


Abstract
Although bacterial gyrase and topoisomerase IV have critical interactions with positively supercoiled DNA, little is known about the actions of these enzymes on overwound substrates. Therefore, the abilities of Bacillus anthracis and Escherichia coli gyrase and topoisomerase IV to relax and cleave positively supercoiled DNA were analyzed. Gyrase removed positive supercoils ∼10-fold more rapidly and more processively than it introduced negative supercoils into relaxed DNA. In time-resolved single-molecule measurements, gyrase relaxed overwound DNA with burst rates of ∼100 supercoils per second (average burst size was 6.2 supercoils). Efficient positive supercoil removal required the GyrA-box, which is necessary for DNA wrapping. Topoisomerase IV also was able to distinguish DNA geometry during strand passage and relaxed positively supercoiled substrates ∼3-fold faster than negatively supercoiled molecules. Gyrase maintained lower levels of cleavage complexes with positively supercoiled (compared with negatively supercoiled) DNA, whereas topoisomerase IV generated similar levels with both substrates. Results indicate that gyrase is better suited than topoisomerase IV to safely remove positive supercoils that accumulate ahead of replication forks. They also suggest that the wrapping mechanism of gyrase may have evolved to promote rapid removal of positive supercoils, rather than induction of negative supercoils.
INTRODUCTION
The topological state of DNA has a dramatic effect on nucleic acid processes in bacterial cells (1–5). DNA is globally underwound (i.e. negatively supercoiled), which enhances the opening of the double helix and facilitates replication and transcription. Conversely, overwound (positively supercoiled) DNA that accumulates ahead of replication and transcription machinery must be removed in order for elongation to progress. Furthermore, fundamental cellular processes such as recombination and replication introduce knots and tangles into the genome, which impede DNA tracking systems and prevent chromosomal segregation during cell division.
Bacteria encode multiple topoisomerases that regulate the topological state of DNA. Among them are two type II topoisomerases, gyrase and topoisomerase IV (6–9). These enzymes function as heterotetramers (GyrA2GyrB2 for gyrase and GrlA2GrlB2 and ParC2ParE2 for topoisomerase IV in Gram-positive and Gram-negative species, respectively) and display sequence homology (6,10). In addition, they both act by creating a transient double-stranded break in one segment of DNA (the gate- or G-segment) and passing a second intact segment (the transport- or T-segment) through the break (3,8,11). In order to maintain the integrity of the genome during this process, gyrase and topoisomerase IV covalently attach to the 5′-terminus of each DNA strand. This ‘cleavage complex’ is a hallmark of enzyme activity (12).
Despite these similarities, differences in the C-terminal domains of GrlA/ParC and GyrA confer each enzyme with a unique array of catalytic activities (7). Because the C-terminal domain of GrlA/ParC allows topoisomerase IV to interact with distal DNA segments, the enzyme uses a ‘canonical’ strand passage mechanism in which it captures existing intra- or intermolecular DNA crossovers (Figure 1) (6). This allows the enzyme to relax (i.e. remove) positive or negative supercoils, which results in a respective decrease or increase in DNA linking number (the number of times the two strands of the double helix cross each other). It also allows the enzyme to remove DNA knots and tangles in a highly efficient manner (6,7).
Figure 1.

Cellular functions and DNA strand passage mechanisms of gyrase and topoisomerase IV. Gyrase removes positively supercoiled DNA ahead of the replication machinery and also introduces negative supercoils into the genome. Topoisomerase IV may assist in the removal of positive supercoils, but primarily acts to resolve precatenanes behind the fork and unlink daughter chromosomes. Topoisomerase IV uses a ‘canonical’ DNA strand passage mechanism and gyrase uses a ‘wrapping’ mechanism to support their decatenation and supercoiling activities, respectively.
In contrast to the canonical mechanism used by all other type II topoisomerases, gyrase uses a mechanism in which the C-terminal domain of the GyrA subunit wraps DNA, inducing a positive crossover between the G- and T-segments that mimics a positive supercoil (Figure 1) (6,13–18). This ‘wrapping’ mechanism has three important implications for gyrase activity. First, the captured G- and T-segments are proximal to one another (19). Therefore, gyrase greatly favors the catalysis of intramolecular strand passage reactions: the enzyme can efficiently alter superhelical density but is very poor at removing knots and tangles (19,20). Second, because gyrase always acts on the induced positive crossover, it works in a unidirectional manner (6,21); in the presence of adenosine triphosphate (ATP), the enzyme can remove positive, but not negative, supercoils and always causes a decrease in DNA linking number. Third, the ability of gyrase to create and subsequently remove positive supercoils allows the enzyme to decrease the linking number beyond that of relaxed DNA. Thus, among all known topoisomerases, gyrase is the only enzyme able to negatively supercoil DNA (7,11).
As a result of their individual properties, gyrase and topoisomerase IV have distinct functions during DNA replication (Figure 1) (7,22–24). Gyrase works primarily ahead of the fork to remove positive supercoils generated by the replicative helicase and to restore the negative superhelicity of the bacterial chromosome (2,3). Although topoisomerase IV can alter superhelical density and help to alleviate torsional stress that accumulates ahead of the fork (25–27), its critical function is to decatenate (i.e. untangle) daughter chromosomes following DNA replication (6,28–30). During DNA synthesis, topoisomerase IV works primarily behind the fork to remove intermolecular DNA crossovers (precatenanes).
Beyond their essential cellular functions, gyrase and topoisomerase IV are targets for the quinolone antibacterials, drugs that act by stabilizing cleavage complexes (8,31–33). As replication forks or other DNA tracking systems encounter these protein-bound DNA roadblocks, transient cleavage complexes are converted to non-ligatable DNA breaks that must be repaired by DNA damage response pathways. Quinolones kill bacteria by overwhelming cells with DNA strand breaks or (potentially) by robbing the cells of the critical activities of gyrase and topoisomerase IV. The relative importance of each enzyme as the primary target for quinolones varies among species (34–39). Gyrase is the primary cellular target for quinolones in Escherichia coli and many other Gram-negative organisms (34,35). It is also the primary target in some Gram-positive species, including B. anthracis (40–42).
Despite the fact that gyrase (potentially assisted by topoisomerase IV) plays an essential role in relaxing overwound DNA ahead of the replication machinery and that quinolone-stabilized cleavage complexes formed ahead of these moving forks are the most dangerous for the cell (12,43–45), little is known about how these enzymes remove positive supercoils or form cleavage complexes on overwound DNA. Therefore, we characterized the activities of B. anthracis and E. coli gyrase and topoisomerase IV on positively supercoiled DNA. Results indicate that gyrase can remove positive supercoils much more rapidly and processively than it can introduce negative supercoils into relaxed DNA. This efficient relaxation of overwound DNA is lost when the GyrA-box, which is required for DNA wrapping (17), is mutated. Topoisomerase IV, using the canonical strand passage mechanism, also acts faster on positively supercoiled DNA. As seen with eukaryotic type II topoisomerases (43), gyrase maintains lower levels of cleavage complexes with positively (as compared with negatively) supercoiled DNA. This is in contrast to topoisomerase IV, which maintains similar levels of cleavage complexes with both substrates. Thus, the ability of gyrase to rapidly remove positive supercoils while maintaining low levels of potentially dangerous cleavage complexes makes it the safer enzyme to work ahead of the replication fork.
MATERIALS AND METHODS
Enzymes, DNA and materials
Wild-type B. anthracis gyrase and topoisomerase IV subunits (GyrA, GyrB, GrlA and GrlB) were expressed and purified using a modification of a previously published protocol (46). Genes were polymerase chain reaction (PCR)-amplified from chromosomal DNA of the Sterne strain of B. anthracis. PCR products were cloned into the pET21b vector, which added an N-terminal 6xHis tag to each protein subunit. The GyrAAla-box construct (in which the seven amino acids of the GyrA-box were replaced with alanine residues) was generated using a QuikChange kit (Stratagene). The identities of all constructs were confirmed by DNA sequencing. Each subunit construct was individually transformed into E. coli strain BL21(DE3) and cells were grown overnight at 37°C with shaking in LB medium containing 100 μg/ml ampicillin. Cultures were diluted 20-fold with fresh medium to yield 500-ml cultures, which were grown at 37°C with shaking at 200 rpm until the optical density at 600 nm was 0.6. Protein expression was induced by adding isopropyl β-D-1-thioglactopyranoside to a final concentration of 0.2 mM and cells were harvested at times optimized for each subunit (GyrA, 3 h; GyrAAla-box, 2 h; GyrB, 15 min; GrlA, 3 h; GrlB, 2 h). Pellets were resuspended in 30 ml of cold CelLytic B buffer (Sigma). Cells were lysed by serial passage through an EmulsiFlex-C5 high-pressure homogenizer at >15 000 psi, cell debris was removed by centrifugation and proteins were purified from the supernatant by passage through a 2-ml Ni-nitrilotriacetic acid affinity column (Qiagen). Columns were washed according to the manufacturer’s instructions. Proteins were eluted using 12 ml of buffer containing 20 mM Tris–HCl pH 7.9, 500 mM NaCl and 300 mM imidazole and exchanged into buffer containing 50 mM Tris–HCl pH 7.5, 200 mM NaCl, 20% glycerol and 5 mM dithiothreitol through 30 kDa molecular weight cut-off centrifugal concentrators (Amicon). Subunits were stored at −80°C.
Escherichia coli topoisomerase IV subunits (ParC and ParE) were purified as described previously (47,48) and stored at −80°C. Escherichia coli gyrase was purchased from New England BioLabs.
Negatively supercoiled pBR322 plasmid DNA was prepared using a Plasmid Mega Kit (Qiagen) as described by the manufacturer. Positively supercoiled pBR322 DNA was prepared by treating negatively supercoiled molecules with recombinant Archaeoglobus fulgidus reverse gyrase (49,50). The number of positive supercoils induced by this process was comparable with the number of negative supercoils in the original pBR322 preparations (49). All experiments with negatively supercoiled DNA used plasmid preparations that were processed identically to the positively supercoiled molecules except that reaction mixtures did not contain reverse gyrase. Relaxed pBR322 plasmid DNA was generated by treating negatively supercoiled pBR322 with calf thymus topoisomerase I (Invitrogen) and purified as described previously (51).
Ciprofloxacin was obtained from LKT Laboratories, stored at 4°C as a 40 mM stock solution in 0.1 M NaOH and diluted 5-fold with 10 mM Tris–HCl pH 7.9 immediately prior to use. Moxifloxacin was obtained from LKT Laboratories and levofloxacin was obtained from Sigma Aldrich. Both drugs were stored at 4°C as 20 mM stock solutions in 100% dimethyl sulfoxide. Spermidine was obtained from Sigma–Aldrich. All other chemicals were analytical reagent grade.
Monitoring DNA supercoiling and relaxation using ensemble experiments
DNA supercoiling and relaxation assays were based on previously published protocols (52,53). For B. anthracis gyrase reactions, GyrA (or GyrAAla-box) and GyrB (tetramer concentration of 400–1000 nM, 1:1 GyrA:GyrB ratio) were incubated for 5 min at 37°C in 100 mM Tris–HCl pH 7.5, 350 mM KGlu and 100 μg/ml bovine serum albumin (BSA, Sigma), then diluted 2-fold with a mixture containing DNA, Mg2+ and ATP for a final reaction volume of 20 μl. The final concentrations of reactants were 200–500 nM gyrase, 5 nM positively or negatively supercoiled or relaxed DNA and 1.5 mM ATP in 50 mM Tris–HCl pH 7.5, 5 mM MgCl2, 175 mM KGlu and 50 μg/ml BSA. Reactions were incubated at 37°C for times indicated and stopped by the addition of 3 μl of a mixture of 0.77% sodium dodecyl sulphate (SDS) and 77.5 mM Na2EDTA. Samples were mixed with 2 μl of agarose loading dye (60% sucrose; 10 mM Tris–HCl pH 7.9, 0.5% bromophenol blue, 0.5% xylene cyanol FF) and subjected to electrophoresis in 1% agarose gels in TBE (100 mM Tris-borate pH 8.3, 2 mM ethylenediaminetetraaceticacid (EDTA)). Gels were stained with 1 μg/ml ethidium bromide for 30 min. DNA bands were visualized with medium-range ultraviolet light on an Alpha Innotech digital imaging system.
Alternatively, reaction products were analyzed by 2D gel electrophoresis as described previously (49). The first dimension was run for 2 h as described in the preceding paragraph. The gel was then soaked in TBE containing 4.5 μg/ml chloroquine for 2 h with gentle shaking followed by electrophoresis in the orthogonal direction (90° clockwise) for 2 h in fresh TBE containing 4.5 μg/ml chloroquine. Gels were stained and DNA bands were visualized as described above.
Escherichia coli gyrase supercoiling assays contained 50 nM gyrase, 5 nM positively supercoiled DNA and 1.5 mM ATP in 50 mM Tris–HCl pH 7.5, 5 mM MgCl2, 175 mM KGlu and 50 μg/ml BSA in a reaction volume of 20 μl. Reactions were incubated, stopped and analyzed as described above.
Bacillus anthracis topoisomerase IV relaxation assays contained 10 nM topoisomerase IV, 5 nM supercoiled pBR322 and 1 mM ATP in 40 mM HEPES pH 7.6, 100 mM KGlu, 10 mM Mg(OAc)2 and 50 mM NaCl in a reaction volume of 20 μl. Reactions were incubated, stopped and analyzed as described above.
Preparation of coilable DNA substrates for single-molecule experiments
An ∼5 kb region of pET-28b was amplified by PCR using primers that incorporated cut sites for BsaI to create DNA with unique, non-palindromic sticky ends as described previously (54). One end of the linear substrate was ligated to an ∼500 bp DNA handle functionalized with multiple biotin moieties, while the other end was ligated to a similar handle containing multiple digoxigenin moieties. An 18 × 5 × 0.08 mm flow chamber was constructed using a pair of glass coverslips joined together by two strips of double-sided adhesive film. The chamber was incubated with DNA at a final concentration of ∼1 pM. The digoxigenin-containing end of the DNA was immobilized to the surface of the chamber with anti-digoxigenin antibodies. The biotin-containing end was attached to an ∼1 μm diameter streptavidin-coated magnetic bead (Dynal). The resulting DNA construct could then be torsionally constrained and manipulated using magnetic tweezers.
Monitoring gyrase-mediated relaxation of positively supercoiled DNA using magnetic tweezers
A magnetic tweezers apparatus similar to that described by Ribeck and Saleh (55) was used to manipulate DNA topology and apply small stretching forces to the substrate. DNA extension was recorded at a rate of 200 frames per second using video-based tracking of bead images at a magnification of 125 nm per pixel. To verify tethering of single DNA molecules, beads were rotated at a force of 3.5 pN. Single coilable DNA molecules denature when underwound and form a plectoneme when overwound, following characteristic extension-rotation curves for both conformational changes (56). The conditions used for magnetic tweezers measurements were consistent with the formation of a single plectoneme on each DNA tether examined (57,58).
DNA relaxation reactions were carried out at 22°C and utilized 1 nM B. anthracis gyrase and 1 mM ATP in 50 mM Tris–HCl pH 7.5, 5 mM MgCl2, 175 mM KGlu, 50 μg/ml BSA and 0.1% Tween-20 (to prevent sticking of the magnetic beads). Positive supercoils were generated at forces <5 pN by rotating the magnetic bead in increments of +50 rotations. Under these conditions, supercoiling reduces DNA extension due to plectoneme formation. Relaxation of the positively supercoiled DNA resulted in extension of the substrate. The magnetic tweezers apparatus was programmed to automatically apply +50 rotations when the DNA extension exceeded a set threshold, typically 20–50 nm below the maximum (fully unwound) extension.
Analysis of burst size and burst rate distributions
A simple detection routine was employed to quantify burst events within recorded data. A candidate point within an activity burst was defined as a point at which the change in mean extension over 100 ms (20 data points) on either side of the point was >100 nm. After the window of the activity burst was defined, it was narrowed by excluding any points that were within one standard deviation of the mean extension 20 ms before or after the burst. The end points of the burst were defined by re-including one data point ahead of and one data point behind this narrowed region. Burst rates were obtained by linear regression of the raw data. Burst sizes were estimated by dividing the change in extension over the burst event by the average extension change resulting from relaxation of a single plectonemic supercoil. The bursting events were independent of force and were therefore pooled in the distributions shown in Figure 5.
Figure 5.

Escherichia coli gyrase removes positive supercoils more rapidly than it introduces negative supercoils into relaxed DNA. A time course is shown for the relaxation of positive supercoils followed by the introduction of negative supercoils. Positively supercoiled [(+)SC] and negatively supercoiled [(−)SC] standards are shown. The gel image is representative of at least three independent experiments.
DNA cleavage
DNA cleavage reactions were based on the procedure of Aldredet al. (52). Bacillus anthracis gyrase reactions contained 500 nM wild-type or 250 nM GyrAAla-box gyrase (1:2 GyrA:GyrB ratio) and 10 nM positively or negatively supercoiled pBR322 in a total volume of 20 μl of 50 mM Tris–HCl pH 7.5, 100 mM KGlu, 5 mM MgCl2 and 50 μg/ml BSA. In some cases, MgCl2 was replaced with CaCl2 and a range of enzyme concentrations was tested. Reactions were incubated at 37°C for 30 min and enzyme–DNA cleavage complexes were trapped by the addition of 2 μl of 5% SDS. Na2EDTA (2 μl of 250 mM) and proteinase K (2 μl of 0.8 mg/ml) were added and samples were incubated at 45°C for 30 min to digest the enzyme. Samples were mixed with 2 μl of agarose loading dye and incubated at 45°C for 2 min before loading on gels. Reaction products were subjected to electrophoresis in 1% agarose gels in 40 mM Tris-acetate pH 8.3 and 2 mM Na2EDTA containing 0.5 μg/ml ethidium bromide and visualized as described above. DNA cleavage was monitored by the conversion of supercoiled plasmid to linear molecules and quantified by comparison to a control reaction in which an equal mass of DNA was digested by EcoRI (New England BioLabs).
DNA cleavage reactions with B. anthracis topoisomerase IV contained 100 nM enzyme (1:2 GrlA:GrlB ratio) and 10 nM positively or negatively supercoiled pBR322 in 20 μl of 40 mM Tris–HCl pH 7.9, 10 mM MgCl2, 50 mM NaCl and 12.5% glycerol. Reactions were incubated at 37°C for 10 min and were stopped, digested and analyzed as described above.
Reactions with E. coli gyrase contained 100 nM enzyme and 10 nM positively or negatively supercoiled pBR322 in 20 μl of 40 mM Tris–HCl pH 7.9, 10 mM MgCl2, 50 mM NaCl and 12.5% glycerol. Reactions with E. coli topoisomerase IV contained 10 nM enzyme (1:1 ParC:ParE ratio) and 10 nM positively or negatively supercoiled pBR322 in 20 μl of 40 mM Tris–HCl pH 7.9, 5 mM MgCl2, 50 mM NaCl and 12.5% glycerol. Reactions with each enzyme were incubated at 37°C for 10 min, then stopped, digested and analyzed as described above.
RESULTS
Bacillus anthracis gyrase relaxes positive supercoils more rapidly than it introduces negative supercoils
As discussed above, gyrase plays two important roles in the cell: it alleviates stress ahead of DNA tracking systems and it generates negative supercoils to help maintain the correct superhelical density of the bacterial chromosome. Although the supercoiling activity of gyrase has been well documented (18,21,59–65), little is known about the ability of the enzyme to remove positive supercoils from DNA. Therefore, we assessed the ability of B. anthracis gyrase to relax a positively supercoiled plasmid and subsequently convert it to a negatively supercoiled molecule (Figure 2A). Enzyme activity was monitored over 45 min in order to observe both the removal of positive supercoils (the relaxation reaction) and the introduction of negative supercoils (the supercoiling reaction) within the same assay. The initial substrate had a superhelical density that was similar to the original plasmid isolated from E. coli, but was opposite in sign (49).
Figure 2.

Bacillus anthracis gyrase removes positive supercoils more rapidly than it introduces negative supercoils into relaxed DNA. (A) Gyrase activity on positively supercoiled DNA. A time course is shown for the relaxation of positive supercoils followed by the introduction of negative supercoils. Positively supercoiled [(+)SC] and negatively supercoiled [(−)SC] standards are shown. (B) Expanded time course for the relaxation of (+)SC DNA by gyrase. (C) Time course for the introduction of negative supercoils into relaxed DNA (Rel) by gyrase. Gel images are representative of at least three independent experiments. (D) Quantification of removal of positive supercoils and introduction of negative supercoils by gyrase shown in parts A–C. The relative amount of (+)SC DNA (white) in each sample was determined by comparison to the (+)SC control. The relative amount of (−)SC DNA (black) in each sample was determined by comparison to the maximum level of (−)SC DNA formed in each experiment. Inset: expanded view of loss of (+)SC DNA. Error bars represent standard deviations of at least three independent experiments.
As shown in the gel in Figure 2A and quantified in Figure 2D, B. anthracis gyrase rapidly relaxed the overwound plasmid and removed all of the positive supercoils within 2 min. To further investigate the speed and processivity of this reaction, we monitored relaxation of positively supercoiled DNA over an expanded 90-s time course (Figure 2B and D). In a fully processive reaction, the enzyme catalyzes the complete relaxation of the substrate with little evidence of intermediate topoisomers. Conversely, in a fully distributive reaction, the enzyme relaxes the entire substrate population synchronously and the complete range of intermediate topoisomers is present. Although the gyrase-catalyzed relaxation of positive supercoils is not entirely processive, relatively few positively supercoiled intermediate topoisomers were observed (further discussed below).
In contrast, the conversion of relaxed plasmid to negatively supercoiled DNA by gyrase occurred much more slowly (Figure 2A and C; quantified in Figure 2D). The enzyme took >15 min to underwind the bulk of the substrate and as much as 45 min to fully underwind the plasmid population. Additionally, as compared to the relaxation reaction, a much higher proportion of reaction products were observed as intermediate topoisomers during the supercoiling reaction, indicating that this latter reaction is considerably more distributive than the removal of positive supercoils. Similar rates for the supercoiling reaction were observed whether the initial DNA substrate was positively supercoiled (Figure 2A) or relaxed (Figure 2C). This highlights the clear distinction between the rates of relaxation and supercoiling.
A number of DNA topoisomers were apparent in the later stages of the relaxation reaction (Figure 2B) and it was difficult to unambiguously discern their supercoil handedness on a one-dimensional gel. These topoisomers may represent positively supercoiled molecules that have yet to be fully relaxed or molecules that have already been relaxed and are partially negatively supercoiled. Therefore, the products of relaxation reactions were analyzed by 2D gel electrophoresis. As seen in Figure 3, all of the positively supercoiled topoisomers were gone by ∼90 s. This confirms the above conclusion that, under the conditions employed, gyrase completely removes all positive supercoils in <2 min. It also indicates that the time required to fully relax positively supercoiled DNA is at least 10-fold shorter than that required for gyrase to fully negatively supercoil the bulk of the relaxed molecules. Despite these rate differences, a small proportion of DNA molecules become negatively supercoiled within 30 s (Figure 3, also see Figure 2A and C), suggesting the presence of a gyrase population that is capable of acting much faster than the majority of enzyme molecules. It is not known whether this reflects an enzyme pool that remains bound and acts in a highly processive manner or uses an alternative DNA strand passage mechanism (66).
Figure 3.

2D gel analysis of Bacillus anthracis gyrase activity on positively supercoiled DNA. (A) Control gel showing mobility of nicked, positively supercoiled [(+)SC], relaxed (Rel) and negatively supercoiled [(−)SC] DNA. (B–D) Activity of gyrase on (+)SC DNA. DNA products generated after 30 s (B), 90 s (C) and 180 s (D) reactions are shown. Gel images are representative of at least three independent experiments.
The removal of positive supercoils and the introduction of negative supercoils were sensitive to the concentration of ATP. Both reactions displayed an apparent Km value of ∼0.15 mM as determined by analysis of a series of time courses carried out at 0.125–1.5 mM ATP (quantification of the effect of [ATP] on the rate of positive supercoil removal is shown in Supplementary Figure S1). Results are consistent with a Michaelis–Menten mechanism in which decreased ATP binding occurs at lower concentrations of the high-energy cofactor. The ATP-dependence of relaxation and supercoiling indicate that both reactions are catalyzed by gyrase. Furthermore, given that standard relaxation/supercoiling reactions were carried out in the presence of 1.5 mM ATP, it is unlikely that differences in rates of relaxation versus supercoiling reflect differential requirements for ATP.
A previous study by Nöllmann et al. compared the abilities of E. coli gyrase to remove positive supercoils and introduce negative supercoils into relaxed DNA (63). On the basis of single-molecule experiments, these authors proposed a mechanochemical model in which the enzyme displayed similar rates for both of these activities at low levels of mechanical force on the DNA substrate [such as those seen in plasmid DNA or in vivo (63,67)]. Indeed, when extrapolated to a 0 pN force on the DNA, <15% difference in reaction rates was predicted. In contrast to their model, Nöllmann et al. observed a greater difference in ensemble experiments: the removal of positive supercoils occurred at rates that were ∼2- to 4-fold faster than those for the introduction of negative supercoils. It was only at increased levels of force in single-molecule experiments, which presumably impaired the ability of E. coli gyrase to wrap DNA and introduce negative supercoils, that substantial differences between the rate of positive supercoil removal and negative supercoil induction were observed.
The reported differences in the relaxation and supercoiling rates at zero force are lower than those seen in the present work with B. anthracis gyrase (>10-fold; see Figure 2). However, unlike our experiments, Nöllmann et al. included spermidine in all of their assays (63). It is notable that this polyamine is often added to gyrase-mediated reactions to enhance rates of DNA supercoiling (63,68–70). Therefore, we examined the effects of 5 mM spermidine, a typical concentration used in gyrase assays (69), on the ability of B. anthracis gyrase to relax positive supercoils and introduce negative supercoils into relaxed DNA. For these reactions, the enzyme concentration was decreased 2.5-fold to spread out the time course. The addition of spermidine enhanced the rate of DNA supercoiling at least 3- to 4-fold (Figure 4A and B). However, as seen in the time courses, the polyamine had little effect on the rate of removal of positive supercoils. This observation was supported by 2D gel analysis (Figure 4C and D). The differential effect of spermidine on gyrase-catalyzed relaxation versus supercoiling may reflect the fact that spermidine promotes plectoneme formation and supercoiling (71), which could favor negative supercoil introduction by gyrase over positive supercoil removal.
Figure 4.

Spermidine enhances the rate of negative supercoiling by Bacillus anthracis gyrase but does not affect the rate of removal of positive supercoils. (A) Gyrase activity on positively supercoiled DNA without spermidine. Positively supercoiled [(+)SC] and negatively supercoiled [(−)SC] standards are shown. (B) Gyrase activity on positively supercoiled DNA in the presence of 5 mM spermidine. (C and D) 2D gel analysis of gyrase activity on positively supercoiled DNA. DNA products generated after 2 min in the absence (C) or presence (D) of 5 mM spermidine. Gel images are representative of at least three independent experiments.
Consistent with our finding that spermidine differentially affects the relaxation and supercoiling rates of B. anthracis gyrase, analysis of the ensemble data from Nöllmann et al. (63) indicates that increasing spermidine from 0.2 to 1.8 mM had virtually no effect on the rate of positive supercoil removal but doubled the rate of negative supercoil introduction. Therefore, to determine whether the intrinsic ability of E. coli gyrase to differentially catalyze DNA relaxation versus supercoiling is comparable to that of the Bacillus enzyme, we used an ensemble assay to assess the relaxation of positive supercoils and the subsequent introduction of negative supercoils by E. coli gyrase in the absence of a polyamine. As seen in Figure 5, the E. coli enzyme removed the positive supercoils within 2 min but took at least 20 min to fully negatively supercoil the substrate. This rate differential (>10-fold) is similar to that observed with B. anthracis gyrase (Figure 2). Thus, it appears that the ability of gyrase to remove positive DNA supercoils substantially faster than it introduces negative supercoils is a fundamental characteristic of both the Gram-positive and Gram-negative enzymes.
Single-molecule measurements of positive supercoil relaxation by B. anthracis gyrase
To further explore the rate at which gyrase removes positive supercoils in the absence of a polyamine, we utilized magnetic tweezers (56) to measure B. anthracis gyrase-catalyzed relaxation of positively supercoiled DNA (Figure 6A). The enzyme typically relaxed positively supercoiled DNA in discrete, rapid bursts (Figure 6A and B). The time between bursts was highly variable, leading to a wide distribution of average relaxation rates (Supplementary Figure S2). In contrast to a previous study with E. coli gyrase (63), relaxation rates for the Bacillus enzyme were independent of force (Supplementary Figure S2). The existence of at least two distinct activity modes further contributed to this variability (Figure 6B and C; Supplementary Figure S3). Gyrase removed multiple supercoils either in rapid bursts (‘burst mode’) or at a steady rate (‘steady mode’). Heterogeneous activity is a well-documented aspect of some enzymes observed in single-molecule studies (72) and is a possible source of plasticity for an enzyme that performs multiple necessary functions.
Figure 6.

Single-molecule measurements of Bacillus anthracis gyrase activity. Data are representative of measurements made on five individual DNA tethers on different days. (A) Representative trace of gyrase activity over time using magnetic tweezers. A cartoon of the experimental setup (not to scale) is shown at right: a single DNA molecule (black line) is torsionally constrained by attachment to a slide and manipulated by the attached magnetic bead (gray sphere). Both the upward force (black arrow) on the bead and its rotation (red arrow) are controlled through an externally applied magnetic field. Counterclockwise bead rotation increases the linking number of the DNA, generating positive supercoils. DNA extension (determined by bead height above slide surface) decreases in proportion to plectoneme supercoiling (yellow arrowhead). Results are shown at left for a single DNA molecule that was extended by a constant upward magnetic force of 1 pN. Upon introduction of 50 positive supercoils (red arrowheads), the DNA extension was reduced as a plectoneme formed. Gyrase removed supercoils in a series of discrete steps and the DNA returned to its initial length, initiating the onset of another measurement cycle. (B and C) Gyrase relaxes positive DNA supercoils in two distinct modes of activity. Representative traces in which the enzyme removed multiple supercoils either in rapid bursts (‘burst mode’ relaxation, B) or at a steady rate (‘steady mode’ relaxation, C), are shown. The DNA was under constant tension of 3.5 and 2.2 pN in B and C, respectively. (D and E) Characterization of burst mode relaxation. Distributions include measurements of 382 individual relaxation cycles in which 50 positive supercoils were removed. In each burst event, gyrase rapidly removed four or more supercoils. The burst size distribution (D) fits a single exponential curve. The burst rate distribution (E) fits an inverse gamma function with shape parameter α = 2.1 ± 0.2 and scale parameter β = 114 ± 11 supercoils/s. Error bars represent the square-root of the number of observed events; these errors were accounted for in the determination of the best-fit parameters (±SE).
Bacillus anthracis gyrase relaxed 50 positive supercoils at an average rate of ≥10 supercoils/s in 319 out of 492 measurements (65%) and ≥20 supercoils/s in 110 measurements (22%) (Supplementary Figure S2). This represents a lower bound for the average relaxation rate, because gyrase frequently removed positive supercoils as rapidly as they were introduced, even up to 60 supercoils per second. However, the enzyme often paused, leading to broadly distributed average velocities that are significantly slower than the burst relaxation rate between pauses.
To characterize gyrase relaxation activity in burst mode, we measured enzyme processivity and speed (Figure 6D and E). The mean burst size of 6.2 ± 0.4 supercoils corresponds to three catalytic cycles executed in rapid succession. The burst size distribution was described by a simple exponential decay function, indicating that the number of sequential catalytic cycles is random. The mean burst rate was 107 ± 23 supercoils/s, which is comparable to the rate of positive supercoil production by E. coli DNA polymerase (73). The burst rate distribution was described by an inverse gamma function, as expected for an enzyme taking a finite number of discrete steps (74).
Gyrase relaxed all 50 positive supercoils introduced during each measurement cycle. This strongly suggests that a single enzyme can process the entire plectoneme (Figure 6A) from a unique binding site at its distal end, which potentially favors binding because it is sterically accessible and presents a preferred binding geometry. A similar placement of E. coli topoisomerase IV at the end of the plectoneme has been concluded from single-molecule experiments (75) and visualized by electron microscopy (76).
The characteristics of the relaxation reaction strongly suggest that a single gyrase enzyme was responsible for the relaxation of each entire plectoneme. First, pauses were often observed between relaxation cycles and between individual activity bursts during relaxation of a single plectoneme (Figure 6A and B; Supplementary Figure S3B). These pauses are inconsistent with the binding of multiple gyrase enzymes per plectoneme, as it is highly unlikely that multiple enzymes would pause synchronously. Second, the rates of relaxation in both the burst and steady modes remained constant over the complete relaxation of a given plectoneme (Figure 6 and Supplementary Figure S3). If multiple enzymes were bound per plectoneme, the relaxation rate would be expected to decrease as the plectoneme shortened because fewer enzymes would remain on the supercoiled portion of the tether and be available to relax the DNA.
As further evidence that the observed relaxation was mediated by gyrase, the rate of positive supercoil relaxation as monitored by magnetic tweezers was sensitive to the concentration of ATP and decreased substantially below 100 μM. At 50 μM ATP, virtually no activity was observed (data not shown). In addition, the fact that the DNA substrates could be continually re-coiled rules out the possibility that the bursts of positive supercoil removal were due to a contaminating nuclease or the breaking of the non-covalent bonds between the DNA and the coverslip. Even if there was ‘breathing’ of the bonds between the DNA and the coverslip (i.e. some bonds dissociated but the DNA remained attached to the coverslip), the maximal possible extension change would be <200 nm. As shown in Figure 6 and Supplementary Figure S3, this value is much smaller than the extension changes observed in each reaction cycle.
As discussed above, B. anthracis gyrase relaxed positive supercoils in at least two modes of activity (Figure 6B and C). The prevalence of the burst mode (319 out of 492 measurements) suggests that it is at least as frequent as the steady mode. Both modes were observed on independent DNA tethers at different force and across samples prepared on different days. The burst size distribution (Figure 6E) points to an underlying stochastic process, which is inconsistent with the regular repetition of single steps and short pauses that would constitute a steady velocity. Thus, the burst and steady modes are recognizably different. Furthermore, switching between these modes was observed even within uninterrupted relaxation events (i.e. the relaxation mode changed without pausing in between) (Supplementary Figure S3C), suggesting dynamic switching between relaxation modes by a single enzyme.
Gyrase requires a wrapping mechanism to efficiently remove positive supercoils
Clearly, gyrase must use a classic DNA-wrapping mechanism in order to introduce negative supercoils. However, on the basis of single-molecule studies, Nöllmann et al. suggested that E. coli gyrase was able to remove positive supercoils by two different strand passage mechanisms depending on the force on the DNA (63). This is despite the fact that the reaction rates remained relatively constant over the entire force range examined (∼0.1–4.5 pN). They proposed that, at low tension, the enzyme acts through the wrapping mechanism, in which only proximal DNA segments are captured. Conversely, they hypothesized that high tension on the DNA prevents wrapping, causing the enzyme to switch to a canonical type II topoisomerase mechanism that favors capture of distal segments. However, these authors did not address this issue experimentally. Thus, it is not obvious how gyrase achieves its rapid rates of positive supercoil relaxation in plasmid DNA [i.e. under conditions of no force (63,67)]. Indeed, canonical type II enzymes that do not wrap DNA, such as human topoisomerase IIα and E. coli topoisomerase IV, have been shown to rapidly remove positive supercoils (27,49,74,75). Thus, gyrase could potentially use a canonical mechanism to efficiently relax overwound DNA. Alternatively, it could rely on the wrapping mechanism for this activity, as positively supercoiled DNA would be an ideal substrate for generating a positive wrap around the GyrA C-terminal domain.
To distinguish between these possibilities, we recapitulated a previously described E. coli GyrA mutant (17) in B. anthracis. In this construct, GyrAAla-box, the seven amino acids of the GyrA-box were replaced with alanine residues. The GyrA-box is necessary for DNA wrapping by gyrase and mutation or removal of this motif in E. coli gyrase resulted in an enzyme that was no longer able to introduce negative supercoils into DNA (17), similar to a gyrase construct in which the entire GyrA C-terminal domain was removed (16). As seen in Figure 7A and B, B. anthracis GyrAAla-box gyrase was unable to convert relaxed to negatively supercoiled DNA and, like canonical type II topoisomerases, gained the ability to relax negatively supercoiled DNA in the presence of ATP. On the basis of these findings, we conclude that disrupting the GyrA-box of B. anthracis gyrase restricts the enzyme to using a canonical type II topoisomerase strand passage mechanism.
Figure 7.

Bacillus anthracis GyrAAla-box gyrase acts as a canonical type II topoisomerase. (A) GyrAAla-box gyrase does not supercoil relaxed DNA. Relaxed (Rel) and negatively supercoiled [(−)SC] DNA standards are shown. (B) GyrAAla-box gyrase slowly relaxes (−)SC DNA. (C) GyrAAla-box gyrase slowly and distributively relaxes positively supercoiled [(+)SC] DNA. Gel images are representative of at least three independent experiments.
As shown in Figure 7C, GyrAAla-box gyrase could still remove positive supercoils, but acted more distributively and much more slowly than the wild-type enzyme. The mutant enzyme took at least 45 min to fully relax overwound DNA (as compared to <2 min for wild-type gyrase, see Figure 2). While these results demonstrate that gyrase can remove positive supercoils using a canonical mechanism at low force, they strongly suggest that the enzyme uses a wrapping mechanism to achieve high rates of processive positive supercoil removal in plasmid DNA.
It is notable that the GyrAAla-box mutant maintains higher levels of DNA cleavage complexes than does the wild-type enzyme (see Figures 9 and 10). As determined by its co-migration with an EcoRI-cleaved DNA standard, the lowest band seen in Figure 7A–C is enzyme-generated linear DNA.
Figure 9.

Bacillus anthracis gyrase maintains lower levels of cleavage complexes on positively supercoiled DNA. (A) Levels of cleavage complexes generated by gyrase on positively supercoiled [(+)SC] DNA (white) or negatively supercoiled [(−)SC] DNA (black) in the presence of ciprofloxacin. Inset: levels of cleavage complexes generated by gyrase in the presence of 10 μM moxifloxacin (Moxi) or levofloxacin (Levo). (B) Levels of cleavage complexes generated by varying concentrations of gyrase on (+)SC DNA (white) or (−)SC DNA (black) in the absence of quinolones. Error bars represent standard deviations for at least three independent experiments.
Figure 10.

Bacillus anthracis GyrAAla-box gyrase maintains lower levels of cleavage complexes on positively supercoiled DNA. (A) Levels of cleavage complexes generated by GyrAAla-box gyrase on positively supercoiled [(+)SC] DNA (white) or negatively supercoiled [(−)SC] DNA (black) in the presence of ciprofloxacin. (B) Levels of cleavage complexes generated by GyrAAla-box gyrase on (+)SC DNA (white) or (−)SC DNA (black) in the absence of quinolones. Error bars represent standard deviations for at least three independent experiments.
Finally, because gyrase cannot remove negative supercoils in the presence of ATP (6), it is impossible to determine whether the wild-type enzyme has an intrinsic ability to distinguish supercoil geometry during strand passage. However, because GyrAAla-box gyrase can relax both substrates under parallel conditions, the mutant enzyme was used to investigate this issue. Although the distributive nature of these reactions makes them difficult to quantify, it is clear from Figure 7B and C that GyrAAla-box gyrase removes positive supercoils from plasmid DNA faster than negative supercoils. Therefore, even in the absence of DNA wrapping, B. anthracis gyrase displays an innate capacity to recognize supercoil geometry during strand passage.
Bacillus anthracis topoisomerase IV recognizes supercoil geometry during strand passage
Previous studies indicate that some, but not all, canonical type II topoisomerases are able to discern the geometry of DNA supercoils during the strand passage reaction. For example, human topoisomerase IIα, but not topoisomerase IIβ or Chlorella virus topoisomerase II, relaxes positively supercoiled DNA ∼10-fold faster than comparably negatively supercoiled plasmid (49,77,78). Furthermore, topoisomerase IV from E. coli also removes positive supercoils ≥10-fold faster (27,75,79). In the case of topoisomerase IV, the difference between rates reflects (at least in part) the ability of the enzyme to carry out a processive reaction on overwound DNA, while its reaction on underwound molecules is distributive.
To determine whether topoisomerase IV from a Gram-positive bacterium can also discern DNA supercoil geometry during strand passage, we examined the ability of B. anthracis topoisomerase IV to remove positive and negative supercoils (Figure 8). As determined by the loss of supercoiled DNA, the enzyme relaxed overwound DNA ∼3-fold faster than comparably underwound molecules. Therefore, we propose that the ability to recognize supercoil geometry during DNA relaxation is conserved among topoisomerase IV from different species. However, there are subtle differences between B. anthracis topoisomerase IV and the E. coli enzyme. First, the disparity in the rates of relaxation of positively versus negatively supercoiled DNA is smaller. Second, the Gram-positive enzyme acted processively on both substrates, which may partly account for the smaller difference in relaxation rates (49,75).
Figure 8.

Bacillus anthracis topoisomerase IV relaxes positively supercoiled DNA faster than negatively supercoiled DNA. (A and B) Relaxation of positively (A) and negatively (B) supercoiled DNA by topoisomerase IV. Positively supercoiled [(+)SC], negatively supercoiled [(−)SC] and relaxed (Rel) standards are shown. Gel images are representative of at least three independent experiments. (C) Quantification of experiments shown in (A) and (B). Relative amounts of relaxed DNA in each experiment were determined by monitoring the loss of the supercoiled band in comparison to supercoiled DNA present in the 0 min sample. Error bars represent standard deviations for at least three independent experiments.
Gyrase can distinguish supercoil geometry during DNA scission and maintains lower levels of cleavage complexes with positively supercoiled DNA
Cleavage complexes formed ahead of replication forks and other DNA tracking systems are most likely to be converted to non-ligatable DNA strand breaks (12,43–45). Because these systems overwind the DNA as they open the double helix, we compared the abilities of B. anthracis and E. coli gyrase to cleave positively supercoiled DNA. Quinolone antibacterials were included in most reactions because of their clinical importance and because they substantially raise levels of cleavage complexes, facilitating quantification (8,31–33).
Bacillus anthracis gyrase recognizes DNA supercoil geometry during cleavage. In the presence of ciprofloxacin, the enzyme maintained ∼3-fold lower levels of cleavage complexes on positively supercoiled as compared with negatively supercoiled DNA (Figure 9A). This geometry recognition also occurred in the presence of two other clinically relevant quinolones, moxifloxacin and levofloxacin (Figure 9A, inset), and in the absence of drug (Figure 9B).
To determine whether DNA wrapping is necessary for gyrase to distinguish supercoil geometry during DNA cleavage, we compared the ability of GyrAAla-box gyrase to cleave positively and negatively supercoiled DNA (Figure 10). Similar to the wild-type enzyme, GyrAAla-box gyrase maintained lower levels of cleavage complexes on positively supercoiled DNA in the presence or absence of ciprofloxacin. Therefore, as was the case for strand passage, the intrinsic ability of gyrase to recognize DNA supercoil geometry during cleavage is independent of wrapping.
We next assessed DNA cleavage mediated by E. coli gyrase to determine whether the ability to discern supercoil geometry during cleavage is a general feature of this type II topoisomerase. Like the Bacillus enzyme, E. coli gyrase also maintained 2- to 3-fold lower levels of cleavage complexes on positively supercoiled DNA (Figure 11).
Figure 11.
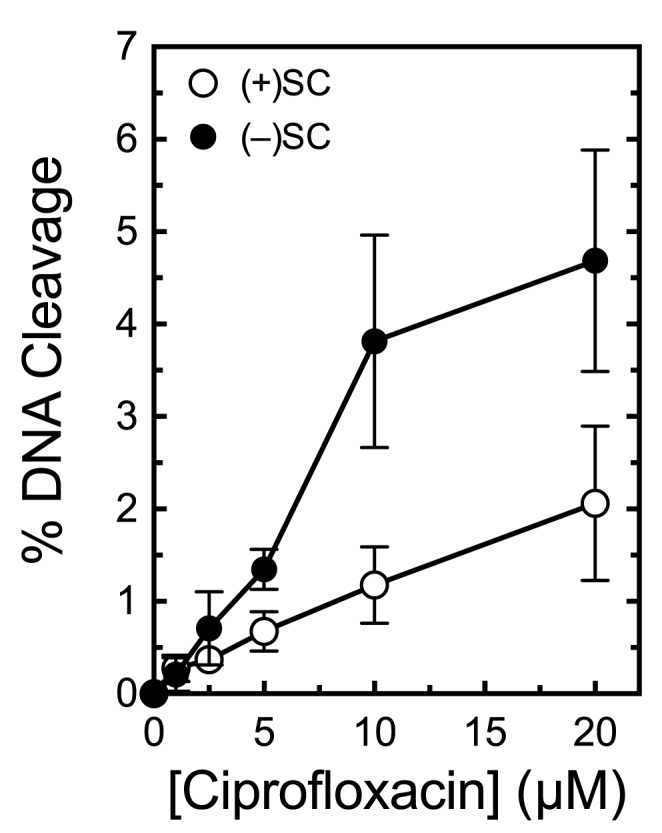
Escherichia coli gyrase maintains lower levels of cleavage complexes on positively supercoiled DNA. Levels of cleavage complexes generated by gyrase on positively supercoiled [(+)SC] DNA (white) or negatively supercoiled [(−)SC] DNA (black) in the presence of ciprofloxacin are shown. Error bars represent standard deviations for at least three independent experiments.
Topoisomerase IV maintains similar levels of cleavage complexes with positively and negatively supercoiled DNA
Results with gyrase are similar to those observed for eukaryotic and viral type II topoisomerases (43,49,77,78,80). To determine whether topoisomerase IV can also discern supercoil geometry during DNA scission, we compared the ability of B. anthracis topoisomerase IV to cleave overwound and underwound plasmid in the presence of ciprofloxacin. As seen in Figure 12A, the enzyme displayed little ability to distinguish between positively and negatively supercoiled substrates and maintained similar levels of cleavage complexes with both.
Figure 12.

Topoisomerase IV maintains similar levels of cleavage complexes on positively and negatively supercoiled DNA. (A) Levels of cleavage complexes generated by Bacillus anthracis topoisomerase IV on positively supercoiled [(+)SC] DNA (white) or negatively supercoiled [(−)SC] DNA (black) in the presence of ciprofloxacin. (B) Levels of cleavage complexes generated by Escherichia coli topoisomerase IV on (+)SC DNA (white) or (−)SC DNA (black) in the presence of ciprofloxacin. Error bars represent standard deviations for at least three independent experiments.
These results are markedly different than those previously reported for E. coli topoisomerase IV (27). An earlier study reported that E. coli topoisomerase IV maintained ∼20-fold higher levels of cleavage complexes on positively supercoiled DNA in the presence of a quinolone (27). Although this disparity may represent divergence between Gram-positive and Gram-negative species, it could also reflect differences in the methodologies used in the present and previous studies. The earlier work used a competition assay that monitored simultaneous cleavage of one negatively and one positively supercoiled plasmid of different lengths. In contrast, we monitored cleavage in parallel experiments that utilized the same plasmid molecules that were equally, but oppositely, supercoiled. Because competition experiments can be inordinately influenced by enzyme–DNA binding affinities, off rates, etc., we re-examined the ability of E. coli topoisomerase IV to discern supercoil geometry using independent DNA cleavage assays. As seen in Figure 12B, results with the E. coli enzyme were similar to those observed with B. anthracis topoisomerase IV. If anything, E. coli topoisomerase IV maintained slightly higher levels of cleavage complexes on negatively supercoiled substrates.
DISCUSSION
Even though the bacterial chromosome is globally underwound by ∼6% (4,81–83), localized regions of overwound DNA are generated by critical nucleic acid processes. For example, DNA becomes positively supercoiled ahead of advancing replication forks, transcription complexes and DNA helicases (1,84–87). In bacterial cells, positive supercoils are removed by gyrase, possibly aided by topoisomerase IV and quinolone-stabilized cleavage complexes most likely to cause permanent genomic damage are formed on overwound DNA (12,43–45). Despite the essential roles of these enzymes on positively supercoiled DNA, little is known about the actions of gyrase or topoisomerase IV on overwound substrates. Results of the present study provide novel insights into these critical enzyme activities.
Although gyrase is typically described as the ‘only known topoisomerase able to generate negative supercoiling’ (11), B. anthracis and E. coli gyrase actually remove positive supercoils considerably faster than they introduce negative supercoils into relaxed DNA. This likely reflects the cellular requirement for rapid removal of positive supercoils during replication.
The two critical cellular functions of gyrase are bounded by distinct temporal constraints. The ‘acute function’ of the enzyme is to remove positive supercoils that rapidly accumulate immediately ahead of replication forks and other DNA tracking systems. This localized activity must occur within a short window of time in order to allow these essential nucleic acid processes to continue. Therefore, it requires a high enzymatic rate. In contrast, the ‘steady-state function’ of gyrase is to maintain the proper superhelical density of the bacterial chromosome. Because local overwinding that occurs as a result of individual nucleic acid events has a relatively minor effect on global supercoiling, the maintenance of topological homeostasis can take place over a protracted time scale.
Despite the fact that gyrase removes positive supercoils much more quickly than it introduces negative supercoils into relaxed DNA, both reactions appear to utilize the same wrapping mechanism to achieve optimal rates. Why, then, does relaxation take place so much faster than supercoiling? One possible explanation is that the relaxation of positively supercoiled DNA is energetically favorable, whereas the introduction of negative supercoils is energetically unfavorable (4). Alternatively, DNA crossovers formed by positive supercoils display higher stability than juxtapositions formed by negative supercoils and therefore could be preferentially used by the enzyme (88). Finally, overwound DNA should intrinsically provide gyrase with the positive supercoils needed to wrap around the C-terminal domain of GyrA. In contrast, when acting on relaxed or negatively supercoiled DNA, the enzyme must first induce a positive supercoil before continuing through its cycle. This necessity could negatively impact both the rate and the processivity of the strand passage reaction.
On the basis of the above, we propose that positively supercoiled DNA is actually the preferred substrate for gyrase. This raises an interesting teleological question: did the initial evolutionary pressure to develop the wrapping mechanism stem from the need to introduce negative supercoils or to remove positive supercoils? Although it is impossible to definitively resolve this issue, we suggest that the latter may be the case. Replication in bacterial systems takes place via two forks moving away from a single origin, each at a rate of 500–1000 nt/s (73). This is in contrast to DNA replication in human cells, in which tens of thousands of origins fire and replication forks move at a much slower rate of ∼30 nt/s (89). As a result, bacterial cells have a unique requirement for a topoisomerase that can remove positive supercoils at a high rate. Conversion of gyrase to a canonical type II topoisomerase by mutating the GyrA-box dramatically reduced the rate of positive supercoil removal. Furthermore, single-molecule experiments indicate that the maximal rate of positive supercoil relaxation by gyrase is several-fold faster than that of topoisomerase IV (27,75,79). Therefore, it appears that a canonical type II enzyme may not be able to act rapidly enough to keep up with the bacterial replication fork. As eukaryotic species evolved, the accompanying slower rates of fork movement, coupled with the emergence of multiple topoisomerases (both type I and type II) that were able to relax positive supercoils, may have dispensed with the need for such a rapidly acting enzyme.
The wrapping mechanism developed for positive supercoil removal concomitantly conferred gyrase with the ability to introduce negative supercoils. As discussed above, this activity plays a critical role in maintaining the topological state of the bacterial chromosome. The requirement for globally underwound DNA endured in eukaryotes, but the advent of histones and higher-order chromatin structures obviated the requirement for an enzyme with an intrinsic ability to underwind DNA.
Although gyrase and topoisomerase IV both catalyze strand passage faster with positively supercoiled substrates, they differ in their ability to distinguish supercoil handedness during DNA cleavage. Whereas gyrase maintains lower levels of cleavage complexes on overwound DNA, topoisomerase IV maintains similar levels of these complexes with over- and underwound molecules. The recognition of DNA geometry by gyrase allows it to act as a safer enzyme ahead of the replication fork. The inability of topoisomerase IV to discern DNA supercoil handedness during cleavage may be a less important consideration, because the enzyme works primarily behind the fork. Thus, the individual abilities of these enzymes to recognize DNA geometry make them well suited for their unique physiological roles.
Finally, these different abilities may impact the efficacy of gyrase and topoisomerase IV as targets for quinolone antibacterials. Functioning ahead of a fork, gyrase is perfectly positioned to create cleavage complexes with the potential to be converted to permanent DNA damage. However, the diminished levels of cleavage complexes generated by the enzyme on positively supercoiled DNA may partially abrogate the cytotoxic effects of quinolones. Conversely, topoisomerase IV maintains high levels of cleavage complexes on overwound substrates, but typically acts behind the fork, where cleavage complexes are less likely to be disrupted by moving tracking systems. Ultimately, it is probable that the physiological locations and the recognition of DNA geometry by gyrase and topoisomerase IV both contribute to the complex relationship between quinolone targeting in purified systems and drug lethality in bacterial cells.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Ethan Tyler of NIH Medical Arts for design work on Figure 1. We also thank Elizabeth G. Gibson and Lorena Infante Lara for critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US Veterans Administration Merit Review Award [I01 Bx002198 to N.O.]; National Institutes of Health (NIH) [GM033944 to N.O., AI81775 to C.L.T.]; Intramural Research Program of the National Heart, Lung and Blood Institute at the National Institutes of Health [HL001056 to K.C.N.]; National Science Foundation [DGE-0909667 to R.E.A.]. Funding for open access charge: NIH [GM033944 to N.O.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang J.C. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 2002; 3:430–440. [DOI] [PubMed] [Google Scholar]
- 2. Koster D.A., Crut A., Shuman S., Bjornsti M.A., Dekker N.H.. Cellular strategies for regulating DNA supercoiling: a single-molecule perspective. Cell. 2010; 142:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vos S.M., Tretter E.M., Schmidt B.H., Berger J.M.. All tangled up: how cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011; 12:827–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bates A.D., Maxwell A.. DNA Topology. 2005; NY: Oxford University Press. [Google Scholar]
- 5. Liu Z., Deibler R.W., Chan H.S., Zechiedrich L.. The why and how of DNA unlinking. Nucleic Acids Res. 2009; 37:661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levine C., Hiasa H., Marians K.J.. DNA gyrase and topoisomerase IV: biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys. Acta. 1998; 1400:29–43. [DOI] [PubMed] [Google Scholar]
- 7. Sissi C., Palumbo M.. In front of and behind the replication fork: bacterial type IIA topoisomerases. Cell. Mol. Life Sci. 2010; 67:2001–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Anderson V.E., Osheroff N.. Type II topoisomerases as targets for quinolone antibacterials: turning Dr. Jekyll into Mr. Hyde. Curr. Pharm. Des. 2001; 7:337–353. [DOI] [PubMed] [Google Scholar]
- 9. Bush N.G., Evans-Roberts K., Maxwell A.. DNA topoisomerases. EcoSal Plus. 2015; 6, doi:10.1128/ecosalplus.ESP-0010-2014. [DOI] [PubMed] [Google Scholar]
- 10. Kato J., Nishimura Y., Imamura R., Niki H., Hiraga S., Suzuki H.. New topoisomerase essential for chromosome segregation in E. coli. Cell. 1990; 63:393–404. [DOI] [PubMed] [Google Scholar]
- 11. Champoux J.J. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 2001; 70:369–413. [DOI] [PubMed] [Google Scholar]
- 12. Deweese J.E., Osheroff N.. The DNA cleavage reaction of topoisomerase II: wolf in sheep's clothing. Nucleic Acids Res. 2009; 37:738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu L.F., Wang J.C.. Micrococcus luteus DNA gyrase: active components and a model for its supercoiling of DNA. Proc. Natl. Acad. Sci. U.S.A. 1978; 75:2098–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu L.F., Wang J.C.. DNA-DNA gyrase complex: the wrapping of the DNA duplex outside the enzyme. Cell. 1978; 15:979–984. [DOI] [PubMed] [Google Scholar]
- 15. Reece R.J., Maxwell A.. The C-terminal domain of the Escherichia coli DNA gyrase A subunit is a DNA-binding protein. Nucleic Acids Res. 1991; 19:1399–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kampranis S.C., Maxwell A.. Conversion of DNA gyrase into a conventional type II topoisomerase. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:14416–14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kramlinger V.M., Hiasa H.. The "GyrA-box" is required for the ability of DNA gyrase to wrap DNA and catalyze the supercoiling reaction. J. Biol. Chem. 2006; 281:3738–3742. [DOI] [PubMed] [Google Scholar]
- 18. Basu A., Parente A.C., Bryant Z.. Structural dynamics and mechanochemical coupling in DNA gyrase. J. Mol. Biol. 2016; 428:1833–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ullsperger C., Cozzarelli N.R.. Contrasting enzymatic acitivites of topoisomerase IV and DNA gyrase from Escherichia coli. J. Biol. Chem. 1996; 271:31549–31555. [DOI] [PubMed] [Google Scholar]
- 20. Marians K.J. DNA gyrase-catalyzed decatenation of multiply linked DNA dimers. J. Biol. Chem. 1987; 262:10362–10368. [PubMed] [Google Scholar]
- 21. Brown P.O., Cozzarelli N.R.. A sign inversion mechanism for enzymatic supercoiling of DNA. Science. 1979; 206:1081–1083. [DOI] [PubMed] [Google Scholar]
- 22. Khodursky A.B., Peter B.J., Schmid M.B., DeRisi J., Botstein D., Brown P.O., Cozzarelli N.R.. Analysis of topoisomerase function in bacterial replication fork movement: use of DNA microarrays. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:9419–9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tadesse S., Graumann P.L.. Differential and dynamic localization of topoisomerases in Bacillus subtilis. J. Bacteriol. 2006; 188:3002–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hsu Y.H., Chung M.W., Li T.K.. Distribution of gyrase and topoisomerase IV on bacterial nucleoid: implications for nucleoid organization. Nucleic Acids Res. 2006; 34:3128–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hiasa H., Marians K.J.. Topoisomerase IV can support oriC DNA replication in vitro. J. Biol. Chem. 1994; 269:16371–16375. [PubMed] [Google Scholar]
- 26. Zechiedrich E.L., Khodursky A.B., Bachellier S., Schneider R., Chen D., Lilley D.M., Cozzarelli N.R.. Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J. Biol. Chem. 2000; 275:8103–8113. [DOI] [PubMed] [Google Scholar]
- 27. Crisona N.J., Strick T.R., Bensimon D., Croquette V., Cozzarelli N.R.. Preferential relaxation of positively supercoiled DNA by E. coli topoisomerase IV in single-molecule and ensemble measurements. Genes Dev. 2000; 14:2881–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang X., Reyes-Lamothe R., Sherratt D.J.. Modulation of Escherichia coli sister chromosome cohesion by topoisomerase IV. Genes Dev. 2008; 22:2426–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Joshi M.C., Magnan D., Montminy T.P., Lies M., Stepankiw N., Bates D.. Regulation of sister chromosome cohesion by the replication fork tracking protein SeqA. PLoS Genet. 2013; 9:e1003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zawadzki P., Stracy M., Ginda K., Zawadzka K., Lesterlin C., Kapanidis A.N., Sherratt D.J.. The localization and action of topoisomerase IV in Escherichia coli chromosome segregation is coordinated by the SMC complex, MukBEF. Cell Rep. 2015; 13:2587–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Drlica K., Hiasa H., Kerns R., Malik M., Mustaev A., Zhao X.. Quinolones: action and resistance updated. Curr. Top. Med. Chem. 2009; 9:981–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aldred K.J., Kerns R.J., Osheroff N.. Mechanism of quinolone action and resistance. Biochemistry. 2014; 53:1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hooper D.C., Jacoby G.A.. Mechanisms of drug resistance: quinolone resistance. Ann. N. Y. Acad. Sci. 2015; 1354:12–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khodursky A.B., Zechiedrich E.L., Cozzarelli N.R.. Topoisomerase IV is a target of quinolones in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:11801–11805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aedo S., Tse-Dinh Y.-C.. Isolation and quantitation of topoisomerase complexes accumulated on Escherichia coli chromosomal DNA. Antimicrob. Agents Chemother. 2012; 56:5458–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pan X.-S., Fisher L.M.. Targeting of DNA gyrase in Streptococcus pneumoniae by sparfloxacin: selective targeting of gyrase or topoisomerase IV by quinolones. Antimicrob. Agents Chemother. 1997; 41:471–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pan X.-S., Fisher L.M.. Streptococcus pneumoniae DNA gyrase and topoisomerase IV: overexpression, purification, and differential inhibition by fluoroquinolones. Antimicrob. Agents Chemother. 1999; 43:1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pan X.-S., Ambler J., Mehtar S., Fisher L.M.. Involvement of topoisomerase IV and DNA gyrase as ciprofloxacin targets in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 1996; 40:2321–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fournier B., Zhao X., Lu T., Drlica K., Hooper D.C.. Selective targeting of topoisomerase IV and DNA gyrase in Staphylococcus aureus: different patterns of quinolone-induced inhibition of DNA synthesis. Antimicrob. Agents Chemother. 2000; 44:2160–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brook I., Elliott T.B., Pryor H.I. 2nd, Sautter T.E., Gnade B.T., Thakar J.H., Knudson G.B.. In vitro resistance of Bacillus anthracis Sterne to doxycycline, macrolides and quinolones. Int. J. Antimicrob. Agents. 2001; 18:559–562. [DOI] [PubMed] [Google Scholar]
- 41. Price L.B., Vogler A., Pearson T., Busch J.D., Schupp J.M., Keim P.. In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob. Agents Chemother. 2003; 47:2362–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grohs P., Podglajen I., Gutmann L.. Activities of different fluoroquinolones against Bacillus anthracis mutants selected in vitro and harboring topoisomerase mutations. Antimicrob. Agents Chemother. 2004; 48:3024–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McClendon A.K., Osheroff N.. The geometry of DNA supercoils modulates topoisomerase-mediated DNA cleavage and enzyme response to anticancer drugs. Biochemistry. 2006; 45:3040–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu L.F., D’Arpa P.. Topoisomerase-targeting antitumor drugs: mechanisms of cytotoxicity and resistance. Important Adv. Oncol. 1992; 1992:79–89. [PubMed] [Google Scholar]
- 45. Li T.K., Liu L.F.. Tumor cell death induced by topoisomerase-targeting drugs. Annu. Rev. Pharmacol. Toxicol. 2001; 41:53–77. [DOI] [PubMed] [Google Scholar]
- 46. Dong S., McPherson S.A., Wang Y., Li M., Wang P., Turnbough C.L. Jr, Pritchard D.G.. Characterization of the enzymes encoded by the anthrose biosynthetic operon of Bacillus anthracis. J. Bacteriol. 2010; 192:5053–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hardin A.H., Sarkar S.K., Seol Y., Liou G.F., Osheroff N., Neuman K.C.. Direct measurement of DNA bending by type IIA topoisomerases: implications for non-equilibrium topology simplification. Nucleic Acids Res. 2011; 39:5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Corbett K.D., Schoeffler A.J., Thomsen N.D., Berger J.M.. The structural basis for substrate specificity in DNA topoisomerase IV. J. Mol. Biol. 2005; 351:545–561. [DOI] [PubMed] [Google Scholar]
- 49. McClendon A.K., Rodriguez A.C., Osheroff N.. Human topoisomerase IIα rapidly relaxes positively supercoiled DNA: implications for enzyme action ahead of replication forks. J. Biol. Chem. 2005; 280:39337–39345. [DOI] [PubMed] [Google Scholar]
- 50. Rodriguez A.C. Studies of a positive supercoiling machine: nucleotide hydrolysis and a multifunctional "latch" in the mechanism of reverse gyrase. J. Biol. Chem. 2002; 277:29865–29873. [DOI] [PubMed] [Google Scholar]
- 51. Aldred K.J., Breland E.J., Vlčková V., Strub M.P., Neuman K.C., Kerns R.J., Osheroff N.. Role of the water-metal ion bridge in mediating interactions between quinolones and Escherichia coli topoisomerase IV. Biochemistry. 2014; 53:5558–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Aldred K.J., McPherson S.A., Wang P., Kerns R.J., Graves D.E., Turnbough C.L., Osheroff N.. Drug interactions with Bacillus anthracis topoisomerase IV: biochemical basis for quinolone action and resistance. Biochemistry. 2012; 51:370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aldred K.J., Blower T.R., Kerns R.J., Berger J.M., Osheroff N.. Fluoroquinolone interactions with Mycobacterium tuberculosis gyrase: enhancing drug activity against wild-type and resistant gyrase. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E839–E846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Seol Y., Neuman K.C.. Single-molecule measurements of topoisomerase activity with magnetic tweezers. Methods Mol. Biol. 2011; 778:229–241. [DOI] [PubMed] [Google Scholar]
- 55. Ribeck N., Saleh O.A.. Multiplexed single-molecule measurements with magnetic tweezers. Rev. Sci. Instrum. 2008; 79:094301. [DOI] [PubMed] [Google Scholar]
- 56. Strick T.R., Allemand J.F., Bensimon D., Croquette V.. Behavior of supercoiled DNA. Biophys. J. 1998; 74:2016–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Marko J.F., Neukirch S.. Competition between curls and plectonemes near the buckling transition of stretched supercoiled DNA. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2012; 85:011908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. van Loenhout M.T., de Grunt M.V., Dekker C.. Dynamics of DNA supercoils. Science. 2012; 338:94–97. [DOI] [PubMed] [Google Scholar]
- 59. Higgins N.P., Peebles C.L., Sugino A., Cozzarelli N.R.. Purification of subunits of Escherichia coli DNA gyrase and reconstitution of enzymatic activity. Proc. Natl. Acad. Sci. U.S.A. 1978; 75:1773–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mizuuchi K., O’Dea M.H., Gellert M.. DNA gyrase: subunit structure and ATPase activity of the purified enzyme. Proc. Natl. Acad. Sci. U.S.A. 1978; 75:5960–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kampranis S.C., Bates A.D., Maxwell A.. A model for the mechanism of strand passage by DNA gyrase. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:8414–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gore J., Bryant Z., Stone M.D., Nollmann M., Cozzarelli N.R., Bustamante C.. Mechanochemical analysis of DNA gyrase using rotor bead tracking. Nature. 2006; 439:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nollmann M., Stone M.D., Bryant Z., Gore J., Crisona N.J., Hong S.C., Mitelheiser S., Maxwell A., Bustamante C., Cozzarelli N.R.. Multiple modes of Escherichia coli DNA gyrase activity revealed by force and torque. Nat. Struct. Mol. Biol. 2007; 14:264–271. [DOI] [PubMed] [Google Scholar]
- 64. Papillon J., Menetret J.F., Batisse C., Helye R., Schultz P., Potier N., Lamour V.. Structural insight into negative DNA supercoiling by DNA gyrase, a bacterial type 2A DNA topoisomerase. Nucleic Acids Res. 2013; 41:7815–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gubaev A., Klostermeier D.. The mechanism of negative DNA supercoiling: a cascade of DNA-induced conformational changes prepares gyrase for strand passage. DNA Repair (Amst). 2014; 16:23–34. [DOI] [PubMed] [Google Scholar]
- 66. Gubaev A., Weidlich D., Klostermeier D.. DNA gyrase with a single catalytic tyrosine can catalyze DNA supercoiling by a nicking-closing mechanism. Nucleic Acids Res. 2016; 44:10354–10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zechiedrich E.L., Cozzarelli N.R.. Roles of topoisomerase IV and DNA gyrase in DNA unlinking during replication in Escherichia coli. Genes Dev. 1995; 9:2859–2869. [DOI] [PubMed] [Google Scholar]
- 68. Bates A.D., Maxwell A.. DNA gyrase can supercoil DNA circles as small as 174 base pairs. EMBO J. 1989; 8:1861–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Williams N.L., Howells A.J., Maxwell A.. Locking the ATP-operated clamp of DNA gyrase: probing the mechanism of strand passage. J. Mol. Biol. 2001; 306:969–984. [DOI] [PubMed] [Google Scholar]
- 70. Hockings S.C., Maxwell A.. Identification of four GyrA residues involved in the DNA breakage-reunion reaction of DNA gyrase. J. Mol. Biol. 2002; 318:351–359. [DOI] [PubMed] [Google Scholar]
- 71. Shao Q., Goyal S., Finzi L., Dunlap D.. Physiological levels of salt and polyamines favor writhe and limit twist in DNA. Macromolecules. 2012; 45:3188–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu B., Baskin R.J., Kowalczykowski S.C.. DNA unwinding heterogeneity by RecBCD results from static molecules able to equilibrate. Nature. 2013; 500:482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McCarthy D., Minner C., Bernstein H., Bernstein C.. DNA elongation rates and growing point distributions of wild-type phage T4 and a DNA-delay amber mutant. J. Mol. Biol. 1976; 106:963–981. [DOI] [PubMed] [Google Scholar]
- 74. Seol Y., Gentry A.C., Osheroff N., Neuman K.C.. Chiral discrimination and writhe-dependent relaxation mechanism of human topoisomerase IIα. J. Biol. Chem. 2013; 288:13695–13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Neuman K.C., Charvin G., Bensimon D., Croquette V.. Mechanisms of chiral discrimination by topoisomerase IV. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:6986–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vologodskii A.V., Zhang W., Rybenkov V.V., Podtelezhnikov A.A., Subramanian D., Griffith J.D., Cozzarelli N.R.. Mechanism of topology simplification by type II DNA topoisomerases. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:3045–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. McClendon A.K., Gentry A.C., Dickey J.S., Brinch M., Bendsen S., Andersen A.H., Osheroff N.. Bimodal recognition of DNA geometry by human topoisomerase IIα: preferential relaxation of positively supercoiled DNA requires elements in the C-terminal domain. Biochemistry. 2008; 47:13169–13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McClendon A.K., Dickey J.S., Osheroff N.. Ability of viral topoisomerase II to discern the handedness of supercoiled DNA: bimodal recognition of DNA geometry by type II enzymes. Biochemistry. 2006; 45:11674–11680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stone M.D., Bryant Z., Crisona N.J., Smith S.B., Vologodskii A., Bustamante C., Cozzarelli N.R.. Chirality sensing by Escherichia coli topoisomerase IV and the mechanism of type II topoisomerases. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:8654–8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lindsey R.H. Jr, Pendleton M., Ashley R.E., Mercer S.L., Deweese J.E., Osheroff N.. Catalytic core of human topoisomerase IIα: insights into enzyme-DNA interactions and drug mechanism. Biochemistry. 2014; 53:6595–65602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bauer W.R., Crick F.H., White J.H.. Supercoiled DNA. Sci. Am. 1980; 243:100–113. [PubMed] [Google Scholar]
- 82. White J.H., Cozzarelli N.R.. A simple topological method for describing stereoisomers of DNA catenanes and knots. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:3322–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vologodskii A.V., Cozzarelli N.R.. Conformational and thermodynamic properties of supercoiled DNA. Annu. Rev. Biophys. Biomol. Struct. 1994; 23:609–643. [DOI] [PubMed] [Google Scholar]
- 84. Schvartzman J.B., Stasiak A.. A topological view of the replicon. EMBO Rep. 2004; 5:256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Liu L.F., Wang J.C.. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:7024–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wu H.Y., Shyy S.H., Wang J.C., Liu L.F.. Transcription generates positively and negatively supercoiled domains in the template. Cell. 1988; 53:433–440. [DOI] [PubMed] [Google Scholar]
- 87. Peter B.J., Ullsperger C., Hiasa H., Marians K.J., Cozzarelli N.R.. The structure of supercoiled intermediates in DNA replication. Cell. 1998; 94:819–827. [DOI] [PubMed] [Google Scholar]
- 88. Timsit Y. Local sensing of global DNA topology: from crossover geometry to type II topoisomerase processivity. Nucleic Acids Res. 2011; 39:8665–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fangman W.L., Brewer B.J.. A question of time: replication origins of eukaryotic chromosomes. Cell. 1992; 71:363–366. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.