecting ISG56 mRNA; results are the means and standard deviation of three technical replicates. (c) As panel (a), but detecting hexon protein using anti-late protein polyclonal antibodies.
Ad5 E4 Orf3 inhibits PML-II function in the IFN response
Ad5 E4 Orf3 binds PML-II directly (Hoppe et al., 2006) and is necessary for Ad5 replication in IFN-treated cells, dependent on the presence of PML (Ullman & Hearing, 2008; Ullman et al., 2007), so we asked whether this interaction also inhibited the natural IFN response to Ad5 infection. To determine whether viral expression of E4 Orf3 had any measurable effect on induction of type 1 IFN during infection, culture media from Ad5-infected HEK293 cells (Fig. 4a) or MRC5 normal human fibroblasts (Fig. 4b) were tested in plaque-reduction assays using Semliki Forest virus, an IFN-sensitive alphavirus. In both cell types, IFN activity was detected from an Orf3-deficient virus infection (inOrf3) while none was detected from wt300 or mock infections. Based on a calibration of the assay with recombinant IFNα, which showed inhibition from 0–100 % by IFN in the range 0.1–100 U ml−1, inOrf3 medium contained ~50 U ml−1 IFN. In separate experiments, IFN levels in infected HEK293 cell culture media were determined using an IFN-responsive reporter assay (Chen et al., 2015). Again, IFN accumulation was detected only in inOrf3-infected cultures (Fig. 4c): amounts were equivalent to about 60 U ml−1, in line with the estimate from the plaque-reduction assay. Thus, the presence of E4 Orf3 causes a measurable reduction in IFN production and secretion stimulated by Ad5 infection.
Fig. 4.

Adenovirus E4 Orf3 inhibits IFN production and IFNβ promoter activation. (a) HEK293 cells were infected at a multiplicity of 10 p.f.u. cell−1 with wild-type Ad5 (wt300) or mutant inOrf3, or were mock-infected. Media was harvested at 8 h post-infection and IFN activity measured by plaque-reduction assay. (b) As panel (a) but using media from Ad5-infected MRC5 fibroblasts harvested at 16 h post-infection. (c) Media from HEK293 cell cultures infected as in (a) was harvested at 6 h post-infection and IFN activity measured using an ISRE-luciferase reporter construct in HEK293 cells. Known amounts of recombinant IFNα were analysed in parallel to provide a standard curve. (d) HEK293 cells were transfected with IFNβ promoter luciferase reporter and β-galactosidase control plasmids together with PML-IIΔRBCC (125 ng) and from 125–625 ng E4 Orf3 plasmid as appropriate and then stimulated with poly(I : C) and reporter activities assayed 8 h later. (e–i) HEK293 cells were transfected with reporter plasmids as in (d) plus 150–600 ng (150–750 ng, panels e, g) of either wild-type E4 Orf3 plasmid (e), or mutant E4 Orf3 R100A (f), N82A (g), L103A (h) or D105A-L106A (i), and then stimulated or not with poly(I : C) as indicated and assayed as in panel (d). Data are the means and standard deviation of three biological replicates.
To test whether E4 Orf3 protein alone was sufficient to inhibit IFN responses, we employed transient expression IFNβ promoter reporter assays. PML-IIΔRBCC is an artificially truncated form of PML-II that does not associate with PML-NB but retains E4 Orf3 binding (Leppard et al., 2009) and has increased ability to potentiate IFNβ promoter activation by inducers such as poly(I : C) (Chen et al., 2015). E4 Orf3 fully reversed the increased response of the IFNβ promoter to poly(I : C) due to PML-IIΔRBCC and further reduced reporter activity to levels below that of poly(I : C) stimulation in the absence of exogenous PML (Fig. 4d). This reduction below baseline reflected the contribution of endogenous PML-II, also an E4 Orf3 target, to the observed IFNβ promoter activation as, in the presence only of endogenous PML, added Orf3 also gave a dose-dependent inhibition of poly(I : C)-stimulated reporter activity (Fig. 4e).
To correlate the activity of E4 Orf3 in regulating IFNβ expression with its ability to bind PML-II, we compared the inhibitory effect of wild-type E4 Orf3 with that of selected Orf3 mutants (Hoppe et al., 2006). Those mutants unable to bind PML-II (N82A, L103A) also failed to inhibit activation of the IFNβ promoter while mutants that retained PML-II binding (R100A, D105-L106A) had inhibitory activity similar to wild-type (Fig. 4e–i). Importantly, mutant D105-L106A uniquely retains PML-NB rearrangement activity whilst lacking the ability to disrupt the location of the MRN protein complex involved in DNA damage repair (Evans & Hearing, 2005). The retention of activity by this mutant thus clearly links the inhibitory effect of E4 Orf3 on IFN induction to its interaction with PML-II.
The inhibitory effect of E4 Orf3 on PML-II function suggested that an Orf3-deficient virus should benefit more from the lack of PML-II in HelaΔII cells than a virus that was able to make Orf3. When wt300 and inOrf3 late protein expression was compared in HelaEV cells (Fig. 5, lanes 2, 4), amounts were very similar, as expected (Huang & Hearing, 1989). As also shown previously, in Vero cells and human fibroblasts (Ullman et al., 2007), IFNα pre-treatment more severely inhibited inOrf3 than wt300 late protein synthesis in HelaEV cells (Fig. 5, lanes 3, 5). Importantly, removal of PML-II in HelaΔII cells largely abolished this difference in viral gene expression (Fig. 5, lanes 8, 10), confirming that PML-II is a significant functional target of E4 Orf3 during infection. However, contrary to expectation, wt300 gene expression benefited more than that of mutant virus inOrf3 from PML-II removal (Fig. 5, lanes 2, 7 and 4, 9), see below. Collectively, our results show that PML-II is inhibitory to Ad5 infection in part through its role in the development of an IFN response and that E4 Orf3 inhibits this function of PML-II.
Fig. 5.
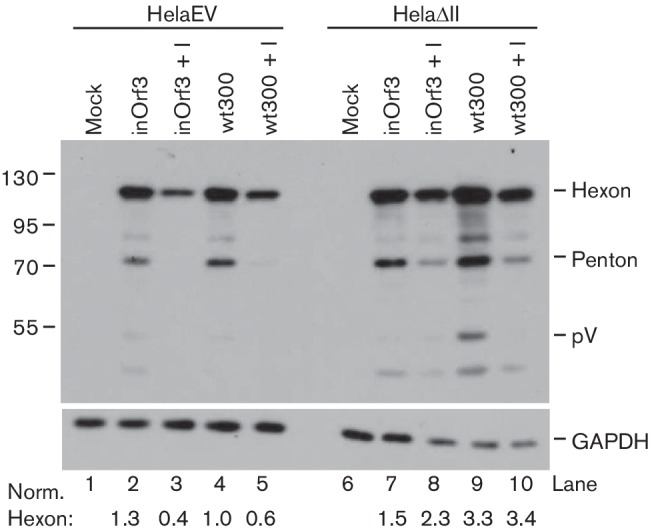
Role of E4 Orf3 in the response of Ad5 to PML-II depletion and IFN-α. HelaEV and HelaΔII cells were either mock-treated (lanes 1, 2, 4, 6, 7 and 9) or treated with 1000 U ml−1 of IFN-α for 24 h (+I; lanes 3, 5, 8 and 10), then mock-infected or infected with Ad5 wt300 or inOrf3 as indicated for 20 h. Total protein lysates were collected and analysed by Western blotting for Ad5 late proteins (upper) or GAPDH (lower). Hexon protein bands were quantified using QuantityOne software, normalized to GAPDH and expressed relative to the value for wt300 in HelaEV cells.
Enhanced growth of Ad5 in HelaΔII cells reflects overexpression of HSP70
A significant part of the benefit to Ad5 of PML-II depletion was independent of IRF3 and hence was not directly related to the IFN response (Fig. 3). Hence, late protein expression by either wt300 or inOrf3 was greater in HelaΔII cells than in equivalently treated HelaEV cells (Fig. 5). When studying stress responses in HelaΔII cells, we fortuitously observed that they displayed elevated levels of HSP70 mRNA and protein under normal growth conditions in comparison with HelaEV cells (Fig. 6a). HSP70 was also induced to a lower level by transient knockdown of PML-II in standard Hela cells (Fig. 6b), suggesting a direct link between loss of PML-II and HSP70 expression. Ad5 infection also induces HSP70 expression (Nevins, 1982) and, since it inhibits many other host genes whose activity is detrimental to infection (Zhao et al., 2003) and pre-existing HSP70 levels correlate with permissivity to Ad infection (Imperiale et al., 1984), we inferred that HSP70 might be the relevant positive factor for Ad5 growth in HelaΔII cells. To test this, hexon expression was compared in cells infected with or without HSP70 knockdown (Fig. 6c, d). Whilst HelaΔII cells showed substantially more hexon mRNA and protein than HelaEV cells when treated with a control siRNA, this difference was abolished by HSP70 siRNA treatment. Moreover, HSP70 siRNA also reduced hexon expression further from its lower base level in HelaEV cells. Thus, HSP70 contributes positively to Ad5 gene expression and the elevated expression of HSP70 in HelaΔII cells is a major factor in the increased efficiency of infection in these cells. The fact that wt300 gene expression benefited more than that of mutant virus inOrf3 from the high HSP70 environment in HelaΔII cells suggests that E4 Orf3 might be involved in the beneficial action of HSP70.
Fig. 6.
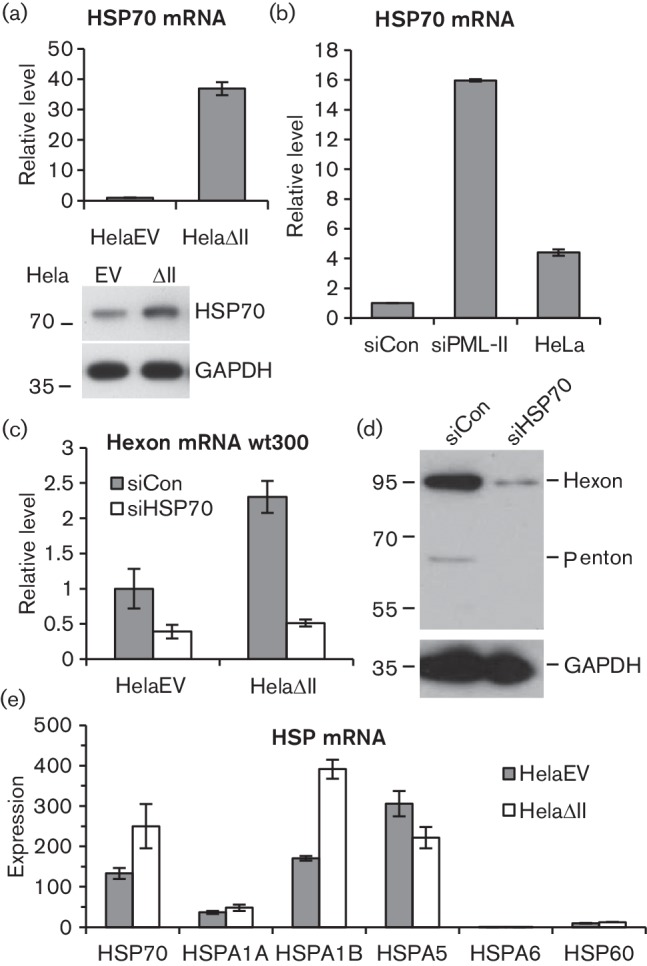
Elevated HSP70 enhances the expression of Ad5 proteins when PML-II is reduced. (a, e) Samples were harvested from HelaEV and HelaΔII cells and analysed for HSP70 mRNA (a) or a selection of HSP mRNAs (e) by RT-qPCR, or (a, lower) for HSP70 protein by Western blot. (b) Hela cells were transfected or not with 125 pmol ml−1 siRNA as indicated and RNA was harvested after 48 h for analysis of HSP70 mRNA by RT-qPCR. (c) HelaEV and HelaΔII cells were transfected with 125 pmol ml−1 HSP70 or control siRNA for 48 h, then infected with Ad5 wt300 for 20 h before RNA was harvested and analysed for hexon mRNA by RT-qPCR. (d) HelaΔII cells were treated with siRNA and infected as in (c), then lysed and analysed by Western blotting as in Fig. 5. In (a–c), data were standardized to an internal control and then normalized to values from: (a) HelaEV; (b) siControl-treated Hela; (c) siControl-treated HelaEV. Panel (e) shows mRNA amounts measured separately for each amplicon, standardized in each case to an internal control. Graphs show the means and standard deviation of three technical replicates.
The assay of HSP70 mRNA shown in Fig. 6a detects transcripts only from the two major heat-inducible loci, HSPA1A and HSPA1B. However, HSP70 encompasses a number of related proteins encoded by the HSPA gene family, only some of which are heat-inducible(Brocchieri et al., 2008). To determine the specificity of HSP70 induction in HelaΔII cells, mRNA levels from several HSPA genes were assessed alongside HSP60 (HSPD gene family). As before, the HSP70 assay detected elevated mRNA levels in HelaΔII cells (Fig. 6e). Interestingly, despite the high level of similarity between the HSPA1A and HSPA1B genes (they encode identical 641 amino acid proteins), elevated HSP70 expression was accounted for almost entirely by HSPA1B mRNA; there was little difference in expression of HSPA1A between the two cell types. In contrast to HSPA1, expression of HSPA5 mRNA, which encodes the endoplasmic reticulum chaperone GRP78 also known as BiP, was if anything slightly reduced by removing PML-II. Another HSP70 family member, HSPA6, which shows no basal expression but is induced by heat stress (Brocchieri et al., 2008), was detected only at low levels and was not induced by removal of PML-II; expression of HSP60 was also unaltered. Thus, the loss of PML-II leads to highly specific induction of the HSPA1B gene, providing HSP70 protein that supports enhanced Ad5 gene expression.
Possible roles of HSP70 during Ad5 infection
HSP70 has been shown previously to inhibit pro-inflammatory NF-κB signalling and hence both the production and the effect of TNF-α (Meng & Harken, 2002). Confirming that HSP70 had this effect in our system, we found that HSP70 knockdown significantly increased the expression of ISG56 in wt300-infected HelaEV cells (Fig. 7a). This suggested that HSP70 might favour Ad5 replication by limiting the induction of innate and inflammatory responses through NF-κB. TNFα is a known activator of NF-κB signalling and is considered to be inhibitory to virus infection (McFadden et al., 2009). Ad5 infection can stimulate NF-κB signalling in several ways (Higginbotham et al., 2002; Pahl et al., 1996; Schmitz et al., 1996) while several viral E3 gene products counteract TNFα activity (Gooding et al., 1988), suggesting NF-κB activation might be inhibitory to Ad5 infection. We therefore tested whether elevated HSP70 in HelaΔII cells enhanced Ad5 infection by inhibiting NF-κB. Reasoning that exogenous TNFα would oppose such an effect and so reduce the benefit of PML-II removal, we analysed Ad5 late gene expression in HelaΔII cells with or without TNFα treatment (Fig. 7b). However, TNFα actually modestly enhanced Ad5 late protein expression in both HelaΔII cells and HelaEV cells. The same effect was seen on hexon mRNA in HelaEV cells and this was potentiated by HSP70 knockdown (Fig. 7c), as expected if HSP70 limits pro-inflammatory signalling that is beneficial to the virus. We also tested the effect of QNZ, an inhibitor of NF-κB activation (Tobe et al., 2003) and found that, consistent with the effect of TNFα treatment, QNZ reduced Ad5 late gene expression in both cell types (Fig. 7d). These data indicate that NF-κB signalling increases rather than inhibits Ad5 gene expression in our system and that HSP70 limits rather than increases this effect. The beneficial effect on Ad5 infection of the high levels of HSP70 in HelaΔII cells must therefore be due to some other function of HSP70.
Fig. 7.
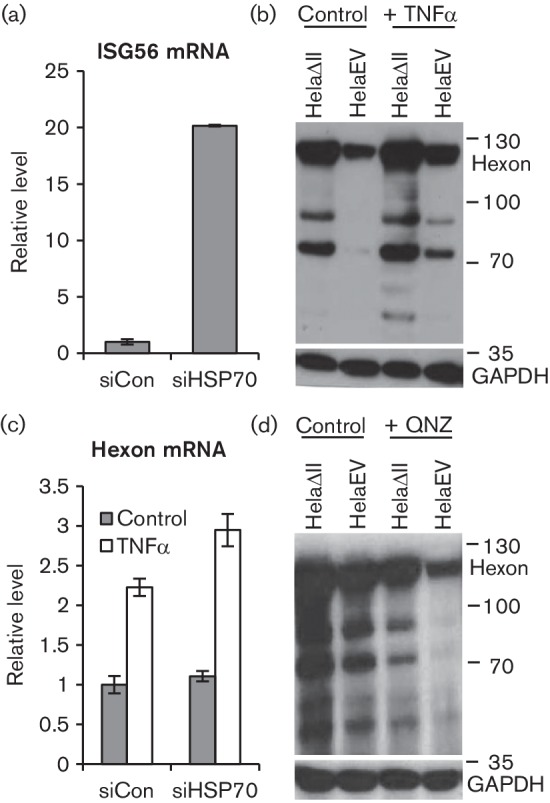
Effects of elevated HSP70 on NF-kB signalling do not cause enhanced Ad5 gene expression. (a) HelaEV cells were treated with HSP70 or control siRNA as in Fig. 6(c), then infected with Ad5 wt300 at m.o.i. of 5 for 20 h and ISG56 mRNA was quantified by RT-qPCR. (b, d) Cells as indicated were treated or not with 50 ng TNFα for 1 h (b) or with 100 nM of the NF-κB inhibitor QNZ for 45 min (d), then infected with Ad5 wt300 at m.o.i. of 5 for 20 h. Protein samples were harvested and analysed for late protein expression by Western blot. (c) HelaEV cells, treated with siRNA as in (a) were treated with TNFα as in (b), infected with Ad5 wt300 as in (a) and hexon mRNA quantified by RT-qPCR. Results in panels (a) and (c) are the means and standard deviation of three technical replicates.
The principal role of HSP70 is as a chaperone: during heat-stress it stabilizes partially denatured proteins to prevent aggregation and facilitate re-folding (Clerico et al., 2015). The Ad5 replication cycle involves both the disassembly and assembly of protein complexes, processes which might be facilitated by HSP70. Indeed, HSP70 interacts both with the hexon shell of Ad2 particles shortly after infection (Niewiarowska et al., 1992) and with fibre protein during the late phase of Ad5 infection (Macejak & Luftig, 1991), and has been implicated in uncoating and import of the genome into the nucleus (Saphire et al., 2000). We therefore examined whether increased HSP70 present in HelaΔII cells altered the subcellular location of hexon protein, as an indicator of possible effects on particle assembly. Prior depletion of HSP70 from these cells, as well as decreasing the overall level of hexon protein as already described, increased more than 2-fold the cytoplasmic : nuclear ratio of hexon (Fig. 8a; quantitation under right panels) whereas it had little effect on the distribution of E1A or E2A DBP. This result suggests that HSP70 overexpression consequent on PML-II depletion may positively affect the assembly of progeny particles in the nuclear compartment and hence could contribute to the increased yield of virus.
Fig. 8.
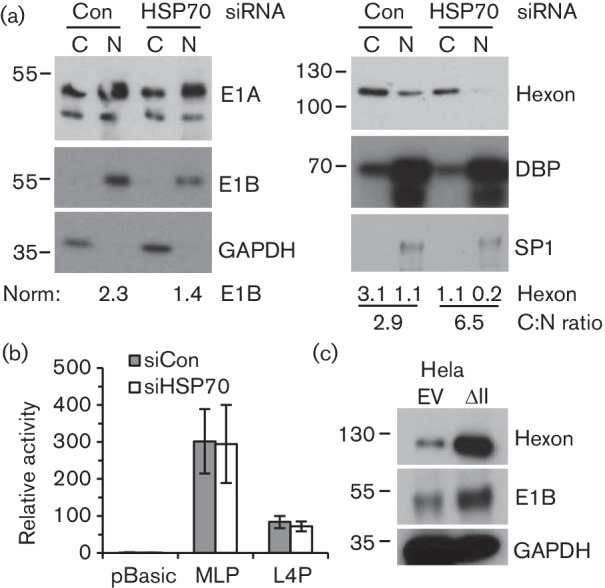
HSP70 promotes nuclear accumulation of Ad5 hexon but has no effect on late promoter activity. (a) HelaΔII cells were treated with HSP70 or control siRNA as in Fig. 6(c), then infected with Ad5 wt300 at m.o.i. of 5 for 16 h. Cytoplasmic and nuclear fractions were analysed by SDS-PAGE and Western blotting. Replicate blots were probed with antibodies to the proteins indicated and bands quantified as in Fig 5, normalized to GAPDH (cytoplasmic) or SP1 (nuclear). (b) Hela cells were treated with siRNA as in (a) and then transfected with MLP or L4P luciferase reporter. Luciferase activity was measured after 20 h and normalized to a β-galactosidase transfection control as described by Wright et al. (2015). (c) HelaEV and HelaΔII cells were infected with wild-type Ad5 at m.o.i. of 5, and total protein extracts analysed by Western blotting at 20 h post-infection for the proteins indicated.
Any impact of HSP70 level on assembly cannot explain the effect of HSP70 on hexon mRNA levels (Fig. 6). This mRNA is produced by processing of transcripts from the major late promoter (MLP), which itself is positively influenced by L4-22K protein expressed from L4P which is activated at the onset of the late phase (Morris et al., 2010). We therefore tested the effect of HSP70 depletion on the activity of MLP and L4P luciferase reporters in Hela cells (Fig. 8b), but found that neither was significantly affected. Thus HSP70 does not increase directly the intrinsic activity of either promoter when taken out of the context of viral infection and must therefore affect late gene expression post-transcriptionally or dependent on the infected cell environment. In this regard, we noted the increase in E1B 55K protein upon PML-II depletion in HelaΔII cells (Fig. 8c), a protein which is known to positively regulate Ad5 late mRNA nucleo-cytoplasmic transport and accumulation (Leppard, 1998). HSP70 depletion in HelaΔII cells reversed this increase in E1B 55K (Fig. 8a), further suggesting that it could be significant in the elevation of late gene expression.
Discussion
Depletion of PML-II from Hela cells, a standard permissive cell line for Ad5, led to a substantial 2–3-fold enhancement in virus yield. This was attributed largely to a general increase in late gene expression, our experiments focusing mainly on the major capsid protein hexon and its mRNA as an example. Thus, PML-II has an overall inhibitory effect on Ad5 infection. Two factors were identified that contributed to the increased infectious productivity upon PML-II depletion: a reduction in IFN response and an increase in HSP70 expression, the latter being the predominant factor. Depletion of HSP70 from HelaΔII cells completely eliminated the advantage to hexon expression of PML-II removal, whilst blocking the IFN response in HelaEV cells by a means other than PML-II depletion only somewhat increased the levels of viral late gene expression.
Previously, Berscheminski and colleagues reported that PML-II was beneficial to Ad5 infection, in apparent contradiction to our findings (Berscheminski et al., 2013). They showed that PML-II potentiated transcriptional activation of a viral early promoter by the E1A 13S protein and that, in a cellular context where all PML proteins were depleted, the addition of exogenous PML-II enhanced virus yield about 3-fold. Comparing these findings with our own, it is important to note the differences in cell environment employed. PML-II function will be a composite of its free and PML-NB associated activities. PML-V is the stable base of PML-NBs (Weidtkamp-Peters et al., 2008), with other isoforms and many other proteins associating with these bodies by protein–protein interactions including SUMO-SIM interactions (Bernardi & Pandolfi, 2007). In a PML-null cell, functions observed for added PML-II will be essentially those of its soluble nuclear form. Indeed, a mutated form of PML-II with reduced ability to associate with PML-NBs was more active than the wild-type in cooperating with E1A in the presence of endogenous PML (Berscheminski et al., 2013), suggesting that interactions with other PML isoforms limit this activity. The overexpressed PML-II will also potentially exceed the capacity of E4 Orf3 expressed upon infection to bind and inactivate it. In contrast, specific depletion of PML-II in an otherwise normal PML background demonstrates the combined net contribution of this protein to the Ad5 lifecycle in all its cellular contexts. Our finding that PML-II removal exerts an overall positive effect on Ad5 growth is therefore not in disagreement with this prior study but instead reveals a new aspect of the functional interaction of PML-II with the virus. Our findings are also consistent with an earlier study showing the importance of PML proteins generally in the inhibition of Ad5 by an established IFN response (Ullman & Hearing, 2008).
Although PML-II is necessary for an efficient IFN response (Chen et al., 2015), Ad5 gained only modest benefit from the loss of this response in HelaΔII cells. This finding is expected since the virus possesses several functions that collectively oppose IFN responses, so allowing infection to succeed even when the cell is capable of launching a response. First, E1A proteins inhibit both the expression of ISGs and the activation of IFNβ transcription (Ackrill et al., 1991; Reich et al., 1988). Second, E1B 55K protein blocks the induction of a number of IFN-inducible genes and is required for efficient replication in normal fibroblasts (Chahal et al., 2012). Third, E4 Orf3 protein is necessary for replication to proceed in permanent cell lines in the face of an established IFN response (Ullman et al., 2007), implying it negatively regulates that response. Fourth, VA RNA I inhibits the induction of an IFN-induced antiviral state by inhibiting protein kinase R (Kitajewski et al., 1986). Finally, activated STAT1 is sequestered in Ad replication centres (Sohn & Hearing, 2011). The further advantage to Ad5 of an inherent lack of IFN response may arise because of the time it otherwise takes for virus-encoded anti-IFN functions to become active. We directly tested the idea that E4 Orf3 would inhibit the IFN response via its targeting of PML-II, and showed that this was the case: wild-type E4 Orf3 inhibited type 1 IFN induction whilst mutant forms unable to bind PML-II could not; E4 Orf3 mutant virus infection elicited more IFN than wild-type; and E4 Orf3 mutant virus late protein expression was more strongly inhibited by prior IFN treatment in cells with functional PML-II. E4 Orf3 is expressed in the early phase but takes time to accumulate. In the period prior to this, our data suggest that inhibition of IFN induction is incomplete.
The principal factor in the enhanced growth of Ad5 in HelaΔII cells was the elevated level of HSP70. Investigating this, we observed a modest effect of HSP70 on Ad5 late protein nuclear accumulation that would favour progeny virus formation but this could not account for the significant enhancement of late gene expression. HSP70 expression/depletion in HelaΔII cells did however also affect the accumulation of the viral E1B 55K protein, which is known to regulate late mRNA accumulation and hence to increase late gene expression (Leppard, 1998). This increased amount of E1B 55K in the presence of elevated HSP70 may contribute to the observed elevation in late gene expression and virus yield under these conditions. HSP70 also opposes inflammatory responses (Meng & Harken, 2002); given the role of PML-II in regulating inflammatory gene expression we considered this to be a plausible basis for the positive effect of HSP70 on Ad5 growth. However, NF-κB activation was actually modestly beneficial to late gene expression. Since, as reported, HSP70 opposed this activation, HSP70 elevation cannot be benefitting Ad5 via effects on NF-κB. The positive effect of NF-κB on Ad5 was unexpected given that the virus encodes functions in its E3 region that inhibit TNFα signalling and hence NF-κB activation (Burgert et al., 2002). However, although these functions will be important in vivo they are known to be dispensable for growth in culture. Thus the small increase in Ad5 gene expression when NF-κB is activated in cell culture should not imply that this response benefits the virus in vivo.
The elevated level of HSP70 in PML-II depleted cells reflected a highly specific increase in mRNA derived from the HSPA1B gene, one of two intronless genes that are strongly heat-inducible members of the HSPA gene family. HSPA1A and HSPA1B are very similar even in their promoter sequences (Brocchieri et al., 2008); their products are not normally distinguished in analyses of heat-induced HSP70 expression. The specific upregulation of HSPA1B by PML-II depletion cannot be due to a general cell stress response, and in particular cannot be attributed to activation of the heat shock transcription factor, HSF, which regulates transcription of HSPA1A and HSPA1B as well as other classes of HSP (Singh et al., 2010). Thus, these results indicate a novel mechanism whereby the HSPA1B promoter is selectively activated. Interestingly, HSPA1A and HSPA1B are located within the MHC III region, between the gene clusters encoding MHC class I and II antigen where specific depletion of individual PML isoforms has been shown to have effects on chromatin architecture and gene expression (Kumar et al., 2007). Further work is needed to test whether HSPA1B induction by PML-II removal reflects a similar mechanism.
HSP70 is also induced during Ad5 infection (Nevins, 1982) and, whilst virus infection might be considered a stress that would lead to generalized activation of HSP expression, this induction is actually specific to HSP70 (Phillips et al., 1991). These studies did not distinguish between HSPA1A and HSPA1B, which were not separately recognized at the time. HSP70 transcription is induced by the virus-coded transactivator, E1A 13S (Wu et al., 1986), which acts via the cellular CCAAT-box factor (CBF) and its binding site in the context of a specific TATA box (Lum et al., 1992; Simon et al., 1988). In this way, induction is independent of HSF. The CBF site is also a target for p53-mediated inhibition of the HSP70 genes (Agoff et al., 1993), suggesting that E1A might disrupt this inhibition. HSP70 expression is also favoured by the viral E1B 55K/E4 Orf6 complex promoting HSP70 mRNA export to the cytoplasm (Moore et al., 1987). Since, as discussed, our study shows that HSP70 levels are also positively linked to E1B 55K accumulation, a feed-forward loop may be established that promotes efficient late gene expression.
The induction of HSP70 during Ad infection may be linked with positive roles for this protein in the virus lifecycle. The best documented of these, in viral uncoating (Saphire et al., 2000), may account for the modest increase in early gene expression seen in HelaΔII cells. However, this action must precede E1A-induced activation of HSP70 synthesis, suggesting that other roles may exist to justify this mechanism. This role also cannot account for the predominant effect on late rather than early gene expression that we observed. A study by White et al. (1988) suggested an involvement of HSP70 in nuclear events linked with PML during Ad2 infection. HSP70 was recruited from the cytoplasm into discrete nuclear structures that co-localized with E1A and which appeared similar to the reorganized PML tracks that are formed by E4 Orf3 (Carvalho et al., 1995; Doucas et al., 1996). Indeed, Carvalho et al. (1995) found a small fraction of E1A and, in a few infected cells, HSP70 located in these Orf3/PML structures, evidence of a physical and/or functional link between HSP70 and PML that might be related to our observations. Interestingly, our work suggests that the presence of E4 Orf3 is required in order for Ad5 to benefit from the elevation of HSP70 that occurs in HelaΔII cells.
Many other viruses induce and/or functionally interact with HSP70 (Santoro et al., 2010) suggesting a general importance of this protein to infection. The avian adenovirus CELO Gam1 protein causes an increase in both HSP70 and HSP40 that is needed for replication, and loss of Gam1 can be complemented by heat shock (Glotzer et al., 2000). Gam1 is also responsible for the loss of PML from infected cells through an inhibition of sumoylation (Colombo et al., 2002), raising the possibility that HSP induction and PML loss are also linked in this system. Human cytomegalovirus, HSV1, vaccinia virus and some paramyxoviruses all induce HSP70 expression (Santoro et al., 2010). For HSV1, heat shock can complement deficiency in ICP0, the protein responsible for PML body disruption and PML degradation (Bringhurst & Schaffer, 2006), while the same is true for E1A deficiency in Ad (Imperiale et al., 1984; Madara et al., 2005).
In conclusion, we have shown that PML-II opposes productive Ad5 infection, in part by supporting innate immune responses but mainly due to a suppressive effect on HSP70 expression. Our study reveals a previously undefined activity for HSP70 in supporting Ad5 late gene expression and demonstrates an inhibitory effect of PML-II on HSP70 expression.
Methods
Generation of HelaΔII and HelaEV cell lines.
Hela cells were transduced with either lentiviral particles encoding an shRNA specific for PML-II or equivalent particles with no shRNA insert. The PML-II shRNA incorporated the active siRNA sequence described by Kumar et al. (2007) which was used previously by our laboratory to achieve functional knockdown of PML-II (Chen et al., 2015). Lentiviral particles were generated using pLKO.1 (Moffat et al., 2006) following protocols supplied by the RNAi consortium (Addgene). Briefly, a double-stranded synthetic oligonucleotide corresponding to the shRNA was cloned into pLKO.1. Specific plasmid clones were verified by sequencing, then transfected with psPAX2 and pMD2.G packaging plasmids into HEK-293T cells using Transit LT-1 (Mirus) to produce VSV-G-pseudotyped particles. Particle stocks were then used to infect Hela cells and transduced cells were selected with 3 µg ml−1 puromycin.
Antibodies and reagents.
Specific primary antibodies were: AdJLB1 rabbit antiserum to Ad5 late proteins (Farley et al., 2004); mouse monoclonal antibodies 2HX-2 to Ad5 hexon (Cepko et al., 1983), B6-8 to Ad5 E2A DNA binding protein (DBP) (Reich et al., 1983), and 2A6 to Ad5 E1B 55K (Sarnow et al., 1982); monospecific anti-peptide sera reactive against PML-II (Xu et al., 2005), kindly provided by Professor K.-S. Chang, M.D. Anderson Cancer Center, University of Texas; FL-425 rabbit anti-IRF3 (SantaCruz); rabbit anti-HSP70 (StressMarq SPC-103C/D); and GA1R mouse anti-GAPDH (Thermo Scientific). Secondary antibodies were: Alexa488-conjugated goat anti-mouse Ig (Life Technologies); horseradish peroxidase (HRP)-conjugated goat anti-mouse Ig (Sigma); and HRP-conjugated goat anti-rabbit Ig (SantaCruz). IFNα was from PBL Assay Science, TNFα from Invitrogen, poly(I : C) from Sigma and 6-amino-4-(phenoxyphenylethylamino)quinazolin (QNZ) from Santa Cruz. siRNAs were: IRF3 (ID 3661; Qiagen); HSP70 (targets HSPA1A and HSPA1B; Ambion); and control B (Chen et al., 2015).
Cell culture and virus infection.
HEK293, HEK293T, Hela and knockdown cell lines were maintained at 37 °C, 5 % CO2 in Dulbecco’s modified Eagle's medium (DMEM) supplemented with 10 % foetal bovine serum (FBS); for maintenance purposes, HelaEV and HelaΔII cells were alternated between media containing or not containing 3 µg ml−1 puromycin. Vero cells were maintained in DMEM supplemented with 5 % FBS and MRC5 cells in 10 % Eagle’s minimal essential medium supplemented with 10 % FBS, 2 mM l-glutamine and 1 % non-essential amino acids. Cells were seeded at the appropriate density 24 h prior to the respective procedure. Light microscope images were recorded on an inverted microscope using a 5× objective. Virus stocks and experimental samples were titred in a fluorescent focus assay. Hela cell monolayers were infected in duplicate with serial dilutions of each stock, incubated at 37 °C, 5 % CO2 for 16 h, then fixed and stained with antibody to E2A DBP to visualize fluorescent cells for counting. Experimental infections were carried out with wild-type Ad5 wt300 or E4 Orf3 mutant inOrf3 (Huang & Hearing, 1989) at a multiplicity of 5 fluorescence focus units (f.f.u.) per cell unless otherwise indicated. siRNA transfections were performed with Lipofectamine 2000 (Invitrogen), using a ratio of 1 µl reagent per 25 pmol siRNA.
Protein and RNA analysis.
For total protein analysis, cells were lysed directly in SDS gel sample buffer. Cytoplasmic and nuclear fractions were generated by lysing cells in 0.67 % (v/v) NP40, 10 mM NaCl, 1.5 mM MgCl2, 10 mM Tris.HCl pH7.5 for 10 min on ice, then nuclei were pelleted by low speed centrifugation; an equal volume of 2× SDS gel sample buffer was added to the supernatant (cytoplasmic fraction). Crude nuclei were washed once in PBS, pelleted as before and then lysed in SDS gel sample buffer (nuclear fraction). Proteins were separated by electrophoresis on 10 % SDS polyacrylamide gels and detected by Western blotting as previously described (Lethbridge et al., 2003). For flow cytometry analysis, single cell suspensions produced by trypsinization were fixed on ice with 10 % (v/v) formalin in PBS for 20 min, permeablized with 0.5 % (v/v) NP40 in PBS for 10 min and then incubated with 1 % (w/v) bovine serum albumin in PBS for 45 min to block nonspecific protein binding. Cells were resuspended in FACS buffer (PBS containing 3 % v/v FBS, 0.07 % w/v NaN3), then incubated with specific primary antibodies to hexon or E2A DBP followed by Alexa488-conjugated secondary antibody. Washed cells in FACS buffer were analysed using a FACSCAN (BectonDickinson) and WinMDI software. Immunofluorescence analysis was performed as previously described (Leppard & Everett, 1999); images were collected with a Leica SP5 confocal microscope system and processed using Leica confocal software. Total RNA was isolated and mRNA quantified by RT-qPCR as previously described (Chen et al., 2015) using the following primers and amplicons: ISG56 and IL-6 (Chen et al., 2015); E1A (113 bp, Ad genome 1422–1534) and hexon (137 bp, 21540–21576) (Schreiner et al., 2013); PML-II 5′AGGCAGAGGAACGCGTTGT-3′ and 5′GGCTCCATGCACGAGTTTTC-3′ (70 bp); HSP70 (Tanaka et al., 2007); HSPA1A, HSPA1B, HSPA5, HSPA6, and HSP60 (www.rtprimerdb.org).
Interferon activity and luciferase reporter assays.
MLP and L4P activity was determined in luciferase reporter assays as described (Morris & Leppard, 2009; Wright et al., 2015). IFNβ promoter activity was measured by transfecting IFNβ-Luc (King & Goodbourn, 1994) in the presence of either wild-type or mutant E4 Orf3 expression plasmids (Hoppe et al., 2006) and pcDNAHisLacZ as an internal control for 24 h and stimulating by transfection with poly(I : C) for a further 8 h, otherwise as previously described (Chen et al., 2015; Morris & Leppard, 2009). IFN activity in cell culture fluids was measured by plaque-reduction assay using infection of Vero cells by Semliki Forest virus (SFV). Subconfluent 12-well cultures were incubated for 24 h with either standard IFNα or with an unknown sample at 1 in 10 dilution, both in normal growth medium. After 24 h, cells were infected with 25 plaque-forming units of SFV, overlaid with agar-solidified medium and then fixed after 48 h incubation and plaques detected with crystal violet. All determinations were made in triplicate. Alternatively, IFN activity was determined by measuring the stimulation of pISRE-Luc by IFN-containing samples for 20 h in a luciferase reporter assay (Chen et al., 2015).
Acknowledgements
Z. A. was supported by a studentship from The Higher Committee for Education Development in Iraq. J. W. was supported by a studentship from the Biotechnology and Biological Sciences Research Council. A. W. was supported by a University of Warwick Undergraduate Research Student Scholarship.
References
- Ackrill A. M., Foster G. R., Laxton C. D., Flavell D. M., Stark G. R., Kerr I. M.(1991). Inhibition of the cellular response to interferons by products of the adenovirus type 5 E1A oncogene. Nucleic Acids Res 194387–4393. 10.1093/nar/19.16.4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agoff S. N., Hou J., Linzer D., Wu B.(1993). Regulation of the human hsp70 promoter by p53. Science 25984–87. 10.1126/science.8418500 [DOI] [PubMed] [Google Scholar]
- Au W. C., Moore P. A., Lowther W., Juang Y. T., Pitha P. M.(1995). Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc Natl Acad Sci U S A 9211657–11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R., Pandolfi P. P.(2007). Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Revs Mol Cell Biol 81006–1016. [DOI] [PubMed] [Google Scholar]
- Berscheminski J., Groitl P., Dobner T., Wimmer P., Schreiner S.(2013). The adenoviral oncogene E1A-13S interacts with a specific isoform of the tumor suppressor PML to enhance viral transcription. J Virol 87965–977. 10.1128/JVI.02023-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berscheminski J., Wimmer P., Brun J., Ip W. H., Groitl P., Horlacher T., Jaffray E., Hay R. T., Dobner T., Schreiner S.(2014). Sp100 isoform-specific regulation of human adenovirus 5 gene expression. J Virol 886076–6092. 10.1128/JVI.00469-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringhurst R. M., Schaffer P. A.(2006). Cellular stress rather than stage of the cell cycle enhances the replication and plating efficiencies of herpes simplex virus type 1 ICP0− viruses. J Virol 804528–4537. 10.1128/JVI.80.9.4528-4537.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocchieri L., Conway de Macario E., Macario A. J. L.(2008). Hsp70 genes in the human genome: Conservation and differentiation patterns predict a wide array of overlapping and specialized functions. BMC Evol Biol 819. 10.1186/1471-2148-8-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgert H. G., Ruzsics Z., Obermeier S., Hilgendorf A., Windheim M., Elsing A.(2002). Subversion of host defense mechanisms by adenoviruses. Curr Top Microbiol Immunol 269273–318. [DOI] [PubMed] [Google Scholar]
- Carvalho T., Seeler J. S., Ohman K., Jordan P., Pettersson U., Akusjärvi G., Carmo-Fonseca M., Dejean A.(1995). Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J Cell Biol 13145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepko C. L., Whetstone C. A., Sharp P. A.(1983). Adenovirus hexon monoclonal antibody that is group specific and potentially useful as a diagnostic reagent. J Clin Microbiol 17360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahal J. S., Qi J., Flint S. J.(2012). The human adenovirus type 5 E1B 55 kDa protein obstructs inhibition of viral replication by type I interferon in normal human cells. PLoS Pathogens 8e1002853 10.1371/journal.ppat.1002853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee A. V., Lopez P., Pandolfi P. P., Roizman B.(2003). Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J Virol 777101–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelbi-Alix M. K., Pelicano L., Quignon F., Koken M. H., Venturini L., Stadler M., Pavlovic J., Degos L., de Thé H.(1995). Induction of the PML protein by interferons in normal and APL cells. Leukemia 92027–2033. [PubMed] [Google Scholar]
- Chen Y., Wright J., Meng X., Leppard K. N.(2015). Promyelocytic leukemia protein isoform II promotes transcription factor recruitment to activate interferon beta and interferon-responsive gene expression. Mol Cell Biol 351660–1672. 10.1128/MCB.01478-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerico E. M., Tilitsky J. M., Meng W., Gierasch L. M.(2015). How Hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J Mol Biol 4271575–1588. 10.1016/j.jmb.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo R., Boggio R., Seiser C., Draetta G. F., Chiocca S.(2002). The adenovirus protein Gam1 interferes with sumoylation of histone deacetylase 1. EMBO Rep 31062–1068. 10.1093/embo-reports/kvf213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchet D., Sykes A., Nicolas A., Orr A., Murray J., Sirma H., Heeren J., Bartelt A., Everett R. D.(2011). PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J Cell Sci 124280–291. 10.1242/jcs.075390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucas V., Ishov A. M., Romo A., Juguilon H., Weitzman M. D., Evans R. M., Maul G. G.(1996). Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev 10196–207. 10.1101/gad.10.2.196 [DOI] [PubMed] [Google Scholar]
- Evans J. D., Hearing P.(2005). Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J Virol 796207–6215. 10.1128/JVI.79.10.6207-6215.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R. D., Murray J.(2005). ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol 795078–5089. 10.1128/JVI.79.8.5078-5089.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R. D., Parada C., Gripon P., Sirma H., Orr A.(2008a). Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J Virol 822661–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R. D., Young D. F., Randall R. E., Orr A.(2008b). Stat-1- and IRF-3-dependent pathways are not essential for repression of ICP0-null mutant herpes simplex virus type 1 in human fibroblasts. J Virol 828871–8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley D. C., Brown J. L., Leppard K. N.(2004). Activation of the early-late switch in adenovirus type 5 major late transcription unit expression by L4 gene products. J Virol 781782–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy M. C., Chelbi-Alix M. K.(2011). Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31145–158. 10.1089/jir.2010.0111 [DOI] [PubMed] [Google Scholar]
- Glotzer J. B., Saltik M., Chiocca S., Michou A. I., Moseley P., Cotten M.(2000). Activation of heat-shock response by an adenovirus is essential for virus replication. Nature 407207–211. 10.1038/35025102 [DOI] [PubMed] [Google Scholar]
- Gooding L. R., Elmore L. W., Tollefson A. E., Brady H. A., Wold W. S.(1988). A 14,700 MW protein from the E3 region of adenovirus inhibits cytolysis by tumor necrosis factor. Cell 53341–346. 10.1016/0092-8674(88)90154-7 [DOI] [PubMed] [Google Scholar]
- Halford W. P., Weisend C., Grace J., Soboleski M., Carr D. J., Balliet J. W., Imai Y., Margolis T. P., Gebhardt B. M.(2006). ICP0 antagonizes Stat 1-dependent repression of herpes simplex virus: implications for the regulation of viral latency. Virol J 344. 10.1186/1743-422X-3-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman Z. C., Black E. P., Amalfitano A.(2007). Adenoviral infection induces a multi-faceted innate cellular immune response that is mediated by the toll-like receptor pathway in A549 cells. Virology 358357–372. 10.1016/j.virol.2006.08.041 [DOI] [PubMed] [Google Scholar]
- Hendrickx R., Stichling N., Koelen J., Kuryk L., Lipiec A., Greber U. F.(2014). Innate immunity to adenovirus. Hum Gene Ther 25265–284. 10.1089/hum.2014.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham J. N., Seth P., Blaese R. M., Ramsey W. J.(2002). The release of inflammatory cytokines from human peripheral blood mononuclear cells in vitro following exposure to adenovirus variants and capsid. Hum Gene Ther 13129–141. 10.1089/10430340152712683 [DOI] [PubMed] [Google Scholar]
- Hoppe A., Beech S. J., Dimmock J., Leppard K. N.(2006). Interaction of the adenovirus type 5 E4 Orf3 protein with promyelocytic leukemia protein isoform II is required for ND10 disruption. J Virol 803042–3049. 10.1128/JVI.80.6.3042-3049.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.-M., Hearing P.(1989). Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J Virol 632605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperiale M. J., Kao H. T., Feldman L. T., Nevins J. R., Strickland S.(1984). Common control of the heat shock gene and early adenovirus genes: evidence for a cellular E1A-like activity. Mol Cell Biol 4867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishov A. M., Maul G. G.(1996). The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J Cell Biol 134815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K., Shiels C., Freemont P. S.(2001). PML protein isoforms and the RBCC/TRIM motif. Oncogene 207223–7233. 10.1038/sj.onc.1204765 [DOI] [PubMed] [Google Scholar]
- Kim Y.-E., Ahn J.-H.(2015). Positive role of promyelocytic leukemia protein in type I interferon response and its regulation by human cytomegalovirus. PLoS Pathog 11e1004785. 10.1371/journal.ppat.1004785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King P., Goodbourn S.(1994). The beta-interferon promoter responds to priming through multiple independent regulatory elements. J Biol Chem 26930609–30615. [PubMed] [Google Scholar]
- Kitajewski J., Schneider R. J., Safer B., Munemitsu S. M., Samuel C. E., Thimmappaya B., Shenk T.(1986). Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 45195–200. 10.1016/0092-8674(86)90383-1 [DOI] [PubMed] [Google Scholar]
- Kumar P. P., Bischof O., Purbey P. K., Notani D., Urlaub H., Dejean A., Galande S.(2007). Functional interaction between PML and SATB1 regulates chromatin-loop architecture and transcription of the MHC class I locus. Nat Cell Biol 945–56. 10.1038/ncb1516 [DOI] [PubMed] [Google Scholar]
- Leib D. A., Harrison T. E., Laslo K. M., Machalek M. A., Moorman N. J., Virgin H. W.(1999). Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med 189663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppard K. N., Dimmock J.(2006). Virus interactions with PML nuclear bodies. Viruses and the Nucleus 213–245. Edited by Hiscox J., Wiley J. [Google Scholar]
- Leppard K. N., Everett R. D.(1999). The adenovirus type 5 E1b 55K and E4 Orf3 proteins associate in infected cells and affect ND10 components. J Gen Virol 80997–1008. 10.1099/0022-1317-80-4-997 [DOI] [PubMed] [Google Scholar]
- Leppard K. N., Wright J.(2012). Targeting of promyelocytic leukaemia proteins and promyelocytic leukaemia nuclear bodies by DNA tumour viruses. Small DNA Tumour Viruses 255–280. Edited by Gaston K.Norfolk, UK: Caister Academic Press. [Google Scholar]
- Leppard K. N.(1998). Regulated RNA processing and RNA transport during adenovirus infection. Semin Virol 8301–307. [Google Scholar]
- Leppard K. N., Emmott E., Cortese M. S., Rich T.(2009). Adenovirus type 5 E4 Orf3 protein targets promyelocytic leukaemia (PML) protein nuclear domains for disruption via a sequence in PML isoform II that is predicted as a protein interaction site by bioinformatic analysis. J Gen Virol 9095–104. 10.1099/vir.0.005512-0 [DOI] [PubMed] [Google Scholar]
- Lethbridge K. J., Scott G. E., Leppard K. N.(2003). Nuclear matrix localization and SUMO-1 modification of adenovirus type 5 E1b 55K protein are controlled by E4 Orf6 protein. J Gen Virol 84259–268. 10.1099/vir.0.18820-0 [DOI] [PubMed] [Google Scholar]
- Lum L. S., Hsu S., Vaewhongs M., Wu B.(1992). The hsp70 gene CCAAT-binding factor mediates transcriptional activation by the adenovirus E1a protein. Mol Cell Biol 122599–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macejak D. G., Luftig R. B.(1991). Association of HSP70 with the adenovirus type 5 fiber protein in infected HEp-2 cells. Virology 180120–125. 10.1016/0042-6822(91)90015-4 [DOI] [PubMed] [Google Scholar]
- Madara J., Krewet J. A., Shah M.(2005). Heat shock protein 72 expression allows permissive replication of oncolytic adenovirus dl1520 (ONYX-015) in rat glioblastoma cells. Mol Cancer 412. 10.1186/1476-4598-4-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul G. G., Everett R. D.(1994). The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J Gen Virol 751223–1233. 10.1099/0022-1317-75-6-1223 [DOI] [PubMed] [Google Scholar]
- McFadden G., Mohamed M. R., Rahman M. M., Bartee E.(2009). Cytokine determinants of viral tropism. Nat Revs Immunol 9645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X. Z., Harken A. H.(2002). The interaction between Hsp70 and TNF-alpha expression: a novel mechanism for protection of the myocardium against post-injury depression. Shock 17345–353. 10.1097/00024382-200205000-00001 [DOI] [PubMed] [Google Scholar]
- Mitchell A. M., Hirsch M. L., Li C., Samulski R. J.(2014). Promyelocytic leukemia protein is a cell-intrinsic factor inhibiting parvovirus DNA replication. J Virol 88925–936. 10.1128/JVI.02922-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J., Grueneberg D. A., Yang X., Kim S. Y., Kloepfer A. M., Hinkle G., Piqani B., Eisenhaure T. M., Luo B., et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 1241283–1298. 10.1016/j.cell.2006.01.040 [DOI] [PubMed] [Google Scholar]
- Moore M., Schaack J., Baim S. B., Morimoto R. I., Shenk T.(1987). Induced heat shock mRNAs escape the nucleocytoplasmic transport block in adenovirus-infected HeLa cells. Mol Cell Biol 74505–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S. J., Leppard K. N.(2009). Adenovirus serotype 5 L4-22K and L4-33K proteins have distinct functions in regulating late gene expression. J Virol 833049–3058. 10.1128/JVI.02455-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S. J., Scott G. E., Leppard K. N.(2010). Adenovirus late-phase infection is controlled by a novel L4 promoter. J Virol 847096–7104. 10.1128/JVI.00107-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins J. R.(1982). Induction of the synthesis of a 70,000 dalton mammalian heat shock protein by the adenovirus E1A gene product. Cell 29913–919. 10.1016/0092-8674(82)90453-6 [DOI] [PubMed] [Google Scholar]
- Niewiarowska J., D'Halluin J. C., Belin M.-T.(1992). Adenovirus capsid proteins interact with HSP70 proteins after penetration in human or rodent cells. Exp Cell Res 201408–416. 10.1016/0014-4827(92)90290-O [DOI] [PubMed] [Google Scholar]
- Ou H. D., Kwiatkowski W., Deerinck T. J., Noske A., Blain K. Y., Land H. S., Soria C., Powers C. J., May A. P.(2012). A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell 151304–319. 10.1016/j.cell.2012.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl H. L., Sester M., Burgert H. G., Baeuerle P. A.(1996). Activation of transcription factor NF-kappaB by the adenovirus E3/19K protein requires its ER retention. J Cell Biol 132511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsalo V., Yondola M. A., Luan B., Shoshani I., Kisker C., Green D. F., Raleigh D. P., Hearing P.(2012). Biophysical and functional analyses suggest that adenovirus E4-ORF3 protein requires higher-order multimerization to function against promyelocytic leukemia protein nuclear bodies. J Biol Chem 28722573–22583. 10.1074/jbc.M112.344234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips B., Abravaya K., Morimoto R. I.(1991). Analysis of the specificity and mechanism of transcriptional activation of the human hsp70 gene during infection by DNA viruses. J Virol 655680–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich N. C., Sarnow P., Duprey E., Levine A. J.(1983). Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 128480–484. 10.1016/0042-6822(83)90274-X [DOI] [PubMed] [Google Scholar]
- Reich N., Pine R., Levy D., Darnell J. E.(1988). Transcription of interferon-stimulated genes is induced by adenovirus particles but is suppressed by E1A gene products. J Virol 62114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro M. G., Amici C., Rossi A.(2010). Role of heat shock proteins in viral infection. Prokaryotic and Eukaryotic Heat Shock Proteins in Infectious Disease 51–84. Edited by Pockley A. G., Calderwood S. K., Santoro M. G.New York: Springer. [Google Scholar]
- Saphire A. C., Guan T., Schirmer E. C., Nemerow G. R., Gerace L.(2000). Nuclear import of adenovirus DNA in vitro involves the nuclear protein import pathway and hsc70. J Biol Chem 275. [DOI] [PubMed] [Google Scholar]
- Sarnow P., Sullivan C. A., Levine A. J.(1982). A monoclonal antibody detecting the adenovirus type 5-E1b-58Kd tumor antigen: characterization of the E1b-58Kd tumor antigen in adenovirus-infected and -transformed cells. Virology 120510–517. 10.1016/0042-6822(82)90054-X [DOI] [PubMed] [Google Scholar]
- Schmitz M. L., Indorf A., Limbourg F. P., Städtler H., Traenckner E. B., Baeuerle P. A.(1996). The dual effect of adenovirus type 5 E1A 13S protein on NF-kappaB activation is antagonized by E1B 19K. Mol Cell Biol 164052–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner S., Bürck C., Glass M., Groitl P., Wimmer P., Kinkley S., Mund A., Everett R. D., Dobner T.(2013). Control of human adenovirus type 5 gene expression by cellular Daxx/ATRX chromatin-associated complexes. Nucleic Acids Res 413532–3550. 10.1093/nar/gkt064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M. C., Fisch T. M., Benecke B. J., Nevins J. R., Heintz N.(1988). Definition of multiple, functionally distinct TATA elements, one of which is a target in the hsp70 promoter for E1A regulation. Cell 52723–729. 10.1016/0092-8674(88)90410-2 [DOI] [PubMed] [Google Scholar]
- Singh I. S., Shah N. G., Almutairy E., Hasday J. D.(2010). Role of HSF1 in infectious disease. Prokaryotic and Eukaryotic Heat Shock Proteins in Infectious Disease 1–31. Edited by Pockley A. G., Calderwood S. K., Santoro M. G.New York: Springer. [Google Scholar]
- Sohn S. Y., Hearing P.(2011). Adenovirus sequesters phosphorylated STAT1 at viral replication centers and inhibits STAT dephosphorylation. J Virol 857555–7562. 10.1128/JVI.00513-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M., Chelbi-Alix M. K., Koken M. H., Venturini L., Lee C., Saïb A., Quignon F., Pelicano L., Guillemin M. C., Schindler C.(1995). Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene 112565–2573. [PubMed] [Google Scholar]
- Tanaka K., Namba T., Arai Y., Fujimoto M., Adachi H., Sobue G., Takeuchi K., Nakai A., Mizushima T.(2007). Genetic evidence for a protective role for heat shock factor 1 and heat shock protein 70 against colitis. J Biol Chem 28223240–23252. 10.1074/jbc.M704081200 [DOI] [PubMed] [Google Scholar]
- Tobe M., Isobe Y., Tomizawa H., Nagasaki T., Takahashi H., Hayashi H.(2003). A novel structural class of potent inhibitors of NF-kappa B activation: structure-activity relationships and biological effects of 6-aminoquinazoline derivatives. Bioorg Med Chem 113869–3878. 10.1016/S0968-0896(03)00438-3 [DOI] [PubMed] [Google Scholar]
- Ullman A. J., Hearing P.(2008). Cellular proteins PML and Daxx mediate an innate antiviral defense antagonized by the adenovirus E4 ORF3 protein. J Virol 827325–7335. 10.1128/JVI.00723-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman A. J., Reich N. C., Hearing P.(2007). Adenovirus E4 ORF3 protein inhibits the interferon-mediated antiviral response. J Virol 814744–4752. 10.1128/JVI.02385-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme E., Laukens K., Dang T. H., Van Ostade X.(2010). A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int J Biol Sci 651–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidtkamp-Peters S., Lenser T., Negorev D., Gerstner N., Hofmann T. G., Schwanitz G., Hoischen C., Maul G., Dittrich P., Hemmerich P.(2008). Dynamics of component exchange at PML nuclear bodies. J Cell Sci 1212731–2743. 10.1242/jcs.031922 [DOI] [PubMed] [Google Scholar]
- White E., Spector D., Welch W.(1988). Differential distribution of the adenovirus E1A proteins and colocalization of E1A with the 70-kilodalton cellular heat shock protein in infected cells. J Virol 624153–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J., Atwan Z., Morris S. J., Leppard K. N.(2015). The human adenovirus type 5 L4 promoter is negatively regulated by TFII-I and L4-33K. J Virol 897053–7063. 10.1128/JVI.00683-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B. J., Hurst H. C., Jones N. C., Morimoto R. I.(1986). The E1A 13S product of adenovirus 5 activates transcription of the cellular human HSP70 gene. Mol Cell Biol 62994–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. X., Zou W. X., Lin P., Chang K. S.(2005). A role for PML3 in centrosome duplication and genome stability. Mol Cell 17721–732. 10.1016/j.molcel.2005.02.014 [DOI] [PubMed] [Google Scholar]
- Zhao H. X., Granberg F., Elfineh L., Pettersson U., Svensson C.(2003). Strategic attack on host cell gene expression during adenovirus infection. J Virol 7711006–11015. 10.1128/JVI.77.20.11006-11015.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Huang X., Yang Y.(2007). Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J Virol 813170–3180. 10.1128/JVI.02192-06 [DOI] [PMC free article] [PubMed] [Google Scholar]