

Abstract
Hypoxia and reoxygenation, ischemia and reperfusion, catecholamine infusion, ouabain, sodium pentobarbital and caffeine, can all be used experimentally to induce ventricular arrhythmias. According to the Lambeth Convention guidelines our experimentally-induced ventricular arrhythmias include but are not limited to: ventricular premature beats (VPB), ventricular salvos (VS), ventricular bigeminy (VB), nonsustained ventricular tachycardia (VTn), sustained ventricular tachycardia (VTs) and ventricular fibrillation (VF, or if the heart is not defibrillated, sudden cardiac death). We have studied these arrhythmias in the absence and presence of adenosine deaminase, methyl xanthines, and more recently, acetaminophen. Our laboratory was the first to discover the anti-arrhythmic properties of acetaminophen an analgesic used in Western medicine for more than 100 years before our publication. We have also identified other cardioprotective properties of acetaminophen, and have begun to work out some of the cellular/molecular mechanisms. For example, we know that acetaminophen protects hypoxic/ischemic cardiac mitochondria, in part, by sustaining function of the mitochondrial permeability transition pore (MPTP, a protein involved in regulating mitochondrial pH). Acetaminophen also attenuates the actions of matrix metalloproteinases that can be harmful to myocardial contractile proteins. Of course, like all science, more work is needed to expand on these and related topics.
Keywords: Arrhythmias, acetaminophen, hypoxia
Introduction
We have invested considerable effort in studying the coronary circulation and ventricular arrhythmias in mammals [1-10]. These experiments have been done in strips and rings of vascular smooth muscle [11,12], in isolated, perfused hearts [13-20], and in anesthetized rabbits [21] and dogs [22-26]. The motivation to study experimental ventricular arrhythmias was initiated in the laboratories of H. Fred Downey and Carl E. Jones when they were my hosts and colleagues at the Texas College of Osteopathic Medicine, Fort Worth, Texas, 1984-1985. During that sabbatical year we studied the influences of hypoxia and administered norepinephrine on the coronary circulation of anesthetized dogs in the absence and presence of adenosine deaminase [22-24]. Use of adenosine deaminase was an alternative approach to the application of methyl xanthines, antagonists of adenosine receptors, to test the hypothesis that adenosine participates in the chemical regulation of coronary blood flow.
As we documented the ability of adenosine deaminase to attenuate coronary blood flow responses to endogenous adenosine, I couldn’t help notice that the enzyme also reduced ventricular arrhythmias caused by hypoxia and norepinephrine. Since returning to Rutgers University in 1985 we have periodically been investigating experimentally-induced ventricular arrhythmias in my laboratory. Our experiments focused on antiarrhythmic and cardioprotective properties of several compounds including: acetaminophen, adenosine deaminase, estrogen, and histamine antagonists.
Currently, we are collecting preliminary data to determine if caffeine, a methyl xanthine and adenosine receptor blocker, predisposes the heart and circulation to arrhythmias and other harmful cardiovascular effects.
Methods and results
Hypoxia with and without adenosine deaminase
Our initial experiments using hypoxia and adenosine deaminase were performed in anesthetized, instrumented mongrel dogs. Dogs were exposed to dual periods of hypoxia, one in the absence and the other in the presence of intracoronary infusions of adenosine deaminase. Coronary blood flow responses and ventricular arrhythmias during a few minutes of severe hypoxia (e.g. 3-5 per cent oxygen in nitrogen), followed by reoxygenation (21 per cent oxygen in nitrogen), were monitored then compared statistically under the two conditions. Adenosine deaminase routinely and significantly attenuated the coronary vasodilation and ventricular arrhythmias caused by hypoxia (Figure 1). We concluded that adenosine plays an important role in the blood flow responses to hypoxia, can contribute to the ventricular arrhythmias of hypoxia, and that adenosine deaminase can be used to test these hypotheses [22-24].
Figure 1.
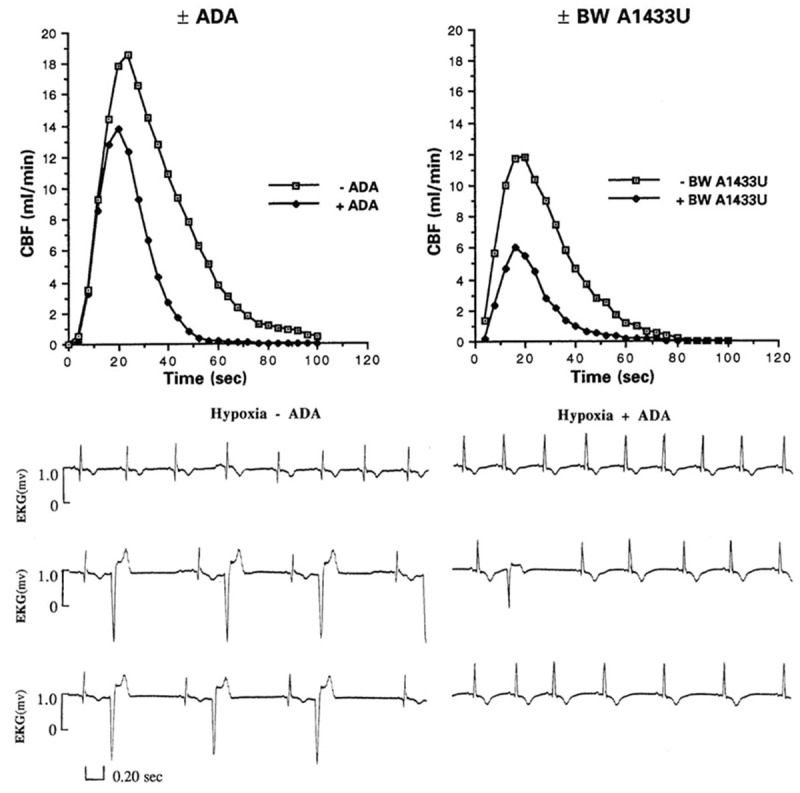
Effects of systemic hypoxia (3% O2, 5% CO2, Bal N2) on cardiac rhythm (Lead I) in the absence (-ADA) and presence (+ADA) of adenosine deaminase (5 U/Kg/min, i.c.). Top row, one minute of hypoxia, middle row 2 minutes of hypoxia, and bottom row, 3 minutes of hypoxia.
Ischemia with and without estrogen
A few years later one of my graduate students became interested in hormone replacement therapy and cardiovascular disease. She decided to investigate the effects of Premarin (conjugated equine estrogen) on ventricular arrhythmias caused by ischemia/reperfusion injury. This was an important question because in the mid-1990s hormone replacement therapy was a debated topic in the clinical treatment of post-menopausal women. Again, anesthetized, instrumented dogs were used. After baseline data were collected, regions of the myocardium perfused by the left anterior descending coronary artery (LAD) were made ischemic for several minutes then reperfused. This was done in the absence and presence of Premarin. Premarin induced some coronary vasomotion in dogs but successfully attenuated the ventricular arrhythmias caused by ischemia/reperfusion injury (Figures 2, 3) [27-30].
Figure 2.
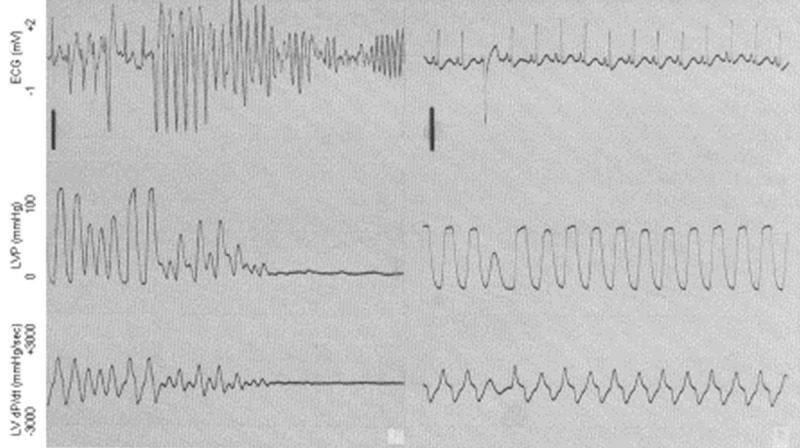
Reperfusion-induced ventricular arrhythmias (including ventricular tachycardia and fibrillation, at left) in the absence and presence (right panel) of conjugated equine estrogen (Premarin). Reperfusion was initiated in both panels at the arrows in the top row (ECG). Note the immediate onset of ventricular tachycardia followed by fibrillation in the absence of estrogen. Modified from McHugh, AJP Heart 268: H2569-H2573, 1995.
Figure 3.
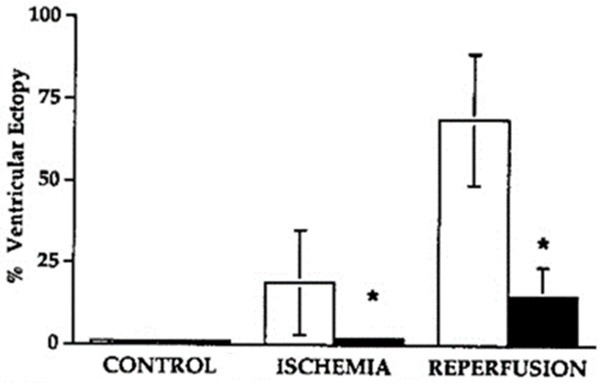
Influence of conjugated equine estrogen (Premarin) on per cent ectopy (black bars) in anesthetized, instrumented dogs during post-ischemia reperfusion. Note the marked and significant reduction in ventricular arrhythmias with estrogen. Modified from McHugh et al, AJP Heart: 268: H2569-H2573, 1995.
Ischemia with and without acetaminophen
In the early 2000s while using isolated, perfused guinea pig hearts (Langendorff preparation) we discovered salutary, cardiovascular effects of acetaminophen that had never been reported [3-7]. These include the abilities of the analgesic to significantly attenuate mechanical and electrical damage caused by myocardial ischemia and reperfusion and other insults. The benefits were seen whether acetaminophen was administered acutely [3-6] or chronically [7]. When our experiments were later extended to anesthetized, instrumented dogs we discovered similar results [2,29] i.e. acetaminophen has cardioprotective properties in both in vitro [3-7] and in vivo preparations [2,26].
For example, Langendorff-perfused hearts never regained complete mechanical ventricular function after a period of 20 minutes of low-flow myocardial ischemia in the presence of vehicle. However, in the presence of acetaminophen, similarly-treated hearts regained complete ventricular mechanical function (e.g. developed left ventricular pressure) within 20-40 minutes of reperfusion [4-10]. In dogs, acetaminophen significantly reduced the size of an infarct caused by occlusion of a major branch of the left anterior descending (LAD) coronary artery [26]. It also reduced the area at risk of further damage beyond the initial insult. Both ischemia and reperfusion and hypoxia and reoxygenation cause potentially-lethal ventricular arrhythmias and acetaminophen significantly attenuated these in the dog [2].
Caffeine and the human cardiovascular system
There is growing and broad concern that caffeine and caffeinated-beverages are harmful if not potentially-lethal in the mammalian cardiovascular system [31-36], see 32, 33 for reviews. In our laboratory we are collecting preliminary data on the question in college-age young single adults. In these experiments volunteers come to the laboratory and are instrumented to collect data on the electrocardiogram (ECG), heart rate (HR), blood flow (finger plethysmography) and blood pressure (cardiomicrophonic analyses of brachial arterial pulsatile blood pressure). This information is collected using modern data acquisition systems (PowerLab model 8/35, ADInstruments, Colorado Springs, CO), physiological transducers, and a PC desktop computer running LabChart software (v.8.0, 2016, ADInstruments, Colorado Springs, CO).
After instrumentation volunteers lie quietly in the supine position on an examination bed for 15-20 minutes while recorded variables achieve their respective steady states (data collected each five minutes until the last two sets of data do not differ). Pre-experimental, baseline data are then collected. The volunteer then quickly ingests a favorite caffeinated beverage (coffee or tea, energy drink, their choice; 220 ± 85 mg), and post-ingestion data are collected at 15 minute intervals for 60 minutes. Lights are dimmed and there is no conversation during the experiment.
We are in the early stages of this investigation. However, from Tables 1 and 2, and Figures 4, 5 and 6, one can see selected, preliminary results. These include: a) consistent reductions in peripheral blood flow within 15-30 minutes post-ingestion of caffeine, b) unstable wandering isoelectric lines in the ECG, c) elevated, high-amplitude T waves, and d) periodic deviations of the ST-segment. We have not yet investigated P-R intervals, QT or QTc segments, or other ECG characteristics. I should also note that these data were collected in volunteer subjects who regularly consume caffeinated beverages (see Table 1).
Table 1.
Personal estimates of caffeine consumption (milligrams) in young adults
| Consume irregularly <100 mg/wk | Consume regularly >100 mg/wk | |||
|---|---|---|---|---|
|
|
||||
| Per day | Per week | Per day | Per week | |
| n | 10 | 10 | 15 | 15 |
| Mean | 6 | 43 | 132 | 926 |
| s.e.m. | 2 | 12 | 34 | 239 |
| t-test | P<0.05 | P<0.05 | ||
n, sample size; mean, average milligrams consumed each day and each week; s.e.m., standard error of the mean; t-test, Students t-test for unequal sample sizes with unequal variances. Data above these P values are significantly different from corresponding data in columns labelled ‘consume irregularly’.
Table 2.
Age (years) volunteers began consuming caffeinated beverages. Also, times of day they begin/end consuming
| Consume irregularly (<100 mg per week) | Consume regularly (>100 mg per week) | |||
|---|---|---|---|---|
| n | 10 | 15 | ||
| Mean | 12±2 years | 12±1 years | ||
| Range | 5-17 years | 4-16 years | ||
| Time of day | 7:30 a.m. | 8:00 p.m. | 7:00 a.m. | 11:00 p.m. |
| Begin | End | Begin | End | |
n, sample size; mg, estimated milligrams of caffeine consumed per day/week.
Figure 4.
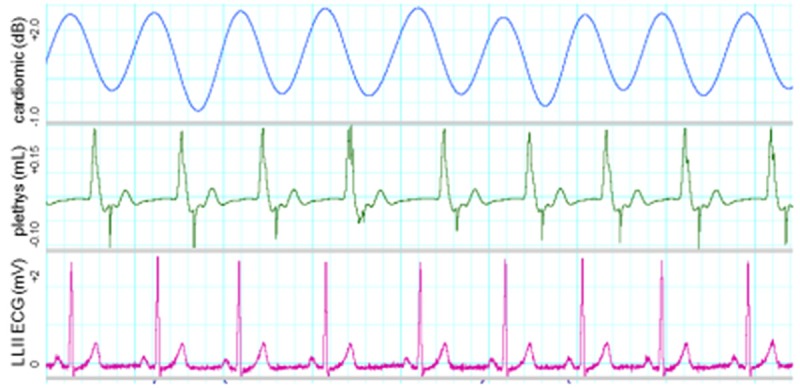
Sample waveforms of one young man who consumes no caffeinated beverages (age 25 years at the time these data were collected). Note the amplitudes of T waves relative to those of QRS complexes.
Figure 5.
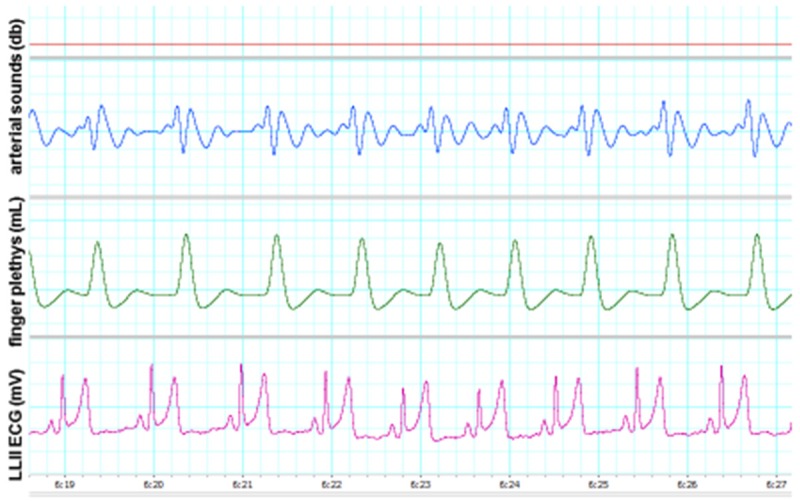
One college-age young adult who consumes caffeinated beverages regularly. Note the amplitudes of T waves compared with those of QRS complexes (i.e. high amplitude T waves). Also note that the ordinate has the same scale as in Figure 4 above.
Figure 6.
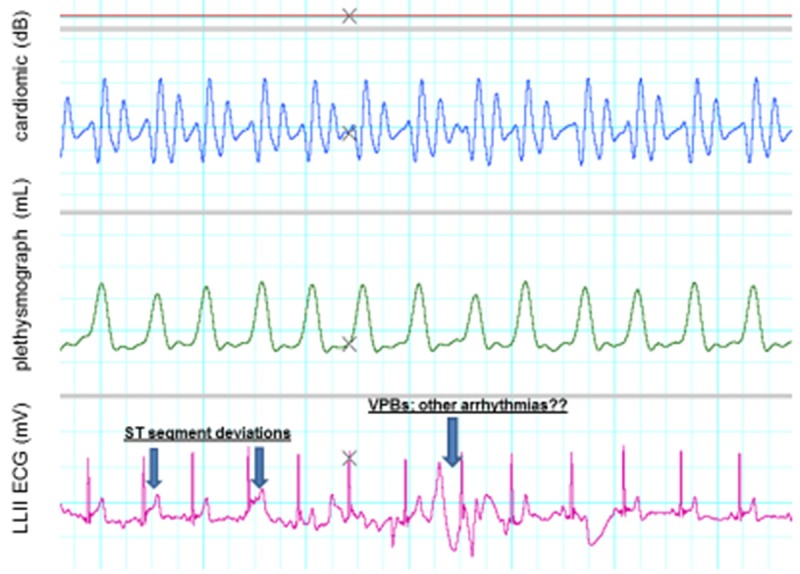
ECG rhythm disturbances and ST changes in a young adult who was conditioned at the time but who consumes caffeinated beverages regularly. Note the elevated ST-segments (first and second arrows). Also note the ventricular premature beat (third arrow). Ordinate has same values as in Figure 4 above.
For comparative purposes Figure 4 is an illustration of a normal ECG in a healthy young adult who does not consume caffeinated beverages (e.g. note the absence of large T waves, ST-segment deviations and unstable isoelectric lines). Figures 5 and 6 illustrate changes in these and other variables in two young adults who consume caffeinated beverages.
Discussion
Before becoming interested in ventricular arrhythmias we spent most of our effort investigating the coronary circulation of mammals [11-25]. Our experiments were done in isolated, perfused Langendorff hearts [13-20], in ex vivo rings and strips of vascular smooth muscle [11,12], and in anesthetized animals [21-26]. From these data we learned, like others before us, that adenosine, histamine, hypercapnia, hypoxia, calcium and potassium channels all influence the coronary vasculature. For example, we found that the pH of physiological perfusate solutions (i.e. indicative of the acid/base environment of the vasculature and interstitial spaces in our preparations) markedly influenced coronary vascular responses to administered adenosine [13]. An acidic pH increased coronary vascular sensitivity to adenosine while a basic pH attenuated it [13]. We also reported that the vascular responsiveness to adenosine, potassium (hyperkalemia) and hypoxia were pH sensitive [14,16,17]. These findings have contributed to subsequent observations including the study of ATP-sensitive potassium channels. Doubtless, more interactions among the above agents and other chemical/metabolic players are still awaiting discovery.
Experimental ventricular arrhythmias
Our interest in experimentally-induced ventricular arrhythmias arose in the mid-1980s and was re-stimulated in the early 2000s. During the period 1984-1985 I spent a sabbatical year studying with H. Fred Downey of the University of Texas Health Sciences Center/Methodist Hospital in Dallas. In the early months of that collaboration, Carl E. Jones, Chair, Department of Physiology, Texas College of Osteopathic Medicine (now part of the North Texas State University of the Health Sciences Center, Denton, TX) recruited Fred Downey to join his physiology faculty in Fort Worth, TX. From that point the three of us enjoyed a fruitful collaboration and enduring friendships. I have always felt indebted to Fred and Carl for the many things I learned from them and their colleagues, and for making me feel like an important member of their departments and activities.
In our experiments in Texas we exposed anesthetized, instrumented dogs to short bouts of severe hypoxia (3% oxygen in air) in the absence and presence of adenosine deaminase [22,23]. Adenosine deaminase is the naturally-occurring enzyme that removes the amine group (NH2) from adenosine producing inosine, a physiologically-inert compound. We also administered intra-coronary boluses of adenosine to compare coronary vascular responses to hypoxia (endogenous adenosine) and exogenous adenosine [22,23]. The enzyme significantly attenuated the coronary vasodilation caused by both of these interventions, as well as those caused by administered norepinephrine [24].
About ten years earlier I had used administered adenosine (250 µg, i.c.) to produce cardiac arrest and maximal coronary vasodilation in isolated, perfused guinea pig hearts (Langendorff) [13]. This amount of adenosine in that experimental preparation causes cardiac arrest within a few cardiac cycles after administration. The cardiac arrest lasts only several seconds but is concurrently accompanied by maximal coronary vasodilation [13]. Quite often adenosine-induced cardiac arrest is preceded by ventricular arrhythmias of the kind defined by the Lambeth Convention guidelines [1].
In the Downey Laboratory I noticed that a few minutes of hypoxia also produced ventricular arrhythmias of the kind defined at Lambeth [1]. These ventricular arrhythmias were consistently attenuated by administered adenosine deaminase. So, not only did endogenous adenosine contribute to the coronary vasodilation of systemic hypoxia in the dog, it also seemed to contribute to ventricular arrhythmogenesis. These findings were consistent with my observations in the mid-1970s using bolus injections of adenosine in perfused guinea pig hearts [11].
Upon returning to my laboratory at Rutgers University, we confirmed our Texas findings by using an adenosine receptor blocker, 8-phenyltheophylline, in similarly-instrumented dogs exposed to systemic hypoxia [25]. In the latter dogs we controlled coronary blood flow at a constant rate during hypoxia, simultaneously measured coronary perfusion pressure, and subsequently calculated changes in coronary vascular resistance with and without 8-phenyltheophylline. In those experiments, 8-phenyltheophylline completely abolished the vasodilation caused by hypoxia.
In my laboratory we continued confirming our earlier findings by exposing isolated, perfused guinea pig hearts to global myocardial hypoxia in the absence and presence of adenosine deaminase [20]. Once again, adenosine deaminase not only attenuated the coronary vasodilation, it also attenuated the hypoxia-induced ventricular arrhythmias.
Acetaminophen and cardioprotection
I took another sabbatical in 1996-97 (Brigham Young University, Provo, UT) and this time returned to an empty (no graduate students) and unfunded laboratory at Rutgers University. In those years my first success at finding extramural funding was through Johnson & Johnson of New Brunswick (COSAT) and McNeil Consumer Healthcare of Fort Washington, PA. They funded our proposal to identify potential cardiovascular effects of acetaminophen in the mammalian myocardium.
In our initial experiments we could not find evidence of direct coronary vascular actions of the monophenol (performed in the isolated, perfused guinea pig heart). However, upon extraction, we observed that acetaminophen-treated hearts stabilized more rapidly than did placebo-treated hearts whether the drug was administered acutely or chronically [2-10]. This was our first clue that acetaminophen possesses salutary, cardioprotective properties that had not previously been reported.
In those experiments our early approach was to examine global, low-flow ischemia and reperfusion, as well as mild-to-severe global hypoxia and reoxygenation in the absence and presence of acetaminophen. In both cases, acetaminophen was cardioprotective [3-7,10]. Moreover, we found that the cardioprotection occurred at the cellular and subcellular levels. For example, in the presence of acetaminophen, hypoxia/ischemia-induced swelling of mitochondria was attenuated or abolished. Moreover, mechanical effects of matrix metalloproteases on contractile proteins inside the cell were inhibited [10], and function of the mitochondrial permeability transition pore (MPTP) at the mitochondrial inner/outer membranes was preserved [9].
Following these experiments in isolated guinea pig hearts, we moved to more physiological, in vivo experiments in dogs and humans. In dog experiments we found that acetaminophen significantly reduced the size of a myocardial infarction (as well as the zone at risk) [26] and was antiarrhythmic [2].
Caffeine and the cardiovascular system
Coffee, after water, is the most-widely consumed liquid in the United States, and is the principal source of intake of caffeine among adults. Coffee is a complex solution containing hundreds of pharmacologically-active compounds, and the health effects of chronic intake of coffee are wide-ranging and incompletely known [31-38].
Caffeine is the most-widely consumed drug and psychoactive substance in the world, with coffee and tea representing its main routes of administration [31-38]. Indeed it can be argued that caffeine is the drug of choice for abuse by the world’s adult population. The physiological mechanism by which caffeine exerts its systemic effects is now largely accepted to be through competitive inhibition of adenosine receptors [33,34,38]. A1 adenosine receptors are more closely associated with neurological effects [33,34,37], and A2A adenosine receptor antagonism leads to cardiovascular effects, including peripheral vasoconstriction and reductions in coronary and cerebral blood flow [13,25,31,32,35,36,38].
In preliminary experiments in young adults we have observed the following cardiovascular events several minutes after ingestion of caffeine: a) elevated, high-amplitude T waves (ECG), b) wandering, unstable isoelectric lines (ECG), c) deviations in the ST-segment (ECG), d) ventricular premature beats (ECG, see Figures 4, 5 and 6), and e) sustained peripheral vasoconstriction with reduced blood flow. This study is not yet complete and the possible advantages of consuming coffee regularly have to be weighed against the risks, which are primarily due to its high content of caffeine.
References
- 1.Walker MJ, Curtis MJ, Hearse DJ, Campbell RW, Janse MJ, Yellon DM, Cobbe SM, Coker SJ, Harness JB, Harron DW, et al. The Lambeth Conventions: guidelines for the study of arrhythmias in ischemia, infarction, and reperfusion. Cardiovasc Res. 1988;22:447–455. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- 2.Merrill GF, Merrill JH, Hadzimichalis N, Baliga S, Jaques K, Golfetti R, Rork T. Antiarrhythmic properties of acetaminophen in the dog. Exp Biol Med. 2007;232:1245–1252. doi: 10.3181/0701-RM-19. [DOI] [PubMed] [Google Scholar]
- 3.Golfetti R, Rork T, Merrill GF. Chronically-administered acetaminophen and the ischemia/reperfused myocardium. Exp Biol Med. 2003;228:674–682. doi: 10.1177/153537020322800605. [DOI] [PubMed] [Google Scholar]
- 4.Golfetti R, VanDyke K, Rork T, Spiler N, Merrill GF. Acetaminophen in the post-ischemia, reperfused myocardium. Exp Biol Med. 2002;227:1031–1038. doi: 10.1177/153537020222701112. [DOI] [PubMed] [Google Scholar]
- 5.Merrill GF. Acetaminophen and low-flow myocardial ischemia: efficacy and antioxidant mechanisms. Am J Physiol Heart. 2002;282:H1341–H1349. doi: 10.1152/ajpheart.00716.2001. [DOI] [PubMed] [Google Scholar]
- 6.Merrill GF, VanDyke K, McConnell P, Powell SR. Coronary and myocardial effects of acetaminophen: protection during ischemia and reperfusion. Am J Physiol Heart. 2001;280:H2631–H2638. doi: 10.1152/ajpheart.2001.280.6.H2631. [DOI] [PubMed] [Google Scholar]
- 7.Merrill GF, Goldberg E. Antioxidant properties of acetaminophen and cardioprotection. Bas Res Cardiol. 2001;96:422–429. doi: 10.1007/s003950170023. [DOI] [PubMed] [Google Scholar]
- 8.Jaques-Robinson KM, Golfetti R, Baliga SS, Hadzimichalis NM, Merrill GF. Acetaminophen is cardioprotective against H2O2-induced injury in vivo . Exp Biol Med. 2008;233:1315–1322. doi: 10.3181/0802-RM-68. [DOI] [PubMed] [Google Scholar]
- 9.Hadzimichalis NM, Baliga SS, Golfetti R, Jaques KM, Firestein B, Merrill GF. Acetaminophen-mediated cardioprotection via inhibition of the mitochondrial permeability transition pore-induced apoptotic pathway. Am J Physiol Heart. 2007;293:H3348–H3355. doi: 10.1152/ajpheart.00947.2007. [DOI] [PubMed] [Google Scholar]
- 10.Rork TH, Hadzimichalis NM, Merrill GF. Acetaminophen attenuates peroxynitrite-activated matrix metalloproteinase-2-mediated troponin I cleavage in the isolated guinea pig myocardium. J Mol Cell Cardiol. 2006;40:553–561. doi: 10.1016/j.yjmcc.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Young MA, Merrill GF. Comparative effects of adenosine and nifedipine in rabbit vascular smooth muscle. Can J Physiol Pharmacol. 1982;61:1057–1062. doi: 10.1139/y83-157. [DOI] [PubMed] [Google Scholar]
- 12.Tozzi CA, Merrill GF. Differential effects of adenosine and verapamil on histamine-induced vascular contractions. Can J Physiol Pharmacol. 1986;64:679–682. doi: 10.1139/y86-112. [DOI] [PubMed] [Google Scholar]
- 13.Merrill GF, Haddy FJ, Dabney JM. Adenosine, theophylline and perfusate pH in the isolated, perfused guinea pig heart. Circ Res. 1978;42:225–229. doi: 10.1161/01.res.42.2.225. [DOI] [PubMed] [Google Scholar]
- 14.Young MA, Merrill GF. Potassium and hydrogen ion interactions in the vasculature of the isolated, perfused guinea pig heart. Blood Vessels. 1980;17:216–224. doi: 10.1159/000158252. [DOI] [PubMed] [Google Scholar]
- 15.Merrill GF, White JT, Krieger LW. Coronary circulation in hearts from hibernating, normothermic, and cold-acclimated hamsters. Am J Physiol. 1981;241:R50–R54. doi: 10.1152/ajpregu.1981.241.1.R50. [DOI] [PubMed] [Google Scholar]
- 16.Merrill GF, Young MA, Tozzi CA, Grosso PC, Marcus KM. Adenosine dilation and adrenergic constriction of coronary blood vessels: PCO2 effects. Artery. 1982;10:395–411. [PubMed] [Google Scholar]
- 17.Young MA, Merrill GF. Differential effects of adenosine and hypoxia on potassium-induced coronary vasodilation in isolated, perfused guinea pig hearts. Blood Vessels. 1982;19:292–301. doi: 10.1159/000158396. [DOI] [PubMed] [Google Scholar]
- 18.Kang YH, Wei HM, Merrill GF. Histamine-induced changes in coronary circulation and myocardial oxygen consumption as influenced by histamine receptor antagonists. FASEB J. 1987;1:483–489. doi: 10.1096/fasebj.1.6.3678703. [DOI] [PubMed] [Google Scholar]
- 19.Merrill GF, Kang YH, Wei HM, Fisher H. Pressure-dependent vasoactive effects of histamine in the coronary circulation. FASEB J. 1987;1:308–311. doi: 10.1096/fasebj.1.4.3653582. [DOI] [PubMed] [Google Scholar]
- 20.Wei HM, Kang YH, Merrill GF. Adenosine’s role in the coronary vasodilation of global myocardial hypoxia: effects of adenosine deaminase. Am J Physiol. 1988;254:H1004–H1009. doi: 10.1152/ajpheart.1988.254.5.H1004. [DOI] [PubMed] [Google Scholar]
- 21.Tozzi CA, Merrill GF. Histamine and myocardial ischemia in the rabbit: potentiation by atherosclerosis and inhibition by chlorpheniramine and verapamil. Cardiovas Res. 1982;19:744–753. doi: 10.1093/cvr/19.12.744. [DOI] [PubMed] [Google Scholar]
- 22.Merrill GF, Downey HF, Jones CE. Adenosine deaminase attenuates canine coronary vasodilation during systemic hypoxia. Am J Physiol. 1986;250:H579–H583. doi: 10.1152/ajpheart.1986.250.4.H579. [DOI] [PubMed] [Google Scholar]
- 23.Merrill GF, Downey HF, Yonekura S, Watanabe N, Jones CE. Adenosine deaminase attenuates canine coronary vasodilation during regional nonischemic myocardial hypoxia. Cardiovas Res. 1988;22:345–350. doi: 10.1093/cvr/22.5.345. [DOI] [PubMed] [Google Scholar]
- 24.Downey HF, Merrill GF, Yonekura S, Watanabe N, Jones CE. Adenosine deaminase attenuates norepinephrine-induced coronary functional hyperemia. Am J Physiol. 1988;254:H417–H424. doi: 10.1152/ajpheart.1988.254.3.H417. [DOI] [PubMed] [Google Scholar]
- 25.Wei HM, Kang YH, Merrill GF. Canine coronary vasodepressor responses to hypoxia are abolished by 8-phenyltheophylline. Am J Physiol. 1989;257:H1043–H1048. doi: 10.1152/ajpheart.1989.257.4.H1043. [DOI] [PubMed] [Google Scholar]
- 26.Merrill GF, Rork T, Spiler N, Golfetti R. Acetaminophen and myocardial infarction in dogs. Am J Physiol Heart. 2004;287:H1913–H1920. doi: 10.1152/ajpheart.00565.2004. [DOI] [PubMed] [Google Scholar]
- 27.McHugh NA, Cook SW, Schairer JA, Merrill GF. Ischemia and reperfusion induced ventricular arrhythmias in the dog: effects of estrogen. Am J Physiol. 1995;268:H2569–H2573. doi: 10.1152/ajpheart.1995.268.6.H2569. [DOI] [PubMed] [Google Scholar]
- 28.McHugh NA, Merrill GF, Powell SR. Estrogen diminishes postischemic production of hydroxyl radical. Am J Physiol. 1998;274:H1950–H1954. doi: 10.1152/ajpheart.1998.274.6.H1950. [DOI] [PubMed] [Google Scholar]
- 29.McHugh NA, Solowiej A, Klabunde RE, Merrill GF. Acute coronary vascular and myocardial perfusion effects of conjugated equine estrogen in the anesthetized dog. Bas Res Cardiol. 1998;93:470–476. doi: 10.1007/s003950050117. [DOI] [PubMed] [Google Scholar]
- 30.McHugh NA, Solowiej A, Sternberg L, Merrill GF. Cardiac and coronary effects of chronically administered estrogen in the dog. Bas Res Cardiol. 1998;93:116–121. doi: 10.1007/s003950050071. [DOI] [PubMed] [Google Scholar]
- 31.O’Keefe JH, Bhatti SK, Patil HR, DiNicolantonio JJ, Lucan SC, Lavie CJ. Effects of habitual coffee consumption on cardiometabolic disease, cardiovascular health, and all-cause mortality. J Am Coll Cardiol. 2013;62:1043–51. doi: 10.1016/j.jacc.2013.06.035. [DOI] [PubMed] [Google Scholar]
- 32.Leal M, Barletta M, Carson S. Maternal-fetal electrocardiographic effects and pharmacokinetics after an acute i. v. administration of caffeine to the pregnant rat. Reprod Toxicol. 1990;4:105–12. doi: 10.1016/0890-6238(90)90004-f. [DOI] [PubMed] [Google Scholar]
- 33.Fredholm BB, Battig K, Holmen J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev. 1999;51:83–133. [PubMed] [Google Scholar]
- 34.Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- 35.Lee JC, Fraser JF, Barnett AG, Johnson LP, Wilson MG, McHenry CM, Walters DL, Warnholtz CR, Khafagi FA. Effect of caffeine on adenosine-induced reversible perfusion defects assessed by automated analysis. J Nucl Cardiol. 2012;19:474–81. doi: 10.1007/s12350-012-9517-x. [DOI] [PubMed] [Google Scholar]
- 36.Hage FG, Iskandrian AE. The effect of caffeine on adenosine myocardial perfusion imaging: time to reassess? J Nucl Cardiol. 2012;19:415–9. doi: 10.1007/s12350-012-9519-8. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Liu T, Gupta A, Spincemaille P, Nguyen TD, Wang Y. Quantitative mapping of cerebral metabolic rate of oxygen (CMRO2) using quantitative susceptibility mapping (QSM) Magn Reson Med. 2015;74:945–52. doi: 10.1002/mrm.25463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fredholm BB. Cardiovascular and renal actions of methylxanthines. Prog Clin Biol Res Review. 1984;158:303–30. [PubMed] [Google Scholar]