

Abstract
The control of insulin release from pancreatic beta cells helps ensure proper blood glucose levels, which is critical for human health. Protein kinase C has been shown to be one key control mechanism for this process. After glucose stimulation, calcium influx into beta cells triggers exocytosis of insulin-containing dense-core granules and activates protein kinase C via calcium-dependent phospholipase C-mediated generation of diacylglycerol. Activated protein kinase C potentiates insulin release by enhancing the calcium sensitivity of exocytosis, likely by affecting two main pathways that could be linked: 1) the reorganization of the cortical actin network, and 2) the direct phosphorylation of critical exocytotic proteins such as munc18, SNAP25, and synaptotagmin. Here, we review what is currently known about the molecular mechanisms of protein kinase C action on each of these pathways and how these effects relate to the control of insulin release by exocytosis. We identify remaining challenges in the field and suggest how these challenges might be addressed to advance our understanding of the regulation of insulin release in health and disease.
Graphical Abstract
Exocytosis and insulin release
Mammalian physiology demands exquisite control over insulin release and blood glucose levels. The debilitating and potentially fatal consequences of dysfunction in the insulin release system are seen in diseases such as diabetes. Careful physiological regulation would be impossible if an endocrine cell’s entire store of insulin was released upon an initial stimulus, rendering the cell unable to respond to subsequent challenges. Instead, there are multiple levels of control to ensure a sustained and balanced release of insulin from beta cells of the pancreas. This review focuses on protein phosphorylation by protein kinase C (PKC), one primary regulatory mechanism which acts at multiple steps in the complex signaling cascade controlling insulin secretion.
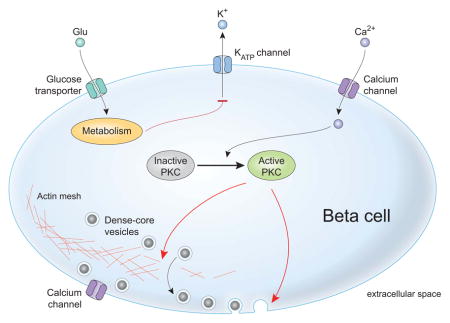
Over fifty years of study of pancreatic beta cells have led to a comprehensive understanding of how glucose initiates the signaling cascade that leads to insulin secretion [1–4] (Figure 1A). At a simplified level, when blood glucose is elevated, transporters move glucose into beta cells of the pancreatic islets. As these sugars are metabolized, the cytoplasmic ratio of ATP/ADP increases, closing ATP-sensitive potassium channels (K-ATP channels) and blocking potassium efflux from the cell [5]. The resulting depolarization of the cell’s plasma membrane opens voltage-gated calcium channels, allowing extracellular calcium to enter the cytoplasm (Figure 1A). Like many regulated secretory systems, elevated intracellular calcium is the ultimate trigger for exocytosis, stimulating the fusion of insulin-containing dense-core granules with the plasma membrane and the release of their luminal contents into the blood [6]. With glucose stimulation, beta cells exhibit a biphasic pattern of insulin release composed of an intense and transient first phase followed by a second sustained phase that builds slowly over time [2, 5]. The pathway described above is active in both phases of biphasic secretion and oscillatory changes in calcium underlie the two-component behavior. In addition to electrogenic effects, glucose metabolism potentiates release in pathways that act downstream of calcium influx. Additionally, other pathways, such as receptor-mediated cAMP elevation contribute to the control of calcium signaling and may directly activate protein kinases [7]. These other pathways integrate extracellular signals from neurotransmitters or hormones and provide additional layers of control over insulin secretion often via cAMP-dependent pathways [7, 8]. In the end, all these signals funnel into the same conserved molecular machinery that drives the fusion of insulin-loaded granules with the plasma membrane.
Figure 1.

The cellular and molecular pathways of glucose-stimulated insulin secretion. A) Glucose enters the cell and is metabolized, which alters the ATP/ADP ratio and closes K-ATP channels. The cell is depolarized which causes calcium influx via voltage-gated calcium channels and activation of PKC. PKC activation is shown schematically here but depends on calcium-mediated activation of phospholipase C, which produces the regulatory lipid diacylglycerol that acts on PKC. One major global function of PKC is to mediate reorganization of the actin cortex (shown bottom center of cell), which allows DCVs to move to the plasma membrane. B) The major molecular components of the exocytotic machinery are shown in schematic form in each stage of the exocytotic cascade. The principle exocytotic protein targets of PKC are highlighted in red in frame 1. 1) DCV translocation to the plasma membrane. 2) DCV docking and priming involves trans-SNARE complex formation along with co-factors like munc13, munc18, and synaptotagmin. 3) DCV fusion occurs upon calcium influx allowing insulin release. 4) Recycling can occur in kiss-and-run or cavicapture exocytosis where dynamin and other canonical endocytotic proteins close the fusion pore to recapture DCV membrane material. This can occur without full cargo release to modulate the amount of cargo released per exocytotic event.
In beta cells the fusion of exocytotic vesicles is driven by a macromolecular complex composed of over a dozen proteins and lipids [6] (Figure 1B). This machinery is broadly conserved in regulated exocytosis from yeast to humans and across diverse secretory systems that are found in endocrine, neuroendocrine, neuronal, cardiac, reproductive and other tissues. The core components are the SNARE proteins: syntaxin, SNAP25, and VAMP [6]. These three proteins are embedded in opposing vesicular (VAMP) and plasma membranes (syntaxin and SNAP25) and interact to form a four-helical bundle when secretory vesicles dock at the plasma membrane. When exocytosis is initiated, the helical bundle zippers completely, which produces mechanical force that pulls the two membranes into close apposition. In vitro, the SNAREs are sufficient to drive membrane fusion [9, 10]. However, in cells additional proteins control the distinct steps in a secretory vesicle’s lifecycle: 1) translocation to and docking with the plasma membrane, 2) priming that activates the vesicle to fuse, and 3) fusion pore formation (Figure 1B). For example, secretory vesicles are coated in small Rab GTPases such as Rab27a and Rab3a, and their effectors, including rabphilin and granuphilin [11]. These proteins play key roles in vesicle trafficking and docking. Priming is believed to involve partial assembly of the SNARE proteins mediated by several modulators including munc13, munc18, CAPS, tomosyn and others [6]. Finally, membrane fusion needs the primary calcium sensor for exocytosis, synaptotagmin, a vesicular membrane protein, which binds calcium and helps to catalyze membrane fusion through still-controversial mechanisms [12–14]. Together these molecules compose the macromolecular assembly that mediates vesicle fusion in a carefully regulated manner. One pathway for regulation of this process is PKC [15].
PKC potentiates insulin release in beta cells; however, it is currently not clear how this is mechanistically accomplished in living cells [15]. First, we discuss PKC structure, regulation, and activation in beta cells. Then, we address two fundamental questions: 1) what is the specific effect of PKC on insulin release, and 2) how does PKC achieve these effects? In the context of these questions, we discuss the current models for PKC action on glucose-stimulated insulin release and further examine three exocytotic proteins that are likely targets of PKC and control vesicle fusion at the plasma membrane.
PKC structure and activation
PKC isozymes are ubiquitous serine-threonine kinases with importance to human health and physiology, as evidenced by their role in many diseases including heart disease, cancer, and diabetes [16]. PKC isozymes are highly regulated; the conventional isozymes require the regulatory lipid diacylglycerol (DAG), negatively charged phospholipids phosphatidylserine and phosphatidylinositide-4,5-bisphosphate (PIP2), and calcium [17]. PKC activity was initially identified in pancreatic islets by Tanigawa et al., and this activity arises from expression of many PKC isoforms in beta cells, including α, βII, ε, λ, and ζ and possibly βI, θ, and η [18, 19].
In beta cells, PKC is activated during glucose stimulated insulin secretion (GSIS) when DAG is produced during calcium influx [20]. DAG production occurs when glucose-induced calcium influx first activates phospholipase C (PLC), which hydrolyzes some of the PIP2 in the membrane to yield DAG and inositol triphosphate (IP3) [21]. In addition to calcium, many PLC isoforms are activated via G-protein coupled receptor pathways, some of which contribute to the regulation of insulin secretion and secretion amplification [22, 23]. Glucose alone can induce both DAG production in islets and subsequent PKC translocation to the plasma membrane, a hallmark of enzyme activation [20, 24–26]. These findings mechanistically link PKC activation to the physiological stimulus that activates beta cells.
There are eight PKC isozymes divided into three classes based on the molecules that activate them [17]. These are 1) conventional PKCs, activated by calcium and DAG; 2) novel PKCs, activated by DAG alone; and 3) the atypical PKCs, activated by neither calcium nor DAG [17]. At the structural level, PKCs have two common domains: the kinase domain, composed of an ATP dependent active site and region for binding the substrate consensus sequence, and the regulatory domain, composed of conserved C1 (binds DAG molecules) and C2 (binds PIP2 via calcium-dependent mechanism) and pseudo-substrate domains [17] (Figure 2A). In its inactive state, the regulatory domain auto-inactivates the kinase domain by positioning the pseudo-substrate sequence in the active site of the kinase domain (Figure 2C-1). Recent structural work on PKC-β suggests a second mechanism of allosteric regulation whereby the C1 domains clamp a critical helix in the kinase domain, which must be released for kinase activation [27, 28] (Figure 2B). In the presence of calcium, the C2 domain binds PIP2 and PS headgroups, tethering the enzyme to the plasma membrane (Figure 2C-2). After membrane association, the C1 domains can interact with DAG. DAG binding then activates the kinase by inducing a conformational change which removes the pseudo-substrate from the active site and releases the C1 clamp. (Figure 2C-3) For conventional PKC, tripartite regulatory interactions provide spatiotemporal control over enzyme activity in the cell by restricting PKC activation to only organelles and regions that simultaneously contain all three of these regulatory modules.
Figure 2.

The structure and activation of conventional PKC. A) The domain organization of conventional PKCs is roughly divided into the kinase domain (chartreuse) and the regulatory domain containing C1 (blue) and C2 (magenta) domains along with the pseudo-substrate domain (light blue). B) The crystal structure of PKCβ-II in its auto-inhibited form, with the C1B domain making contacts with the kinase domain (the C1 clamp). The PS and C1A domains and several linker regions were not visible in the structure (PDB: 3PFQ). The authors note that the extended arrangement of the C2 domain distal to the kinase domain is likely to be an artifact of crystal packing [27]. C) The activation of conventional PKC by calcium and lipids. 1) Cytosolic PKC is auto-inhibited by pseudo-substrate domain binding to the active site of the kinase along with the C1 clamp engaged with a critical helix on the kinase. 2) Calcium binds the C2 domain which mediates its interaction with PS and PIP2 lipids (red) in the membrane. 3) After translocation to the membrane, the C1 domains bind DAG (orange) which releases the C1 clamp and fully disengages the pseudo-substrate from the active site, thereby activating the enzyme.
What is the effect of PKC on insulin release?
For close to 40 years PKC has been known to potentiate insulin release from beta cells [29, 30]. The foundational work on PKC’s effects on exocytosis relied on the application of phorbol esters (phorbol 12-myristate 13-acetate (PMA or also abbreviated TPA)). Phorbol esters act as analogs to the lipid DAG that, together with calcium, activate conventional PKCs [31–33]. Consequently, enhanced insulin release upon PMA treatment was taken as evidence that PKC stimulates insulin secretion [34–37]. The use of PKC inhibitors supported this conclusion [38–41]. Additionally, in several cell types, the application of exogenous PKC to permeabilized cells enhanced exocytosis, providing further evidence for a direct effect of the enzyme on secretion [42, 43]. PKC activation and translocation to the membrane are linked, and thus in addition to correlating enhanced secretion with PMA, the necessary translocation of PKC to the plasma membrane upon PMA treatment has also been observed [24, 25, 38, 44–49]. These experiments first took the form of enzymatic assays of PKC activity that was associated with the plasma membrane [38]. Further biochemical and imaging studies showed that PKC isozymes α, β, ε, and θ all translocate to the plasma membrane or insulin granules upon activation with several compounds (glucose, PMA, or potassium-driven depolarization) [24, 25, 44–49]. Additionally, there is evidence that atypical PKC isozymes may play a role in secretion [50]. Overall, there is strong consensus that PKC activation enhances insulin release.
PKC does not, however, directly stimulate secretion by initiating calcium influx into the cell [51]. Work from multiple groups measuring calcium currents has shown that PMA alone or coupled with glucose does not modulate intracellular calcium influx [34, 51–53]. Instead of contributing to calcium influx, the application of PMA shifts the calcium sensitivity of exocytosis to lower calcium concentrations. This was originally shown in chromaffin cells [54, 55] and then in beta cells [56–59] using a variety of cracked or patched cell systems where the calcium concentration can be clamped. In pioneering work from Zawalich et al. (1983), it was shown that the combination of a calcium ionophore and PMA could reproduce the classic biphasic insulin release pattern [36]. Calcium ionophores produced the initial spike in insulin release, and PMA treatment supported the second phase of insulin release in the continued presence of the ionophore. Indeed, PKC activation alone is not required for the first phase of insulin secretion after glucose stimulus [60, 61]. Together, these early studies suggested PKC potentiates insulin released in response to glucose stimulation, but PKC itself cannot initiate release [62]. PKC likely increases the calcium sensitivity of the exocytotic machinery, thereby potentiating the overall amount of insulin secreted in response to a given calcium stimulus.
Aside from studies in model systems of insulin secretion, there is evidence that PKC dysfunction could play a role in diabetes. Aberrant PKC activity, perhaps through misregulation of PKC isozymes, might underlie the diabetic phenotype in Goto-Kakizaki rats, a common animal model for type 2 diabetes [63–65]. Furthermore, PKC-ε knockout in a mouse model prevented glucose intolerance and inhibition of PKC-ε increased insulin secretion in diabetic mice, suggesting a role of this isozyme in disease [66]. In new work, the immediately-releasable pool of vesicles, composed of vesicles tightly associated with calcium channels, was observed to be absent in insulin-secreting cells from human donors with type II diabetes [67]. As discussed below, interaction between L-type calcium channels and insulin granules may be modulated by PKC.
A word of caution before we consider how PKC enhances insulin release. Although a useful pharmacologic tool, the broad activity of phorbol esters likely has contributed to significant confusion in the field. It was noted at least as early as 1985 by Tamagawa et al. that other targets of PMA could be contributing to the drug’s effects on insulin release [59, 68]. For example, munc13, an essential factor in vesicle priming and exocytosis, is a phorbol ester receptor [69, 70]. Studies have identified munc13 as being crucial for the second phase of insulin release, the phase in which PKC was originally postulated to act [36, 71, 72]. Though more recent work in neurons has developed an integrated model in which both PKC-dependent (possibly via munc18) and PKC-independent (possibly via munc13) pathways are necessary for exocytosis [73, 74], the wide-ranging targets of PMA make deconvoluting the specific effects of PKC challenging.
How does PKC globally enhance insulin release?
Consistent with its ubiquitous activity, PKC likely acts on many targets to regulate insulin release. Here, we distinguish between global cell-wide PKC effects (Figure 1A) and local effects on protein targets directly involved in exocytosis of dense core vesicles (Figure 1B). First, we will cover PKC modulation at global sites:
PKC could regulate proteins that control the structure and dynamics of the cortical actin network and thus allow more insulin granules to dock at the plasma membrane.
PKC’s effect on calcium channels has been controversial, but a direct means to regulate insulin granule exocytosis could be to directly modulate calcium channel activity or location.
PKC helps to control many global cellular processes including transcription and cell metabolism. Because insulin secretion is linked to cellular ATP levels, effects on cell metabolism could indirectly affect exocytosis.
One global effect of PKC activation is actin rearrangements that allow vesicles to move towards the plasma membrane. Many secretory cells have a ~100 nm-thick dense cortical network of actin near the plasma membrane that is thought to act as a physical barrier to vesicles [75]. PKC-induced reorganization of this actin layer has been observed in multiple systems: beta cells, chromaffin cells, neuronal systems, and others [47, 53, 76–79]. Rearrangement of the actin cortex has been interpreted as having two effects. First vesicle pools near the plasma membrane are larger (in the case of pre-stimulus PKC activation), which accounts for increased initial rates of exocytosis from the readily-releasable pool (RRP) [53, 80]. The second effect is to enhance RRP refilling after initial stimulation to potentiate sustained release or cell adaptation [53, 81]. The sustained phase of insulin release must include the recruitment of additional insulin granules from the reserve pool to the plasma membrane, consequently it seems likely PKC-mediated cortical actin rearrangement could play a prominent role in potentiating sustained insulin release.
One PKC target that controls cortical actin structure is a well-known actin crosslinking protein, myristoylated alanine-rich C-kinase substrate (MARCKS). MARCKS cross-links actin and binds PIP2, which is regulated by PKC phosphorylation [82]. Activation of PKC releases MARCKS from the plasma membrane and has been directly correlated with cortical actin disassembly in chromaffin cells [47, 76]. In insulin-secreting cells, the translocation of PKC to the plasma membrane and release of MARCKS has been directly visualized [46]. These studies demonstrate that a global effect of PKC activation is the re-organization of the cortical actin cytoskeleton through loss of MARCKS from the plasma membrane.
In addition to MARCKS, several other proteins directly modified by PKC are likely playing a role in actin rearrangement and vesicle movement. Vesicle-localized myosin may be regulated via PKC, perhaps via myosin light chain kinase (MLCK), which is a PKC target [83]. 14-3-3 protein was originally identified as playing a PKC-dependent role in exocytosis as well [42, 84]. Subsequent work has shown 14-3-3 likely acts as a scaffolding factor between vesicle-bound Rab-effector proteins and other molecules, such as myosin, however, it may be dependent on other kinase systems [85, 86].
Another possible target of PKC is the plasma membrane calcium channels necessary for insulin release. Modulation of these channels could provide a direct mechanism to increase the calcium response to glucose metabolism and consequently increase the exocytotic response. Beta cells have L-type, R-type, and P/Q-type calcium channels [87]. L-type channels are the primary drivers, though are not required, for GSIS [87–90]. Early reports indicated PKC activation could cause calcium influx and inhibition of PKC could block exocytosis [33, 91–93], however, these effects may have been caused by intracellular calcium stores [93, 94]. The current weight of evidence shows no change of intracellular calcium levels, or indeed inhibition of calcium currents, upon PKC activation leading to the conclusion that PKC does not directly modulate calcium levels in the cell [34, 51–53, 59, 95]. Additional confusion likely also arose due to phorbol ester PKC-independent effects on channel behavior [96, 97]. PKC’s primary effect is to enhance the calcium sensitivity of exocytosis rather than to amplify or modulate calcium currents via channel activity.
To the extent that PKC may influence calcium channels, it could regulate interactions between calcium channels and binding partners, which could affect exocytosis without causing large changes in calcium currents. Yokohama et al. demonstrated that Cav2.2 channels are direct PKC targets, and that phosphorylation modulates interaction of syntaxin with the channel [98]. Indeed, deletion of the syntaxin interaction site on these channels or expression of a peptide competitor for the interaction site blocks exocytosis, pointing to an important role for PKC’s modulation of this interaction [99, 100]. If syntaxin interaction with Cav2.2 primarily serves to localize the channel to the exocytotic machinery [101], rather than modulating channel activity, this may reconcile the findings that PKC does not affect calcium channel currents but that channel modification by PKC is an important regulatory mechanism. Proximity to calcium channels would provide syntaxin and the exocytotic machinery with high local calcium concentration, which is hypothesized to be a mechanism for generating the immediately-releasable pool of exocytotic vesicles [102]. Importantly, it was recently found that this pool of vesicles is absent in cells from human donors with type II diabetes [67], which strongly suggests that modulation of the L-type channel-syntaxin interaction by PKC could be critically important in diabetes.
Finally, PKC can readily affect cell metabolism, which has an indirect impact on insulin secretion [103]. Several studies have shown some PKC isoforms target transcription factors [24, 104, 105]. These factors regulate the expression of metabolic genes, such as hexokinase, and consequently influence calcium currents and insulin release along the standard GSIS pathway by affecting ATP levels in the cell [22]. For example, generation of a PKC-ε knockout-mouse showed that this specific PKC likely plays a role in regulating metabolic pathways in beta cells [66]. Insulin protein synthesis is not necessary for continued insulin secretion from islets for several hours [2, 4], so it is unlikely that any PKC effects on protein synthesis would be relevant to insulin release over this timescale. Overall, the global effects of PKC regulation on cell metabolism must be very complex and more work is needed to understand PKC’s contribution to cellular ATP levels.
What are the local exocytotic protein targets of PKC?
A direct method for PKC to potentiate insulin release would be to phosphorylate and activate components of the exocytotic machinery. Bulk measurements show that potentiation occurs by enhancement of the calcium sensitivity of exocytosis [56–59]. At a basic level this results in more insulin released per calcium stimulus. Several possible pathways could work to achieve this:
PKC could directly alter the ability of single vesicles to fuse by modifying proteins involved in vesicle priming and fusion, thus increasing the total number of exocytotic events per cell [15, 78].
PKC could enhance vesicle docking, by modifying proteins involved in docking interactions, to increase the size of the docked vesicle pool. More docked vesicles with the same probability of fusion per vesicle could produce an enhancement of total insulin release [52].
PKC could modulate the amount of insulin cargo released from single vesicles in kiss-and-run type exocytotic events [106]. Far less work has been done to directly study this phenomenon, and kiss-and-run exocytosis appears to be a minor pathway in beta cells [107].
Here, we discuss several exocytotic protein targets that could mediate these possible mechanisms.
Many exocytotic proteins are targets of post-translational modification by a host of kinases [15, 108–110]. In vitro, PKC phosphorylates syntaxin [108, 110]. Also, PKC can phosphorylate VAMP [108, 111], though other kinases are likely to be more relevant [15]. NSF is a PKC target and phosphorylation appears to reduce its affinity for SNARE complexes [112]. There is some evidence in neurons that NSF and PKC-ε control trafficking of receptors to the cell surface [113]. However, although many of these proteins can be phosphorylated in vitro, there remains little direct evidence for these phosphorylation events playing a dominant role in insulin secretion. In contrast, the most well studied PKC targets in the exocytotic machinery are munc18, synaptotagmin, and SNAP25 [15].
Munc18 is a syntaxin-binding protein that plays a critical docking and priming role in exocytosis [114] (Figure 1B1-3). Munc18 binds to the closed form of syntaxin, but also interacts with the ternary SNARE complex via a binding site on syntaxin’s N-terminus [114]. Munc18 is phosphorylated by PKC at residues Ser306 and Ser313 [115, 116]. In vivo munc18 phosphorylation has been observed in many cell systems: islets, chromaffin cells, neurons, and others [117–122]. In neuronal synapses, munc18 phosphorylation is kept low by phosphatases, such that upon PKC activation a large dynamic change in phosphorylation state can be achieved [118, 123]. Munc18 appears to be important in RRP refilling, which could be attributed to a syntaxin trafficking role or to an increase in SNARE ternary complex assembly [119, 120]. Overall there is good evidence that munc18 is regulated by PKC in vivo and that the protein’s phosphorylation state is dynamically controlled, suggesting it could be an important regulatory module.
How does munc18 phosphorylation relate to insulin release? A longstanding hypothesis is that phosphorylation of munc18 causes opening of a closed form of syntaxin and allows for more vesicles to dock to the plasma membrane [115]. This would increase the number of calcium-sensitive vesicles at the plasma membrane. Another hypothesis comes from work by Mandic et al. who looked more closely at munc18-1 and munc18-2, two isoforms expressed in beta cells, and their function in insulin secretion [124]. Interestingly, these authors show that munc18-1 and munc18-2 support exocytosis with distinct kinetics and that the two isoforms affect the calcium sensitivity of vesicles at the membrane. Phosphorylation of both is important for their function. Differential phosphorylation and activation of these isoforms could be the molecular correlate of the PKC-dependent calcium sensitivity changes of exocytosis in beta cells. Ultimately, the mechanistic details of munc18 phosphorylation with respect to insulin release remains unclear.
Synaptotagmin is the primary calcium sensor of exocytosis, making it an ideal candidate for PKC-mediated calcium sensitization of the exocytotic apparatus [125] (Figure 1B3). However, phosphorylation of synaptotagmin does not appear to directly affect its calcium affinity. The protein is phosphorylated by PKC at Thr112, and phosphorylation levels increase when PKC is activated by PMA [126]. There are many synaptotagmin isoforms, with Syt-1 and Syt-7 playing major roles in neuronal and endocrine secretion, respectively [125]. Synaptotagmin isoforms have different affinities for calcium, suggesting that they could mediate exocytosis of distinct vesicle pools in vivo [125]. Sorting of Syt-1 and Syt-7 onto distinct vesicle pools occurs in chromaffin cells and affects exocytotic behavior [127].
Evidence suggests that phosphorylation increases synaptotagmin affinity for the SNARE complex [15, 128]. It is, however, unclear how this might affect release. Synaptotagmin has been identified as part of the minimal vesicle docking machinery in chromaffin cells [129]. Thus, phosphorylation of synaptotagmin could modulate docking and because vesicle fusogenicity is heterogeneous, docking of different vesicle populations could lead to changes in apparent calcium sensitivity of exocytosis. For example, phosphorylation of synaptotagmin-7 by PKC would enhance its affinity for the SNARE complex and consequently enhance docking of synaptotagmin-7-labeled vesicles with the plasma membrane. This could lead to a change in the apparent calcium sensitivity of exocytosis, as synaptotagmin-7 has a higher calcium affinity than synaptotagmin-1 [127]. Consistent with a scenario under which different synaptotagmin isoforms are relevant PKC targets in different cellular systems, Nagy et al. showed in chromaffin cells that overexpression of synaptotagmin-1 phosphomutants and phosphomimetics did not appear to alter exocytosis, whereas in another system, hippocampal neurons, phosphomimetics of synaptotagmin-1 regulate exocytosis [73, 130]. Synaptotagmin isoform-specific phosphorylation has yet to be demonstrated, but it represents an exciting hypothesis for the molecular mechanisms underlying the sensitization of the exocytotic machinery by PKC.
Finally, SNAP-25 appears to be a PKC substrate in vivo, though the role of this modification in insulin secretion is still controversial. The most compelling evidence for physiologically relevant phosphorylation-dependent control of SNAP-25 is that knock-in mice expressing a SNAP-25 mutant that cannot be phosphorylated display neurological defects including anxiety and epilepsy [131, 132]. SNAP-25 phosphorylation was first observed in vitro and was quickly shown to occur in response to PMA treatment in PC12 cells, correlating with enhanced exocytosis [109, 133]. The primary site for PKC-mediated phosphorylation of SNAP-25 is Ser187 [133]. Further studies in insulin-secreting, neuroendocrine, and neuronal systems have been at odds over whether PKC-mediated phosphorylation of SNAP25 directly affects or is only correlated with exocytosis [134–139]. It is currently thought that phosphorylation of SNAP-25 increases its interaction with syntaxin and that this increases the population of the highly-calcium sensitive pool of vesicles [139, 140]. More recent work suggests that endogenous SNAP-25 may have complicated earlier studies on phosphomimetic SNAP-25 mutants [138, 139]. Despite phenotypic effects in neurons of model organisms, SNAP-25’s physiological role in insulin secretion remains controversial.
In addition to the exocytotic proteins discussed above, PKC could affect insulin release by modulating the amount of cargo released at single vesicles (Figure 1B3-4). More complete release of insulin from single vesicles could enhance the overall amount of insulin released without changing the total number of fusion events per cell. Early evidence for a role for PKC modulating fusion pore behavior in neuromuscular junctions came from the observation that staurosporine, a PKC inhibitor, affected fusion pore behavior and vesicle collapse into the plasma membrane [141]. In neurons, PKC was shown to affect exocytosis after calcium binding to synaptotagmin, suggesting that fusion pore regulation could play a role [142]. In chromaffin cells, amperometry has been used to examine the kinetics of the fusion pore at very high time resolution. PKC activation appears to accelerate fusion pore expansion [117, 143, 144], however, some studies have also shown that PKC tends to decrease the average quantal size of fusion events [117, 143]. Quantal size could be linked to the probability of kiss-and-run exocytosis, which may be a function of the stimulation protocol, making it challenging to interpret these findings with respect to physiological glucose stimulation.
PKC’s effect on quantal size may involve regulation of endocytotic protein components responsible for mediating kiss-and-run exocytosis. There is a growing body of evidence to suggest that canonical endocytotic proteins—dynamin, amphiphysin, syndapin, and others—may play a role in modulating fusion pore expansion or collapse via a kiss-and-run mechanism [127, 145–149]. Dynamin and other endocytotic proteins are known to be modulated by PKC activity [150, 151]. By targeting these proteins, PKC could be modulating fusion pore dynamics or closure and thus affecting the amount of insulin release. This hypothesis has not been fully explored and future work with insulin granules are an ideal system to determine if kiss-and-run or sub-quantal cargo release could play an important role in physiological insulin levels.
PKC may also modulate fusion pore expansion and kinetics through its control of exocytotic protein factors. Barclay et al. suggest munc18 phosphorylation has a direct effect on fusion pore kinetics, though munc18 itself is unlikely to have direct effects on the membrane [117]. Instead munc18 could be either allowing more trans-SNARE complex assembly or enabling more efficient zippering of SNAREs [152, 153]. In addition to modulating the amount of SNARE complex formation, munc18 could directly affect syntaxin-mediated regulation of fusion pore diameter and behavior [154]. Munc18 also appears to help to regulate the selection of larger diameter vesicles for docking and fusion, and vesicle size directly contributes to fusion pore size [154]. Recent work has implicated synaptotagmin in fusion pore kinetic control [127], though it is not clear how its phosphorylation might affect this putative function. Obviously, lipids play a central role in fusion pore structure and dynamics [155], and Xue et al. invoke a lipid-mediated mechanism to account for PKC effects on fusion pore dynamics [144]. PKC activates phospholipase D, an enzyme which produces phosphatidic acid via the cleavage of phosphatidylcholine in the membrane. Phosphatidic acid has a small headgroup and high spontaneous negative curvature that would help to accommodate the high membrane curvatures at the nascent fusion pore. Overall, it seems possible PKC could exert an effect at the nascent fusion pore itself, though the mechanism of this remains unclear and requires further study, particularly with respect to insulin granules.
Bringing it all together
At the organismal level, activation of PKC enhances the amount of insulin that islets release into the blood upon glucose challenge. Potentiation of exocytosis occurs primarily in the second sustained phase of insulin release. PKC likely mediates this process by acting on two pathways that are connected: increasing the size of the RRP and sensitizing the exocytotic apparatus to calcium.
The transient first phase of GSIS corresponds to fusion and depletion of the RRP, so refilling this pool is necessary for the second, sustained phase of GSIS. RRP refilling requires the partial disassembly or re-organization of the cortical actin network, which allows more insulin granules to translocate to the plasma membrane. PKC may regulate this step via phosphorylation of MARCKS, an actin-crosslinking protein directly implicated in actin disassembly.
PKC also potentiates insulin release by enhancing the calcium sensitivity of the exocytotic machinery, but it remains unclear how this is achieved. No known modifications of exocytotic proteins directly increases their calcium affinity, which would be the most straightforward mechanism to enhance the calcium sensitivity of exocytosis. One mechanism to achieve calcium sensitization could involve isoform specific phosphorylation of munc18 or synaptotagmin. As reserve vesicles translocate to the plasma membrane, PKC may help to determine which vesicles successfully dock. Different isoforms of both munc18 and synaptotagmin display distinct calcium affinities that could be used to modulate exocytotic calcium sensitivity [124, 125]. Such a model is consistent with observations that PKC acts to increase the size of the highly calcium-sensitive vesicle pool [156–158]. Several studies have shown newcomer vesicles are more likely to fuse and are more calcium sensitive, which is consistent with this model [159, 160]. Another possible mechanism to enhance the calcium sensitivity of exocytosis is to increase the size of the RRP [52]. Phosphorylation of all three of the exocytotic proteins we discussed—munc18, synaptotagmin, and SNAP25—could enhance vesicle docking by either providing more docking sites or increasing the affinity of the docking machinery for vesicles. A larger pool of docked vesicles with a homogeneous calcium sensitivity for each vesicle could yield a larger exocytotic response to a given calcium stimulus [52].
It is possible that both PKC effects on cortical actin as well as specific exocytotic proteins could contribute to a unified mechanism of enhance insulin release. Cortical actin disassembly enables vesicle translocation from the reserve pool and phosphorylation of exocytotic proteins with known roles in vesicle docking would then ensure that newly arrived vesicles dock with the plasma membrane. Both mechanisms contribute to enlarging the docked, potentially active vesicle pool and thus giving rise to a larger insulin release upon a calcium stimulus.
Challenges and open questions
Despite ongoing characterization of PKC modifications to exocytotic proteins, we still lack a unified understanding linking these changes to PKC’s effect on bulk insulin release. This is due in part to the complexity of GSIS. Insulin secretion is several steps removed from the introduction of glucose. Each step is regulated, and PKC could act at several of them. Other regulatory enzymes act in concert with PKC. Additionally, PKC isozymes act in different signaling pathways and the isolation of their individual effects on exocytosis is challenging. Compounding the difficulties in sorting out these pathways, past tools for modulating the activity of PKC necessary to gain mechanistic insight into their roles in GSIS have been relatively non-specific.
Deepening our understanding of PKC’s role in GSIS will require harnessing recently developed techniques and developing new tools. Here we describe three challenges remaining in the field and offer suggestions on experimental approaches to address them.
Despite good evidence that munc18, synaptotagmin, and SNAP-25 are physiological PKC targets, we do not yet have a unified theory of their roles in specific cell types. In isolation, it appears that each protein plays a role in exocytotic regulation, but their combined effects have not been studied. This question is likely an arena where advances in quantitative proteomics coupled with crosslinking methods could provide clarity. With APEX2 or similar methods combined with phosphoproteomics, an organelle specific, quantitative examination of the modifications of munc18, synaptotagmin, and SNAP-25 could be performed from the same sample [161]. Another powerful technique to compare modifications of exocytotic proteins in living cells is the use of fluorescent biosensors for specific kinase activity on exocytotic proteins of interest [162]. The introduction of environmentally-sensitive fluorophores using mutagenesis on endogenous (transfection or CRISPR-based expression of labeling constructs of exocytotic proteins of interest) or exogenous (recombinant protein microinjected into cells) proteins could report on their phosphorylation state in real time in living cells after insulin secretion is stimulated.
In addition to the exocytotic proteins, we have described several cellular targets of PKC that are likely to be playing a role in GSIS potentiation. It is not currently clear if multiple PKC isozymes target these cellular locations, or if there is overlap in isozyme function. Specific pharmacologic activators and inhibitors of PKCs would first enable further unraveling of how individual isozymes might have specific roles to play in GSIS. To some extent, specific peptide-based inhibitors have been used in the past, and more isoform-specific activator and inhibitors would be illuminating. Additionally, though distinct isozyme translocation in time and space has been observed, it is not clear where exactly PKC is being localized in each case. Multiple sites-of-action—the actin cortex, vesicles, and the plasma membrane—are all found within the same diffraction limited space. Super-resolution fluorescence microscopy or electron microscopy could resolve isozyme localization between these sites.
Lastly, we suggest that different isoforms of synaptotagmin could be differentially regulated by PKC which would provide the cell a means to control the calcium sensitivity of exocytosis. It would be of great interest to investigate if these different vesicle populations are subject to PKC regulation and respond uniquely to PKC activation. Targeted phosphoproteomics and live cell imaging could be used to identify which isoforms of synaptotagmin reside on vesicles at the membrane and which vesicles are most fusogenic upon stimulation.
PKC plays an important role in modulating insulin secretion from pancreatic beta cells. Dysfunctional PKC signaling may be one mechanistic cause of diabetic phenotypes in model organisms for the disease, which underscores the importance of fully understanding PKC’s contribution to the complex regulation of insulin release. PKC appears to act in multiple steps of GSIS, importantly both at the cortical actin network and at the level of individual exocytotic proteins that play key roles in mediating membrane fusion. With advances in targeted phospho-proteomics and live-cell super resolution fluorescence microscopy, we can move towards a unified model for how PKC acts at multiple sites in vivo to potentiate insulin secretion. Future work on PKC could lead to novel therapies or drugs to target exocytosis in the hopes of curing diabetes and other endocrine diseases.
Highlights.
Activation of protein kinase C potentiates insulin secretion from beta cells.
Glucose stimulation produces calcium influx that activates conventional protein kinase C.
Protein kinase C regulates cortical actin rearrangement that facilitates insulin release.
Munc18, SNAP25, and synaptotagmin are exocytotic proteins likely regulated by protein kinase C.
Acknowledgments
We thank Ethan Tyler from the NIH Medical Arts Design Section for preparation of Figure 1. We thank Thaddeus Davenport and Agila Somasundaram for critical reading of the manuscript and all members of the Taraska lab for helpful discussions. JWT is supported by the Intramural Research Program of the National Heart Lung and Blood Institute, National Institutes of Health.
List of abbreviations
- PKC
Protein kinase C
- DAG
diacylglycerol
- PIP2
phosphatidylinositol-4,5-bisphosphate
- GSIS
glucose stimulated insulin secretion
- PLC
phospholipase C
- IP3
inositol triphosphate
- PMA
phorbol 12-myristate 13-acetate
- TPA
tetradecanoylphorbol acetate (synonym of PMA)
- RRP
readily-releaseable pool
- MARCKS
myristoylated alanine-rich C-kinase substrate
- MLCK
myosin light chain kinase
- DCV
dense-core vesicle
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Coore HG, Randle PJ. Regulation of insulin secretion studied with pieces of rabbit pancreas incubated in vitro. Biochem J. 1964;93(1):66–78. doi: 10.1042/bj0930066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curry DL, Bennett LL, Grodsky GM. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology. 1968;83(3):572–84. doi: 10.1210/endo-83-3-572. [DOI] [PubMed] [Google Scholar]
- 3.Dean PM, Matthews EK. Electrical activity in pancreatic islet cells. Nature. 1968;219(5152):389–90. doi: 10.1038/219389a0. [DOI] [PubMed] [Google Scholar]
- 4.Grodsky GM, et al. Effects of Carbohydrates on Secretion of Insulin from Isolated Rat Pancreas. Am J Physiol. 1963;205:638–44. doi: 10.1152/ajplegacy.1963.205.4.638. [DOI] [PubMed] [Google Scholar]
- 5.Rorsman P, et al. The Cell Physiology of Biphasic Insulin Secretion. News Physiol Sci. 2000;15:72–77. doi: 10.1152/physiologyonline.2000.15.2.72. [DOI] [PubMed] [Google Scholar]
- 6.Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490(7419):201–7. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furman B, Ong WK, Pyne NJ. Cyclic AMP signaling in pancreatic islets. Adv Exp Med Biol. 2010;654:281–304. doi: 10.1007/978-90-481-3271-3_13. [DOI] [PubMed] [Google Scholar]
- 8.Tang SH, Sharp GW. Atypical protein kinase C isozyme zeta mediates carbachol-stimulated insulin secretion in RINm5F cells. Diabetes. 1998;47(6):905–12. doi: 10.2337/diabetes.47.6.905. [DOI] [PubMed] [Google Scholar]
- 9.Kreutzberger AJ, et al. Assembly and Comparison of Plasma Membrane SNARE Acceptor Complexes. Biophys J. 2016;110(10):2147–50. doi: 10.1016/j.bpj.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pobbati AV, Stein A, Fasshauer D. N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science. 2006;313(5787):673–6. doi: 10.1126/science.1129486. [DOI] [PubMed] [Google Scholar]
- 11.Izumi T. Physiological roles of Rab27 effectors in regulated exocytosis. Endocr J. 2007;54(5):649–57. doi: 10.1507/endocrj.kr-78. [DOI] [PubMed] [Google Scholar]
- 12.Honigmann A, et al. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat Struct Mol Biol. 2013;20(6):679–86. doi: 10.1038/nsmb.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316(5828):1205–8. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, et al. Calcium sensitive ring-like oligomers formed by synaptotagmin. Proc Natl Acad Sci U S A. 2014;111(38):13966–71. doi: 10.1073/pnas.1415849111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snyder DA, Kelly ML, Woodbury DJ. SNARE complex regulation by phosphorylation. Cell Biochem Biophys. 2006;45(1):111–23. doi: 10.1385/CBB:45:1:111. [DOI] [PubMed] [Google Scholar]
- 16.Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov. 2012;11(12):937–57. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88(4):1341–78. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanigawa K, et al. Calcium-activated, phospholipid-dependent protein kinase in rat pancreas islets of langerhans. Its possible role in glucose-induced insulin release. FEBS Lett. 1982;138(2):183–6. doi: 10.1016/0014-5793(82)80436-5. [DOI] [PubMed] [Google Scholar]
- 19.Wuttke A, Yu Q, Tengholm A. Autocrine Signaling Underlies Fast Repetitive Plasma Membrane Translocation of Conventional and Novel Protein Kinase C Isoforms in beta Cells. J Biol Chem. 2016;291(29):14986–95. doi: 10.1074/jbc.M115.698456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zawalich WS, Zawalich KC. Regulation of insulin secretion by phospholipase C. Am J Physiol. 1996;271(3 Pt 1):E409–16. doi: 10.1152/ajpendo.1996.271.3.E409. [DOI] [PubMed] [Google Scholar]
- 21.Thore S, et al. Feedback activation of phospholipase C via intracellular mobilization and store-operated influx of Ca2+ in insulin-secreting beta-cells. J Cell Sci. 2005;118(Pt 19):4463–71. doi: 10.1242/jcs.02577. [DOI] [PubMed] [Google Scholar]
- 22.Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52(5):739–51. doi: 10.1007/s00125-009-1314-y. [DOI] [PubMed] [Google Scholar]
- 23.Wuttke A, Idevall-Hagren O, Tengholm A. P2Y(1) receptor-dependent diacylglycerol signaling microdomains in beta cells promote insulin secretion. FASEB J. 2013;27(4):1610–20. doi: 10.1096/fj.12-221499. [DOI] [PubMed] [Google Scholar]
- 24.Yedovitzky M, et al. Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic beta-cells. J Biol Chem. 1997;272(3):1417–20. doi: 10.1074/jbc.272.3.1417. [DOI] [PubMed] [Google Scholar]
- 25.Ganesan S, et al. Immunocytochemical localization of alpha-protein kinase C in rat pancreatic beta-cells during glucose-induced insulin secretion. J Cell Biol. 1992;119(2):313–24. doi: 10.1083/jcb.119.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biden TJ, et al. Ca2+-mediated generation of inositol 1,4,5-triphosphate and inositol 1,3,4,5-tetrakisphosphate in pancreatic islets. Studies with K+, glucose, and carbamylcholine. J Biol Chem. 1987;262(8):3567–71. [PubMed] [Google Scholar]
- 27.Leonard TA, et al. Crystal structure and allosteric activation of protein kinase C betaII. Cell. 2011;144(1):55–66. doi: 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lucic I, Truebestein L, Leonard TA. Novel Features of DAG-Activated PKC Isozymes Reveal a Conserved 3-D Architecture. J Mol Biol. 2016;428(1):121–41. doi: 10.1016/j.jmb.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 29.Malaisse WJ, et al. Insulinotropic effect of the tumor promoter 12-O-tetradecanoylphorbol-13-acetate in rat pancreatic islets. Cancer Res. 1980;40(10):3827–31. [PubMed] [Google Scholar]
- 30.Virji MA, Steffes MW, Estensen RD. Phorbol myristate acetate: effect of a tumor promoter on insulin release from isolated rat islets of Langerhans. Endocrinology. 1978;102(3):706–11. doi: 10.1210/endo-102-3-706. [DOI] [PubMed] [Google Scholar]
- 31.Castagna M, et al. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982;257(13):7847–51. [PubMed] [Google Scholar]
- 32.Takai Y, et al. Unsaturated diacylglycerol as a possible messenger for the activation of calcium-activated, phospholipid-dependent protein kinase system. Biochem Biophys Res Commun. 1979;91(4):1218–24. doi: 10.1016/0006-291x(79)91197-5. [DOI] [PubMed] [Google Scholar]
- 33.Petersen OH, Wollheim CB. Diacylglycerol as messenger. Nature. 1990;344(6264):300. doi: 10.1038/344300c0. [DOI] [PubMed] [Google Scholar]
- 34.Ammala C, et al. Activation of protein kinases and inhibition of protein phosphatases play a central role in the regulation of exocytosis in mouse pancreatic beta cells. Proc Natl Acad Sci U S A. 1994;91(10):4343–7. doi: 10.1073/pnas.91.10.4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arkhammar P, et al. Effects of protein kinase C activation on the regulation of the stimulus-secretion coupling in pancreatic beta-cells. Biochem J. 1989;264(1):207–15. doi: 10.1042/bj2640207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zawalich W, Brown C, Rasmussen H. Insulin secretion: combined effects of phorbol ester and A23187. Biochem Biophys Res Commun. 1983;117(2):448–55. doi: 10.1016/0006-291x(83)91221-4. [DOI] [PubMed] [Google Scholar]
- 37.Hutton JC, Peshavaria M, Brocklehurst KW. Phorbol ester stimulation of insulin release and secretory-granule protein phosphorylation in a transplantable rat insulinoma. Biochem J. 1984;224(2):483–90. doi: 10.1042/bj2240483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Easom RA, et al. Comparison of effects of phorbol esters and glucose on protein kinase C activation and insulin secretion in pancreatic islets. Biochem J. 1989;264(1):27–33. doi: 10.1042/bj2640027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terbush DR, Holz RW. Activation of protein kinase C is not required for exocytosis from bovine adrenal chromaffin cells. The effects of protein kinase C(19–31), Ca/CaM kinase II(291–317), and staurosporine. J Biol Chem. 1990;265(34):21179–84. [PubMed] [Google Scholar]
- 40.Hughes SJ, Ashcroft SJ. Effects of a phorbol ester and clomiphene on protein phosphorylation and insulin secretion in rat pancreatic islets. Biochem J. 1988;249(3):825–30. doi: 10.1042/bj2490825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zawalich WS, Zawalich KC. Effects of protein kinase C inhibitors on insulin secretory responses from rodent pancreatic islets. Mol Cell Endocrinol. 2001;177(1–2):95–105. doi: 10.1016/s0303-7207(01)00422-1. [DOI] [PubMed] [Google Scholar]
- 42.Morgan A, Burgoyne RD. Exo1 and Exo2 proteins stimulate calcium-dependent exocytosis in permeabilized adrenal chromaffin cells. Nature. 1992;355(6363):833–6. doi: 10.1038/355833a0. [DOI] [PubMed] [Google Scholar]
- 43.Naor Z, et al. Induction of exocytosis in permeabilized pituitary cells by alpha- and beta-type protein kinase C. Proc Natl Acad Sci U S A. 1989;86(12):4501–4. doi: 10.1073/pnas.86.12.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ganesan S, et al. Glucose-induced translocation of protein kinase C in rat pancreatic islets. Proc Natl Acad Sci U S A. 1990;87(24):9893–7. doi: 10.1073/pnas.87.24.9893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mendez CF, et al. Rapid association of protein kinase C-epsilon with insulin granules is essential for insulin exocytosis. J Biol Chem. 2003;278(45):44753–7. doi: 10.1074/jbc.M308664200. [DOI] [PubMed] [Google Scholar]
- 46.Mogami H, et al. Decoding of short-lived Ca2+ influx signals into long term substrate phosphorylation through activation of two distinct classes of protein kinase C. J Biol Chem. 2003;278(11):9896–904. doi: 10.1074/jbc.M210653200. [DOI] [PubMed] [Google Scholar]
- 47.Park YS, et al. Involvement of protein kinase C-epsilon in activity-dependent potentiation of large dense-core vesicle exocytosis in chromaffin cells. J Neurosci. 2006;26(35):8999–9005. doi: 10.1523/JNEUROSCI.2828-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H, et al. Bimodal role of conventional protein kinase C in insulin secretion from rat pancreatic beta cells. J Physiol. 2004;561(Pt 1):133–47. doi: 10.1113/jphysiol.2004.071241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinton P, et al. Dynamics of glucose-induced membrane recruitment of protein kinase C beta II in living pancreatic islet beta-cells. J Biol Chem. 2002;277(40):37702–10. doi: 10.1074/jbc.M204478200. [DOI] [PubMed] [Google Scholar]
- 50.Harris TE, Persaud SJ, Jones PM. Atypical isoforms of pKc and insulin secretion from pancreatic beta-cells: evidence using Go 6976 and Ro 31-8220 as Pkc inhibitors. Biochem Biophys Res Commun. 1996;227(3):672–6. doi: 10.1006/bbrc.1996.1567. [DOI] [PubMed] [Google Scholar]
- 51.Bozem M, Nenquin M, Henquin JC. The ionic, electrical, and secretory effects of protein kinase C activation in mouse pancreatic B-cells: studies with a phorbol ester. Endocrinology. 1987;121(3):1025–33. doi: 10.1210/endo-121-3-1025. [DOI] [PubMed] [Google Scholar]
- 52.Gillis KD, Mossner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16(6):1209–20. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 53.Vitale ML, Seward EP, Trifaro JM. Chromaffin cell cortical actin network dynamics control the size of the release-ready vesicle pool and the initial rate of exocytosis. Neuron. 1995;14(2):353–63. doi: 10.1016/0896-6273(95)90291-0. [DOI] [PubMed] [Google Scholar]
- 54.Knight DE, Baker PF. The phorbol ester TPA increases the affinity of exocytosis for calcium in ‘leaky’ adrenal medullary cells. FEBS Lett. 1983;160(1–2):98–100. doi: 10.1016/0014-5793(83)80944-2. [DOI] [PubMed] [Google Scholar]
- 55.Bittner MA, Holz RW. Protein kinase C and clostridial neurotoxins affect discrete and related steps in the secretory pathway. Cell Mol Neurobiol. 1993;13(6):649–64. doi: 10.1007/BF00711564. [DOI] [PubMed] [Google Scholar]
- 56.Hughes SJ, Christie MR, Ashcroft SJ. Potentiators of insulin secretion modulate Ca2+ sensitivity in rat pancreatic islets. Mol Cell Endocrinol. 1987;50(3):231–6. doi: 10.1016/0303-7207(87)90021-9. [DOI] [PubMed] [Google Scholar]
- 57.Jones PM, Salmon DM, Howell SL. Protein phosphorylation in electrically permeabilized islets of Langerhans. Effects of Ca2+, cyclic AMP, a phorbol ester and noradrenaline. Biochem J. 1988;254(2):397–403. doi: 10.1042/bj2540397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pocotte SL, et al. Effects of phorbol ester on catecholamine secretion and protein phosphorylation in adrenal medullary cell cultures. Proc Natl Acad Sci U S A. 1985;82(3):930–4. doi: 10.1073/pnas.82.3.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tamagawa T, Niki H, Niki A. Insulin release independent of a rise in cytosolic free Ca2+ by forskolin and phorbol ester. FEBS Lett. 1985;183(2):430–2. doi: 10.1016/0014-5793(85)80825-5. [DOI] [PubMed] [Google Scholar]
- 60.Carpenter L, et al. PKC alpha is activated but not required during glucose-induced insulin secretion from rat pancreatic islets. Diabetes. 2004;53(1):53–60. doi: 10.2337/diabetes.53.1.53. [DOI] [PubMed] [Google Scholar]
- 61.Hii CS, et al. A re-assessment of the role of protein kinase C in glucose-stimulated insulin secretion. Biochem J. 1987;246(2):489–93. doi: 10.1042/bj2460489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morgan A, et al. Regulation of exocytosis by protein kinase C. Biochem Soc Trans. 2005;33(Pt 6):1341–4. doi: 10.1042/BST0331341. [DOI] [PubMed] [Google Scholar]
- 63.Guenifi A, et al. Carbachol restores insulin release in diabetic GK rat islets by mechanisms largely involving hydrolysis of diacylglycerol and direct interaction with the exocytotic machinery. Pancreas. 2001;22(2):164–71. doi: 10.1097/00006676-200103000-00009. [DOI] [PubMed] [Google Scholar]
- 64.Rose T, Efendic S, Rupnik M. Ca2+-secretion coupling is impaired in diabetic Goto Kakizaki rats. J Gen Physiol. 2007;129(6):493–508. doi: 10.1085/jgp.200609604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Warwar N, et al. Dynamics of glucose-induced localization of PKC isoenzymes in pancreatic beta-cells: diabetes-related changes in the GK rat. Diabetes. 2006;55(3):590–9. doi: 10.2337/diabetes.55.03.06.db05-0001. [DOI] [PubMed] [Google Scholar]
- 66.Schmitz-Peiffer C, et al. Inhibition of PKCepsilon improves glucose-stimulated insulin secretion and reduces insulin clearance. Cell Metab. 2007;6(4):320–8. doi: 10.1016/j.cmet.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 67.Gandasi NR, et al. Ca2+ channel clustering with insulin-containing granules is disturbed in type 2 diabetes. J Clin Invest. 2017 doi: 10.1172/JCI88491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deeney JT, et al. Reversible Ca2+-dependent translocation of protein kinase C and glucose-induced insulin release. J Biol Chem. 1996;271(30):18154–60. doi: 10.1074/jbc.271.30.18154. [DOI] [PubMed] [Google Scholar]
- 69.Betz A, et al. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21(1):123–36. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- 70.Sheu L, et al. Regulation of insulin exocytosis by Munc13-1. J Biol Chem. 2003;278(30):27556–63. doi: 10.1074/jbc.M303203200. [DOI] [PubMed] [Google Scholar]
- 71.Kang L, et al. Munc13-1 is required for the sustained release of insulin from pancreatic beta cells. Cell Metab. 2006;3(6):463–8. doi: 10.1016/j.cmet.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 72.Rhee JS, et al. Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell. 2002;108(1):121–33. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- 73.de Jong AP, et al. Phosphorylation of synaptotagmin-1 controls a post-priming step in PKC-dependent presynaptic plasticity. Proc Natl Acad Sci U S A. 2016;113(18):5095–100. doi: 10.1073/pnas.1522927113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wierda KD, et al. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron. 2007;54(2):275–90. doi: 10.1016/j.neuron.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 75.Porat-Shliom N, et al. Multiple roles for the actin cytoskeleton during regulated exocytosis. Cell Mol Life Sci. 2013;70(12):2099–121. doi: 10.1007/s00018-012-1156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rose SD, et al. Chromaffin cell F-actin disassembly and potentiation of catecholamine release in response to protein kinase C activation by phorbol esters is mediated through myristoylated alanine-rich C kinase substrate phosphorylation. J Biol Chem. 2001;276(39):36757–63. doi: 10.1074/jbc.M006518200. [DOI] [PubMed] [Google Scholar]
- 77.Roth D, Burgoyne RD. Stimulation of catecholamine secretion from adrenal chromaffin cells by 14-3-3 proteins is due to reorganisation of the cortical actin network. FEBS Lett. 1995;374(1):77–81. doi: 10.1016/0014-5793(95)01080-x. [DOI] [PubMed] [Google Scholar]
- 78.Vaughan PF, Walker JH, Peers C. The regulation of neurotransmitter secretion by protein kinase C. Mol Neurobiol. 1998;18(2):125–55. doi: 10.1007/BF02914269. [DOI] [PubMed] [Google Scholar]
- 79.Vitale ML, Rodriguez Del Castillo A, Trifaro JM. Protein kinase C activation by phorbol esters induces chromaffin cell cortical filamentous actin disassembly and increases the initial rate of exocytosis in response to nicotinic receptor stimulation. Neuroscience. 1992;51(2):463–74. doi: 10.1016/0306-4522(92)90330-5. [DOI] [PubMed] [Google Scholar]
- 80.Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21(4):885–93. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- 81.Smith C. A persistent activity-dependent facilitation in chromaffin cells is caused by Ca2+ activation of protein kinase C. J Neurosci. 1999;19(2):589–98. doi: 10.1523/JNEUROSCI.19-02-00589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hartwig JH, et al. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature. 1992;356(6370):618–22. doi: 10.1038/356618a0. [DOI] [PubMed] [Google Scholar]
- 83.Yu W, et al. Synergism of protein kinase A, protein kinase C, and myosin light-chain kinase in the secretory cascade of the pancreatic beta-cell. Diabetes. 2000;49(6):945–52. doi: 10.2337/diabetes.49.6.945. [DOI] [PubMed] [Google Scholar]
- 84.Morgan A, Burgoyne RD. Interaction between protein kinase C and Exo1 (14-3-3 protein) and its relevance to exocytosis in permeabilized adrenal chromaffin cells. Biochem J. 1992;286(Pt 3):807–11. doi: 10.1042/bj2860807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brozzi F, et al. MyRIP interaction with MyoVa on secretory granules is controlled by the cAMP-PKA pathway. Mol Biol Cell. 2012;23(22):4444–55. doi: 10.1091/mbc.E12-05-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sun L, Bittner MA, Holz RW. Rim, a component of the presynaptic active zone and modulator of exocytosis, binds 14-3-3 through its N terminus. J Biol Chem. 2003;278(40):38301–9. doi: 10.1074/jbc.M212801200. [DOI] [PubMed] [Google Scholar]
- 87.Rorsman P, Braun M, Zhang Q. Regulation of calcium in pancreatic alpha- and beta-cells in health and disease. Cell Calcium. 2012;51(3–4):300–8. doi: 10.1016/j.ceca.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vasseur M, Debuyser A, Joffre M. Sensitivity of pancreatic beta cell to calcium channel blockers. An electrophysiologic study of verapamil and nifedipine. Fundam Clin Pharmacol. 1987;1(2):95–113. doi: 10.1111/j.1472-8206.1987.tb00549.x. [DOI] [PubMed] [Google Scholar]
- 89.Malaisse WJ, Boschero AC. Calcium antagonists and islet function. XI. Effect of nifedipine. Horm Res. 1977;8(4):203–9. doi: 10.1159/000178801. [DOI] [PubMed] [Google Scholar]
- 90.Schulla V, et al. Impaired insulin secretion and glucose tolerance in beta cell-selective Ca(v)1.2 Ca2+ channel null mice. EMBO J. 2003;22(15):3844–54. doi: 10.1093/emboj/cdg389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Arkhammar P, et al. Protein kinase C modulates the insulin secretory process by maintaining a proper function of the beta-cell voltage-activated Ca2+ channels. J Biol Chem. 1994;269(4):2743–9. [PubMed] [Google Scholar]
- 92.Velasco JM, Petersen JU, Petersen OH. Single-channel Ba2+ currents in insulin-secreting cells are activated by glyceraldehyde stimulation. FEBS Lett. 1988;231(2):366–70. doi: 10.1016/0014-5793(88)80851-2. [DOI] [PubMed] [Google Scholar]
- 93.Wollheim CB, Ullrich S, Pozzan T. Glyceraldehyde, but not cyclic AMP-stimulated insulin release is preceded by a rise in cytosolic free Ca2+ FEBS Lett. 1984;177(1):17–22. doi: 10.1016/0014-5793(84)80972-2. [DOI] [PubMed] [Google Scholar]
- 94.Parfitt KD, Madison DV. Phorbol esters enhance synaptic transmission by a presynaptic, calcium-dependent mechanism in rat hippocampus. J Physiol. 1993;471:245–68. doi: 10.1113/jphysiol.1993.sp019900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zaitsev SV, et al. Dissociation between changes in cytoplasmic free Ca2+ concentration and insulin secretion as evidenced from measurements in mouse single pancreatic islets. Proc Natl Acad Sci U S A. 1995;92(21):9712–6. doi: 10.1073/pnas.92.21.9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nakamura J, et al. Protein kinase C-dependent and -independent inhibition of Ca(2+) influx by phorbol ester in rat pancreatic beta-cells. Cell Signal. 2001;13(3):199–205. doi: 10.1016/s0898-6568(01)00136-x. [DOI] [PubMed] [Google Scholar]
- 97.Suga S, et al. Phorbol ester impairs electrical excitation of rat pancreatic beta-cells through PKC-independent activation of KATP channels. BMC Pharmacol. 2001;1:3. doi: 10.1186/1471-2210-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yokoyama CT, et al. Mechanism of SNARE protein binding and regulation of Cav2 channels by phosphorylation of the synaptic protein interaction site. Mol Cell Neurosci. 2005;28(1):1–17. doi: 10.1016/j.mcn.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 99.Harkins AB, et al. Deletion of the synaptic protein interaction site of the N-type (CaV2.2) calcium channel inhibits secretion in mouse pheochromocytoma cells. Proc Natl Acad Sci U S A. 2004;101(42):15219–24. doi: 10.1073/pnas.0401001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barg S, et al. Fast exocytosis with few Ca(2+) channels in insulin-secreting mouse pancreatic B cells. Biophys J. 2001;81(6):3308–23. doi: 10.1016/S0006-3495(01)75964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang SN, et al. Syntaxin 1 interacts with the L(D) subtype of voltage-gated Ca(2+) channels in pancreatic beta cells. Proc Natl Acad Sci U S A. 1999;96(18):10164–9. doi: 10.1073/pnas.96.18.10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Barg S, et al. A subset of 50 secretory granules in close contact with L-type Ca2+ channels accounts for first-phase insulin secretion in mouse beta-cells. Diabetes. 2002;51(Suppl 1):S74–82. doi: 10.2337/diabetes.51.2007.s74. [DOI] [PubMed] [Google Scholar]
- 103.Biden TJ, et al. The diverse roles of protein kinase C in pancreatic beta-cell function. Biochem Soc Trans. 2008;36(Pt 5):916–9. doi: 10.1042/BST0360916. [DOI] [PubMed] [Google Scholar]
- 104.Hashimoto N, et al. PKClambda regulates glucose-induced insulin secretion through modulation of gene expression in pancreatic beta cells. J Clin Invest. 2005;115(1):138–45. doi: 10.1172/JCI22232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yang SN, Berggren PO. CaV2.3 channel and PKClambda: new players in insulin secretion. J Clin Invest. 2005;115(1):16–20. doi: 10.1172/JCI23970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Turner KM, Burgoyne RD, Morgan A. Protein phosphorylation and the regulation of synaptic membrane traffic. Trends Neurosci. 1999;22(10):459–64. doi: 10.1016/s0166-2236(99)01436-8. [DOI] [PubMed] [Google Scholar]
- 107.Hanna ST, et al. Kiss-and-run exocytosis and fusion pores of secretory vesicles in human beta-cells. Pflugers Arch. 2009;457(6):1343–50. doi: 10.1007/s00424-008-0588-0. [DOI] [PubMed] [Google Scholar]
- 108.Foster LJ, et al. Binary interactions of the SNARE proteins syntaxin-4, SNAP23, and VAMP-2 and their regulation by phosphorylation. Biochemistry. 1998;37(31):11089–96. doi: 10.1021/bi980253t. [DOI] [PubMed] [Google Scholar]
- 109.Hirling H, Scheller RH. Phosphorylation of synaptic vesicle proteins: modulation of the alpha SNAP interaction with the core complex. Proc Natl Acad Sci U S A. 1996;93(21):11945–9. doi: 10.1073/pnas.93.21.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Risinger C, Bennett MK. Differential phosphorylation of syntaxin and synaptosome-associated protein of 25 kDa (SNAP-25) isoforms. J Neurochem. 1999;72(2):614–24. doi: 10.1046/j.1471-4159.1999.0720614.x. [DOI] [PubMed] [Google Scholar]
- 111.Nielander HB, et al. Phosphorylation of VAMP/synaptobrevin in synaptic vesicles by endogenous protein kinases. J Neurochem. 1995;65(4):1712–20. doi: 10.1046/j.1471-4159.1995.65041712.x. [DOI] [PubMed] [Google Scholar]
- 112.Matveeva EA, et al. Phosphorylation of the N-ethylmaleimide-sensitive factor is associated with depolarization-dependent neurotransmitter release from synaptosomes. J Biol Chem. 2001;276(15):12174–81. doi: 10.1074/jbc.M007394200. [DOI] [PubMed] [Google Scholar]
- 113.Chou WH, et al. GABAA receptor trafficking is regulated by protein kinase C(epsilon) and the N-ethylmaleimide-sensitive factor. J Neurosci. 2010;30(42):13955–65. doi: 10.1523/JNEUROSCI.0270-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Han GA, et al. Munc18-1 as a key regulator of neurosecretion. J Neurochem. 2010;115(1):1–10. doi: 10.1111/j.1471-4159.2010.06900.x. [DOI] [PubMed] [Google Scholar]
- 115.Fujita Y, et al. Phosphorylation of Munc-18/n-Sec1/rbSec1 by protein kinase C: its implication in regulating the interaction of Munc-18/n-Sec1/rbSec1 with syntaxin. J Biol Chem. 1996;271(13):7265–8. doi: 10.1074/jbc.271.13.7265. [DOI] [PubMed] [Google Scholar]
- 116.Sassa T, et al. The synaptic protein UNC-18 is phosphorylated by protein kinase C. Neurochem Int. 1996;29(5):543–52. doi: 10.1016/0197-0186(96)00009-5. [DOI] [PubMed] [Google Scholar]
- 117.Barclay JW, et al. Phosphorylation of Munc18 by protein kinase C regulates the kinetics of exocytosis. J Biol Chem. 2003;278(12):10538–45. doi: 10.1074/jbc.M211114200. [DOI] [PubMed] [Google Scholar]
- 118.de Vries KJ, et al. Dynamics of munc18-1 phosphorylation/dephosphorylation in rat brain nerve terminals. Eur J Neurosci. 2000;12(1):385–90. doi: 10.1046/j.1460-9568.2000.00931.x. [DOI] [PubMed] [Google Scholar]
- 119.Liu J, et al. Fluorescence resonance energy transfer reports properties of syntaxin1a interaction with Munc18-1 in vivo. J Biol Chem. 2004;279(53):55924–36. doi: 10.1074/jbc.M410024200. [DOI] [PubMed] [Google Scholar]
- 120.Nili U, et al. Munc18-1 phosphorylation by protein kinase C potentiates vesicle pool replenishment in bovine chromaffin cells. Neuroscience. 2006;143(2):487–500. doi: 10.1016/j.neuroscience.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 121.Uchida T, et al. Protein kinase Cdelta plays a non-redundant role in insulin secretion in pancreatic beta cells. J Biol Chem. 2007;282(4):2707–16. doi: 10.1074/jbc.M610482200. [DOI] [PubMed] [Google Scholar]
- 122.Fu J, et al. Protease-activated receptor-1 activation of endothelial cells induces protein kinase Calpha-dependent phosphorylation of syntaxin 4 and Munc18c: role in signaling p-selectin expression. J Biol Chem. 2005;280(5):3178–84. doi: 10.1074/jbc.M410044200. [DOI] [PubMed] [Google Scholar]
- 123.Genc O, et al. Munc18-1 is a dynamically regulated PKC target during short-term enhancement of transmitter release. Elife. 2014;3:e01715. doi: 10.7554/eLife.01715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mandic SA, et al. Munc18-1 and Munc18-2 proteins modulate beta-cell Ca2+ sensitivity and kinetics of insulin exocytosis differently. J Biol Chem. 2011;286(32):28026–40. doi: 10.1074/jbc.M111.235366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pinheiro PS, Houy S, Sorensen JB. C2-domain containing calcium sensors in neuroendocrine secretion. J Neurochem. 2016;139(6):943–958. doi: 10.1111/jnc.13865. [DOI] [PubMed] [Google Scholar]
- 126.Hilfiker S, et al. Regulation of synaptotagmin I phosphorylation by multiple protein kinases. J Neurochem. 1999;73(3):921–32. doi: 10.1046/j.1471-4159.1999.0730921.x. [DOI] [PubMed] [Google Scholar]
- 127.Rao TC, et al. Distinct fusion properties of synaptotagmin-1 and synaptotagmin-7 bearing dense core granules. Mol Biol Cell. 2014;25(16):2416–27. doi: 10.1091/mbc.E14-02-0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Verona M, et al. Changes of synaptotagmin interaction with t-SNARE proteins in vitro after calcium/calmodulin-dependent phosphorylation. J Neurochem. 2000;74(1):209–21. doi: 10.1046/j.1471-4159.2000.0740209.x. [DOI] [PubMed] [Google Scholar]
- 129.de Wit H, et al. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138(5):935–46. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 130.Nagy G, et al. Different effects on fast exocytosis induced by synaptotagmin 1 and 2 isoforms and abundance but not by phosphorylation. J Neurosci. 2006;26(2):632–43. doi: 10.1523/JNEUROSCI.2589-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kataoka M, et al. A single amino acid mutation in SNAP-25 induces anxiety-related behavior in mouse. PLoS One. 2011;6(9):e25158. doi: 10.1371/journal.pone.0025158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Watanabe S, et al. Epileptogenesis and epileptic maturation in phosphorylation site-specific SNAP-25 mutant mice. Epilepsy Res. 2015;115:30–44. doi: 10.1016/j.eplepsyres.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 133.Shimazaki Y, et al. Phosphorylation of 25-kDa synaptosome-associated protein. Possible involvement in protein kinase C-mediated regulation of neurotransmitter release. J Biol Chem. 1996;271(24):14548–53. doi: 10.1074/jbc.271.24.14548. [DOI] [PubMed] [Google Scholar]
- 134.Finley MF, Scheller RH, Madison DV. SNAP-25 Ser187 does not mediate phorbol ester enhancement of hippocampal synaptic transmission. Neuropharmacology. 2003;45(6):857–62. doi: 10.1016/s0028-3908(03)00283-1. [DOI] [PubMed] [Google Scholar]
- 135.Gonelle-Gispert C, et al. Phosphorylation of SNAP-25 on serine-187 is induced by secretagogues in insulin-secreting cells, but is not correlated with insulin secretion. Biochem J. 2002;368(Pt 1):223–32. doi: 10.1042/BJ20020896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hepp R, Cabaniols JP, Roche PA. Differential phosphorylation of SNAP-25 in vivo by protein kinase C and protein kinase A. FEBS Lett. 2002;532(1–2):52–6. doi: 10.1016/s0014-5793(02)03629-3. [DOI] [PubMed] [Google Scholar]
- 137.Nagy G, et al. Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J Neurosci. 2002;22(21):9278–86. doi: 10.1523/JNEUROSCI.22-21-09278.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shu Y, et al. Phosphorylation of SNAP-25 at Ser187 mediates enhancement of exocytosis by a phorbol ester in INS-1 cells. J Neurosci. 2008;28(1):21–30. doi: 10.1523/JNEUROSCI.2352-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yang Y, et al. Phosphomimetic mutation of Ser-187 of SNAP-25 increases both syntaxin binding and highly Ca2+-sensitive exocytosis. J Gen Physiol. 2007;129(3):233–44. doi: 10.1085/jgp.200609685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xu NJ, et al. Inhibition of SNAP-25 phosphorylation at Ser187 is involved in chronic morphine-induced down-regulation of SNARE complex formation. J Biol Chem. 2004;279(39):40601–8. doi: 10.1074/jbc.M406896200. [DOI] [PubMed] [Google Scholar]
- 141.Henkel AW, Betz WJ. Staurosporine blocks evoked release of FM1-43 but not acetylcholine from frog motor nerve terminals. J Neurosci. 1995;15(12):8246–58. doi: 10.1523/JNEUROSCI.15-12-08246.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu XS, Wu LG. Protein kinase c increases the apparent affinity of the release machinery to Ca2+ by enhancing the release machinery downstream of the Ca2+ sensor. J Neurosci. 2001;21(20):7928–36. doi: 10.1523/JNEUROSCI.21-20-07928.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Graham ME, Fisher RJ, Burgoyne RD. Measurement of exocytosis by amperometry in adrenal chromaffin cells: effects of clostridial neurotoxins and activation of protein kinase C on fusion pore kinetics. Biochimie. 2000;82(5):469–79. doi: 10.1016/s0300-9084(00)00196-6. [DOI] [PubMed] [Google Scholar]
- 144.Xue R, Zhao Y, Chen P. Involvement of PKC alpha in PMA-induced facilitation of exocytosis and vesicle fusion in PC12 cells. Biochem Biophys Res Commun. 2009;380(2):371–6. doi: 10.1016/j.bbrc.2009.01.105. [DOI] [PubMed] [Google Scholar]
- 145.Bittner MA, Aikman RL, Holz RW. A nibbling mechanism for clathrin-mediated retrieval of secretory granule membrane after exocytosis. J Biol Chem. 2013;288(13):9177–88. doi: 10.1074/jbc.M113.450361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tsuboi T, McMahon HT, Rutter GA. Mechanisms of dense core vesicle recapture following “kiss and run” (“cavicapture”) exocytosis in insulin-secreting cells. J Biol Chem. 2004;279(45):47115–24. doi: 10.1074/jbc.M408179200. [DOI] [PubMed] [Google Scholar]
- 147.Chiang HC, et al. Post-fusion structural changes and their roles in exocytosis and endocytosis of dense-core vesicles. Nat Commun. 2014;5:3356. doi: 10.1038/ncomms4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Trexler AJ, Sochacki KA, Taraska JW. Imaging the recruitment and loss of proteins and lipids at single sites of calcium-triggered exocytosis. Mol Biol Cell. 2016;27(15):2423–34. doi: 10.1091/mbc.E16-01-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Anantharam A, Axelrod D, Holz RW. Real-time imaging of plasma membrane deformations reveals pre-fusion membrane curvature changes and a role for dynamin in the regulation of fusion pore expansion. J Neurochem. 2012;122(4):661–71. doi: 10.1111/j.1471-4159.2012.07816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Robinson PJ, et al. Dynamin GTPase regulated by protein kinase C phosphorylation in nerve terminals. Nature. 1993;365(6442):163–6. doi: 10.1038/365163a0. [DOI] [PubMed] [Google Scholar]
- 151.Slepnev VI, et al. Role of phosphorylation in regulation of the assembly of endocytic coat complexes. Science. 1998;281(5378):821–4. doi: 10.1126/science.281.5378.821. [DOI] [PubMed] [Google Scholar]
- 152.Baker RW, et al. A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science. 2015;349(6252):1111–4. doi: 10.1126/science.aac7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ma L, et al. Munc18-1-regulated stage-wise SNARE assembly underlying synaptic exocytosis. Elife. 2015:4. doi: 10.7554/eLife.09580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jorgacevski J, et al. Munc18-1 tuning of vesicle merger and fusion pore properties. J Neurosci. 2011;31(24):9055–66. doi: 10.1523/JNEUROSCI.0185-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chasserot-Golaz S, et al. Lipid dynamics in exocytosis. Cell Mol Neurobiol. 2010;30(8):1335–42. doi: 10.1007/s10571-010-9577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wan QF, et al. Protein kinase activation increases insulin secretion by sensitizing the secretory machinery to Ca2+ J Gen Physiol. 2004;124(6):653–62. doi: 10.1085/jgp.200409082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yang Y, Gillis KD. A highly Ca2+-sensitive pool of granules is regulated by glucose and protein kinases in insulin-secreting INS-1 cells. J Gen Physiol. 2004;124(6):641–51. doi: 10.1085/jgp.200409081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yang Y, et al. A highly Ca2+-sensitive pool of vesicles is regulated by protein kinase C in adrenal chromaffin cells. Proc Natl Acad Sci U S A. 2002;99(26):17060–5. doi: 10.1073/pnas.242624699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kasai K, et al. Docking is not a prerequisite but a temporal constraint for fusion of secretory granules. Traffic. 2008;9(7):1191–203. doi: 10.1111/j.1600-0854.2008.00744.x. [DOI] [PubMed] [Google Scholar]
- 160.Pedersen MG, Sherman A. Newcomer insulin secretory granules as a highly calcium-sensitive pool. Proc Natl Acad Sci U S A. 2009;106(18):7432–6. doi: 10.1073/pnas.0901202106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lam SS, et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods. 2015;12(1):51–4. doi: 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gonzalez-Vera JA, Morris MC. Fluorescent Reporters and Biosensors for Probing the Dynamic Behavior of Protein Kinases. Proteomes. 2015;3(4):369–410. doi: 10.3390/proteomes3040369. [DOI] [PMC free article] [PubMed] [Google Scholar]