

ABSTRACT
K-Ras must localize to the plasma membrane (PM) for biological activity. We show here that multiple acid sphingomyelinase (ASM) inhibitors, including tricyclic antidepressants, mislocalized phosphatidylserine (PtdSer) and K-RasG12V from the PM, resulting in abrogation of K-RasG12V signaling and potent, selective growth inhibition of mutant K-Ras-transformed cancer cells. Concordantly, in nude mice, the ASM inhibitor fendiline decreased the rate of growth of oncogenic K-Ras-expressing MiaPaCa-2 tumors but had no effect on the growth of the wild-type K-Ras-expressing BxPC-3 tumors. ASM inhibitors also inhibited activated LET-60 (a K-Ras ortholog) signaling in Caenorhabditis elegans, as evidenced by suppression of the induced multivulva phenotype. Using RNA interference against C. elegans genes encoding other enzymes in the sphingomyelin (SM) biosynthetic pathway, we identified 14 enzymes whose knockdown strongly or moderately suppressed the LET-60 multivulva phenotype. In mammalian cells, pharmacological agents that target these enzymes all depleted PtdSer from the PM and caused K-RasG12V mislocalization. These effects correlated with changes in SM levels or subcellular distribution. Selected compounds, including sphingosine kinase inhibitors, potently inhibited the proliferation of oncogenic K-Ras-expressing pancreatic cancer cells. In conclusion, these results show that normal SM metabolism is critical for K-Ras function, which may present therapeutic options for the treatment of K-Ras-driven cancers.
KEYWORDS: K-Ras, sphingomyelin, acid sphingomyelinase
INTRODUCTION
Ras proteins are small guanine nucleotide binding proteins that cycle between inactive GDP-bound and active GTP-bound states. These states are controlled by guanine nucleotide exchange factors that promote Ras activation and GTPase-activating proteins that inactivate Ras. Ras-GTP activates multiple downstream effectors in order to transmit signals for cell proliferation and cell survival (1–4). Missense gain-of-function mutations in three Ras genes have been detected in 15% of all human cancers, with G12, G13, and Q61 being most common (1). Of the three ubiquitously expressed Ras isoforms, H-Ras, N-Ras, and K-Ras, oncogenic mutations in K-Ras are most prevalent, occurring in ∼95% of pancreatic, ∼45% of colorectal, and ∼35% of lung cancers (1).
In order to function, Ras proteins must localize to the inner leaflet of the plasma membrane (PM). This is achieved through a set of three posttranslational modifications of the C-terminal CAAX motif, common to all Ras proteins, that generates a C-terminal cysteine farnesyl carboxy-methyl ester (5). In the case of H-Ras, N-Ras, and K-Ras4A, the farnesylated C-terminal cysteine operates in concert with palmitoylation of adjacent cysteine residues to generate the complete PM anchor, whereas in the case of K-Ras4B (referred to as K-Ras herein), the farnesylated C-terminal cysteine operates in concert with a C-terminal polybasic domain for PM localization (5, 6).
Multiple strategies have been employed to block Ras function in cancer. For example, there was a large effort to block the first step in CAAX processing, farnesylation, which is absolutely required for Ras biological activity (5). However, K-Ras and N-Ras alternatively can be prenylated by geranylgeranyl transferase 1 (GGTase1) when cells are treated with farnesyltransferase inhibitors (FTIs) and anchor efficiently to the PM (7, 8). Since geranylgeranylated oncogenic K-Ras and N-Ras are equally transforming as the cognate farnesylated proteins, FTIs have no value as anti-K-Ras and -N-Ras drugs (9, 10). After the clinical failure of FTIs, Ras drug discovery efforts largely switched toward developing inhibitors of downstream Ras targets, including CRAF, BRAF, phosphatidylinositol 3-kinase, and MEK (11–19). Alternative approaches to anti-K-Ras drug design have included the development of small molecules that covalently modify a specific oncogenic point mutation, G12C, that accounts for 15% of K-Ras mutant tumors (20, 21).
Preventing localization of Ras to the PM completely abrogates biological activity; thus, small molecules with this capacity have significant potential therapeutic value (2, 11, 22, 23). One example is the development of inhibitors of PDEδ, which is required to maintain K-Ras on the PM after attachment of the PM anchor (24). Another example is fendiline, an obsolete L-type calcium channel blocker that mislocalizes both splice variants of K-Ras (4A and 4B) from the PM to endomembranes, disrupts their nanoclustering on the PM, and effectively inhibits the proliferation of K-Ras-positive cancer cells (25, 26). Fendiline mislocalizes K-Ras4A and K-Ras4B by off-target inhibition of acid sphingomyelinase (ASM), an enzyme that hydrolyzes sphingomyelin (SM) to ceramide (Cer) (25, 27). ASM inhibition causes SM loading and aberrant endolysosomal function that in turn depletes the PM of phosphatidylserine (PtdSer) and cholesterol. In this study, we demonstrate that other inhibitors of ASM mimic the effects of fendiline and show that perturbing cellular SM levels through inhibition of many other enzymes in the SM metabolic pathway results in redistribution of both PtdSer and K-Ras. We therefore show that SM metabolism is an important regulator of K-Ras function that can be manipulated for the treatment of various K-Ras-driven cancers.
RESULTS
ASM inhibition redistributes PM PtdSer and mislocalizes K-RasG12V.
We showed previously that fendiline mislocalizes K-Ras from the PM by depleting PtdSer from the inner leaflet of the PM (25, 26). This is a direct consequence of fendiline acting as a functional inhibitor of ASM (28, 29), because supplementing fendiline-treated cells with exogenous ASM rescues K-Ras mislocalization. Therefore, in this study we first examined if other ASM inhibitors, which include the FDA-approved tricyclic antidepressants desipramine, imipramine, and amitriptyline (27, 28), could mimic the effects of fendiline on PtdSer and K-Ras localization. First, we determined the abundance and subcellular distribution of SM in cells treated with tricyclic antidepressants. To visualize SM localization, a nontoxic recombinant fragment of lysenin tagged with green fluorescent protein (GFP-Lys) was incubated with live cells or fixed permeabilized cells. In live control cells, weak GFP-Lys staining was mostly evident on the PM. In the ASM inhibitor-treated cells, GFP-Lys staining was greatly enhanced but largely confined to intracellular vesicular structures (Fig. 1A). Staining of intracellular SM with GFP-Lys following permeabilization was also substantially increased by all of the drugs (Fig. 1B). These data recapitulate results with fendiline, which dose dependently increased total GFP-Lys staining that was also largely confined to intracellular vesicles (25). In contrast, the calcium channel blocker nifedipine, which does not inhibit ASM, had no effect on SM content or distribution, indicating that the effect of fendiline on SM abundance and subcellular localization is independent of its activity as a calcium channel blocker.
FIG 1.

ASM inhibitors cause intracellular accumulation of SM and mislocalize K-RasG12V from the PM. (A) Representative confocal images of wild-type BHK cells treated with vehicle (DMSO), fendiline (10 μM), imipramine (10 μM), desipramine (10 μM), amitriptyline (1 μM), or nifedipine (10 μM) for 48 h and incubated with 20 μg/ml MBP-GFP-lysenin and DAPI for 15 min at room temperature in serum-free medium. (B) Representative confocal images of wild-type BHK cells treated with vehicle (DMSO), fendiline (10 μM), imipramine (10 μM), desipramine (10 μM), amitriptyline (1 μM), or nifedipine (10 μM) for 48 h and fixed, permeabilized, and stained with GFP-lysenin and DAPI. (C) Representative confocal images taken in the green channel of MDCK cells stably coexpressing mCherry-CAAX and mGFP–K-RasG12V treated with vehicle (DMSO), imipramine (10 μM), desipramine (10 μM), amitriptyline (1 μM), or nifedipine (10 μM) for 48 h. (D) K-RasG12V mislocalization was quantified using Manders coefficients as the fraction of mCherry-CAAX colocalizing with mGFP–K-RasG12V. The Manders coefficient for DMSO-treated control was 0.244 ± 0.01.
We next tested whether the tricyclic antidepressants inhibit the PM localization of oncogenic K-Ras. Madin-Darby canine kidney (MDCK) cells stably coexpressing mGFP–K-RasG12V and mCherry-CAAX, a general endomembrane marker (30), were treated with drugs for 48 h and analyzed by confocal microscopy. The extent of K-RasG12V mislocalization was calculated using Manders coefficients, which quantify the fraction of mCherry-CAAX colocalizing with mGFP–K-RasG12V. The greater the value of the Manders coefficient, the more extensive the displacement of mGFP–K-RasG12V from the PM. The results show that treatment with tricyclic antidepressants dose dependently mislocalized K-Ras from the PM, with amitriptyline being the most potent (Fig. 1C and D). To quantify the effect of ASM inhibitors on K-RasG12V nanoclustering, intact basal PM sheets from MDCK cells expressing mGFP–K-RasG12V treated with drugs for 24 h were labeled with gold-conjugated anti-GFP antibodies and analyzed by electron microscopy (EM). Spatial mapping of K-RasG12V on the PM revealed significant decreases in the peak values of the L(r) − r clustering statistic, Lmax (Fig. 2A), indicating a reduction in the amount of nanoclustered K-RasG12V on the PM. We also observed a significant reduction in anti-GFP immunogold labeling after ASM inhibitor treatment, indicating that the tricyclic antidepressants significantly deplete K-Ras from the inner leaflet of the PM (Fig. 2B), concordant with the confocal imaging results (Fig. 1C and D).
FIG 2.
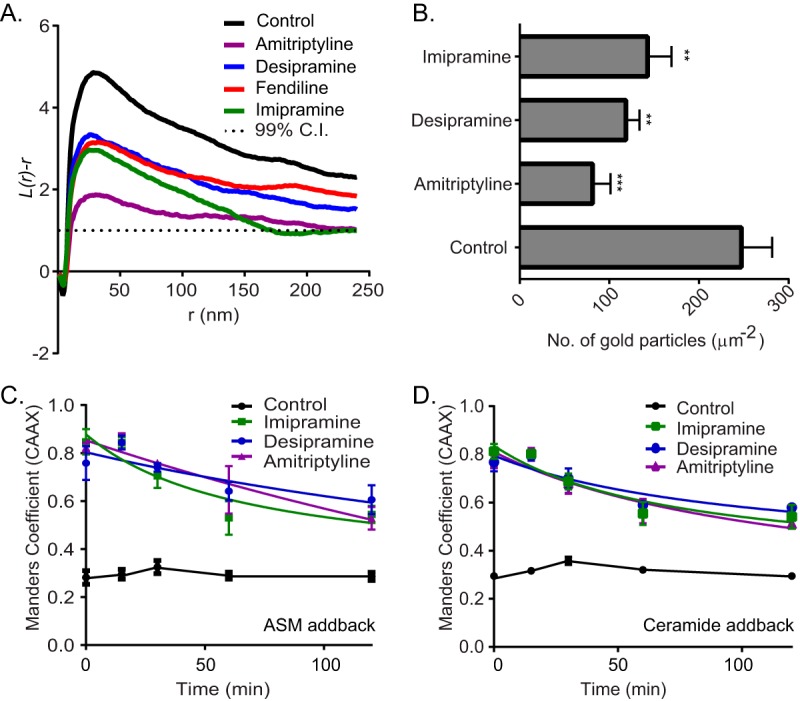
ASM inhibitors reduce PM binding and nanoclustering of K-RasG12V. (A) Basal PM sheets prepared from MDCK cells expressing mGFP–K-RasG12V after treatment with vehicle (DMSO), imipramine (10 μM), desipramine (10 μM), or amitriptyline (1 μM) for 24 h were labeled with anti-GFP-conjugated gold particles. The spatial distribution of the gold particles visualized by EM was analyzed using univariate K functions. Plots of the weighted mean standardized univariate K functions are shown. (B) The graph shows mean numbers of gold particles in PM sheets ± standard errors of the means (n = 20). Significant differences were assessed by using Student's t tests (**, P < 0.01; ***, P < 0.001). (C and D) MDCK cells stably coexpressing mGFP–K-RasG12V and mCherry-CAAX were treated with vehicle (DMSO), imipramine (10 μM), desipramine (10 μM), or amitriptyline (1 μM) for 48 h and then incubated with recombinant ASM or 10 μM Cer in the continued presence of drugs and imaged by confocal microscopy. Cells were fixed at various time points, and PM localization of K-RasG12V was quantified by using Manders coefficients after imaging with a confocal microscope.
To confirm that K-Ras mislocalization occurred as a consequence of ASM inhibition, we supplemented ASM inhibitor-treated cells with exogenous recombinant ASM. This supplementation technique works because ASM is taken up by endocytosis and macropinocytosis and is delivered to the lumen of the late endolysosomal system, where it operates. One direct consequence of ASM inhibition is a reduction in the cellular levels of Cer. We therefore also analyzed the effects of acutely supplementing ASM inhibitor-treated cells with exogenous Cer. MDCK cells were imaged at different time points, and the mislocalization of K-RasG12V was quantified using Manders coefficients. As shown in Fig. 2C and D, ASM or Cer supplementation partially corrected the mislocalization of K-RasG12V from the PM induced by the ASM inhibitors. These results recapitulate earlier observations with fendiline (25).
Fendiline causes K-Ras mislocalization by depleting PtdSer from the inner leaflet of the PM (25). To determine if the same mechanism operates with the tricyclic antidepressants, MDCK cells stably coexpressing mCherry-CAAX and mGFP-LactC2, a probe for PtdSer (30), were treated with the drugs for 48 h and analyzed by quantitative confocal microscopy. The results show that the tricyclic antidepressants disrupt the PM localization of mGFP-LactC2 and, by inference PtdSer, did so with potencies (measured as 50% inhibitory concentrations [IC50s]) very similar to their respective potencies for mislocalizing K-RasG12V from the PM (Fig. 3A and B). EM analysis also showed reduced PM PtdSer content on the inner leaflet of the PM, measured as immunogold labeling density for GFP-LactC2 in the drug-treated cells; the spatial organization of PtdSer was also disrupted (Fig. 3C). Similar to the effect of fendiline, all of the ASM inhibitors also depleted the PM of cholesterol, as evidenced by mislocalization of mCherry-D4H, a cholesterol probe, from the PM to intracellular vesicular structures (Fig. 3D). Note that in contrast to fendiline, nifedipine, a calcium channel blocker that does not inhibit ASM, had no effect on K-RasG12V or LactC2 PM localization (Fig. 1A to C and 3A).
FIG 3.
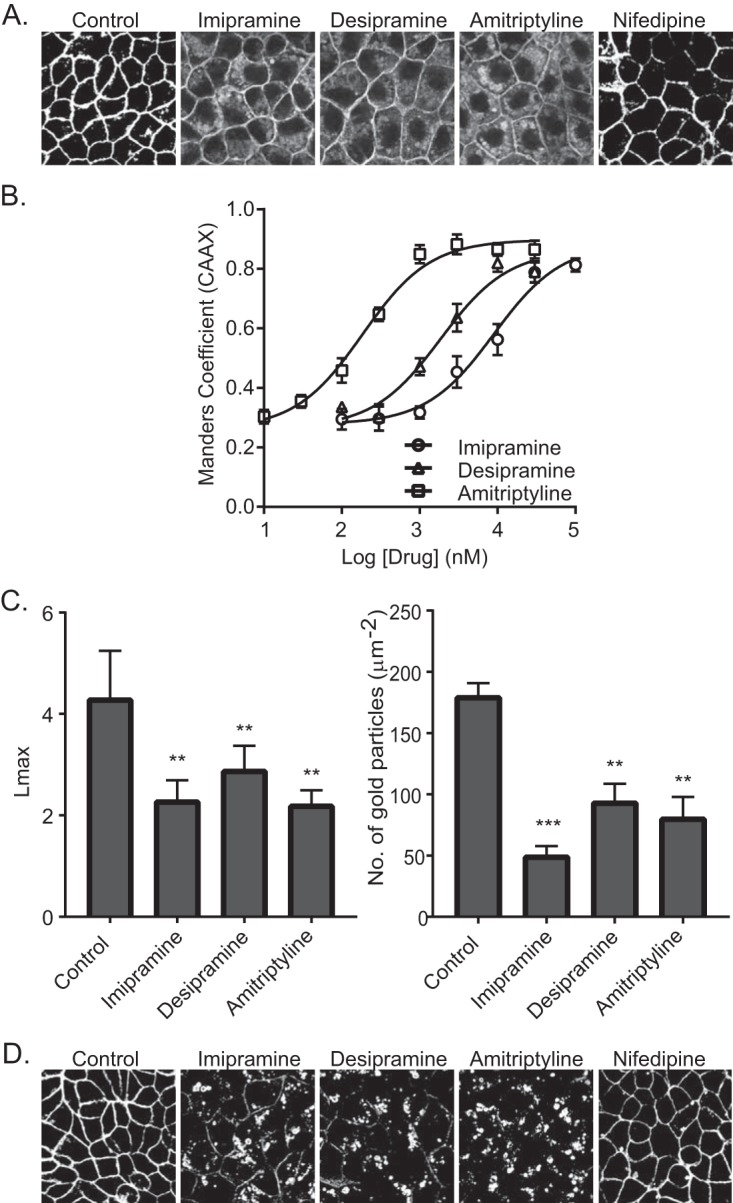
ASM inhibitors cause mislocalization of PtdSer and cholesterol from the PM. (A) Representative confocal images taken in the green channel of MDCK cells stably coexpressing mCherry-CAAX and mGFP-LactC2 treated with vehicle (DMSO), imipramine (10 μM), desipramine (10 μM), amitriptyline (1 μM), or nifedipine (10 μM) for 48 h. (B) LactC2 mislocalization was quantified by using Manders coefficients as the fraction of mCherry-CAAX colocalizing with mGFP-LactC2. The Manders coefficient for DMSO-treated control was 0.295 ± 0.02. (C) Basal PM sheets prepared from MDCK cells expressing mGFP-LactC2 after treatment with vehicle (DMSO), imipramine (10 μM), desipramine (10 μM), or amitriptyline (1 μM) for 24 h were labeled with anti-GFP-conjugated gold and visualized by EM. The peak values, Lmax, of the weighted mean K function L(r) − r curves and the number of gold particles per μm2 area are shown as bar graphs (n = 20). Significant differences were evaluated in bootstrap tests (**, P < 0.01) or Student's t tests (**, P < 0.01; ***, P < 0.001). (D) Representative confocal images of MDCK cells stably expressing the cholesterol probe mCherry-D4H treated with vehicle (DMSO), imipramine (10 μM), desipramine (10 μM), amitriptyline (1 μM), or nifedipine (10 μM) for 48 h.
ASM inhibition abrogates K-Ras function.
Concordant with the induced K-Ras mislocalization, the tricyclic antidepressants significantly reduced phosphorylated extracellular signal-regulated kinase (ppERK) levels in MDCK cells expressing constitutively active K-RasG12V (Fig. 4A). Treatment with these drugs also potently inhibited proliferation of pancreatic cancer cell lines with an oncogenic K-Ras mutation more so than a pancreatic cancer cell line expressing wild-type (WT) K-Ras. A similar selective inhibition of proliferation of oncogenic mutant K-Ras-expressing cells was also observed in endometrial, lung, and colon cancer cell lines following treatment with ASM inhibitors (Fig. 4B). We then determined the effect of fendiline on the growth of the WT K-Ras-expressing pancreatic cancer cell line BxPC-3 and oncogenic K-Ras-expressing MiaPaCa-2 cells implanted subcutaneously into the flanks of nu/nu immunosuppressed mice. The animals were randomized into control and fendiline-treated groups (10 mice per group). Treatment was initiated when the tumors reached a mean volume of 100 mm3. Fendiline (12.5 mg/kg of body weight) was given once daily intraperitoneally for 5 days, with 2 days of no treatment in between, for the duration of the experiment. Administration of fendiline strikingly decreased the rate of growth of MiaPaCa-2 cells xenografted in nude mice but had no effect on the growth of the BxPC-3 tumors (Fig. 4C). There was no toxicity detected in any of the fendiline-treated groups during the experiment, and accordingly, there was no significant difference in the body weight of the animals in the different groups. These results demonstrate that fendiline can selectively reduce the growth of oncogenic K-Ras-transformed tumors in vivo.
FIG 4.
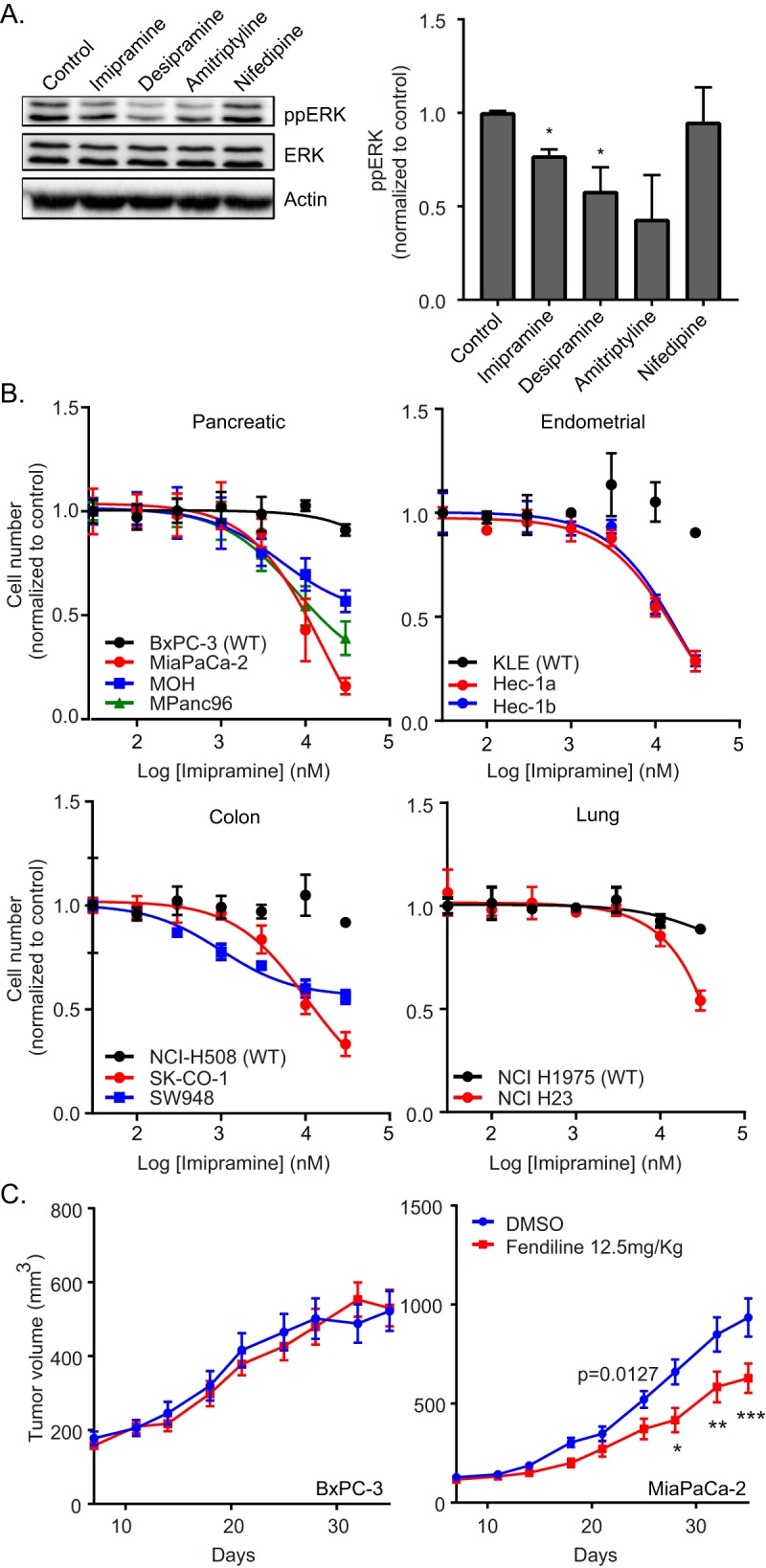
ASM inhibitors abrogate K-Ras signaling and function in vitro and in vivo. (A) MDCK cells stably expressing mGFP–K-RasG12V were treated with vehicle (DMSO) or 10 μM imipramine, desipramine, amitriptyline, or nifedipine for 48 h. Following treatment, whole-cell lysates were prepared and ppERK levels were measured by quantitative Western blotting. ppERK levels were normalized to the control sample. Significant differences were assessed by using Student's t tests (*, P < 0.05) (n = 3). (B) A panel of wild-type (BxPC-3, KLE, NCI-H508, and NCI H1975) or oncogenic mutant K-Ras-expressing (MiaPaCa-2, MOH, MPanc96, Hec-1a, Hec-1b, SK-CO-1, SW948, and NCI H23) pancreatic, endometrial, colon, and lung tumor cells were seeded in 96-well plates and treated for 72 h with vehicle (DMSO) or various concentrations of imipramine. The number of viable cells was quantified using the CyQuant cell proliferation assay kit (Molecular Probes). (C) nu/nu mice implanted with BxPC-3 or MIAPaCa-2 tumors were treated with vehicle (DMSO) or fendiline (12.5 mg/kg) intraperitoneally, and tumor sizes were measured with an external caliper every 3 to 4 days. Time and fendiline treatment affected MiaPaCa2 xenograft growth (P = 0.027 by 2-way ANOVA). Posttest analysis, using the Bonferroni correction for multiple comparisons, showed that xenografts treated with fendiline were significantly smaller on days 28, 32, and 35 (*, P < 0.05; **, P < 0.01; ***, P < 0.001). No significant effects due to drug treatment were observed in BxPC-3 tumors.
Assessing the importance of the SM biosynthetic pathway to K-Ras function in C. elegans.
We used the well-validated invertebrate model system Caenorhabditis elegans, which has a single Ras gene, let-60, that is a K-Ras ortholog (31–34) to test the role of ASM in maintaining K-Ras function. Activating mutations in let-60 such as LET-60 G13D (n1046) induces a readily quantifiable multivulva phenotype (32). We therefore determined if treatment of these worms with the ASM inhibitors would suppress the multivulva phenotype. L1 larvae were cultured in M9 buffer containing the E. coli strain OP50 in the presence of dimethyl sulfoxide (DMSO) or drugs. After 4 to 5 days, worms that reached the adult stage were scored for the presence of the multivulva phenotype. The MEK inhibitor U0126 was used as a positive control. The MEK inhibitor and all of the ASM inhibitors robustly reverted the multivulva phenotype to a single-vulva WT phenotype, whereas the control drug nifedipine had no effect (Fig. 5A and C).
FIG 5.

Genetic and pharmacological perturbation of Cer-SM biosynthetic pathway alters K-Ras function in C. elegans. (A) Strain let-60(n1046) L1 larvae were cultured in M9 buffer containing the E. coli strain OP50 in the presence of vehicle (DMSO) or 30 μM fendiline, imipramine, desipramine, amitriptyline, or U0126. After 4 to 5 days, when worms reached the adult stage, they were scored for the presence of the multivulva phenotype using differential interference contrast (DIC)/Nomarski microscopy. Representative images of worms with the multivulva phenotype (shown by arrowheads) and suppression of this phenotype with fendiline are shown. (B) RNAi was induced by feeding let-60(n1046) L1 worms through adult stage with E. coli strain HT115, producing dsRNA to target genes. The presence of the multivulva phenotype was scored using DIC/Nomarski microscopy. (C) Strain let-60(n1046) L1 larvae were cultured in M9 buffer containing E. coli strain OP50 in the presence of vehicle (DMSO), 20 μM fumonisin B1, 50 nM myriocin, 100 μM D609, 175 μM 2-OHOA, 30 μM l-threo-DHS, 30 μM K1, 10 μM GT11, or 10 μM nifedipine. After 4 to 5 days, when worms reached the adult stage, they were scored for the presence of the multivulva phenotype using DIC/Nomarski microscopy. (D) lin-1(sy254) strain L1 larvae were cultured in M9 buffer containing the E. coli strain OP50 in the presence of vehicle (DMSO) or 30 μM fendiline, 20 μM fumonisin B1, 30 μM K1, 10 μM GT11, or 10 μM nifedipine. After 4 to 5 days, when worms reached the adult stage, they were scored for the presence of the multivulva phenotype using DIC/Nomarski microscopy. (E) RNAi was induced by feeding lin-1(sy254) L1 worms through adult stage with E. coli strain HT115, producing dsRNA to target genes. The presence of the multivulva phenotype was scored using DIC/Nomarski microscopy. One hundred to 200 worms were assayed per treatment or RNAi knockdown. Significant differences were evaluated in Student's t tests (*, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant).
Our data thus far show that multiple inhibitors of ASM mislocalize K-Ras by increasing SM levels, depleting Cer, and depleting PM PtdSer. Staurosporine also depletes PM PtdSer and mislocalizes K-Ras (35). Recent work has shown that staurosporine also increases SM levels by reducing ORMDL3 expression, which negatively regulates serine-palmitoyl-transferase (SPT) as the first and rate-limiting step for Cer and SM biosynthesis (36). We reasoned that if simply perturbing cellular SM and/or Cer levels can cause redistribution of PtdSer and K-Ras mislocalization, then inhibition of many other enzymes in the complex, spatially distributed SM biosynthetic pathway also may abrogate K-Ras function and therefore serve as potential drug targets for K-Ras inhibitors. We therefore used RNA interference (RNAi) against 18 validated C. elegans Cer-SM biosynthetic enzyme orthologs to identify genes whose silencing would suppress the multivulva phenotype associated with expression of activated LET-60 G13D (n1046). We observed that knockdown of 4 genes, cgt-3 (one of 3 orthologues of the glucosylceramide synthase gene), sphk-1 (sphingosine kinase [SK] gene), hyl-2 (CER synthase gene), and asm-1 (acid sphingomyelinase gene), were equipotent with two previously reported potent suppressors of the LET-60 G13D multivulva phenotype (heo-1 and riok-1) (Fig. 5B) (37, 38). Knockdown of 10 further genes, lagr-1 (ortholog of the CER synthase gene), degs-1 (dihydroceramide desaturase gene), sptl-1 (serine palmitoyl transferase gene), sms-1 (sphingomyelin synthase 1 gene), asah-1 (acid ceramidase gene), smpd-2 (neutral sphingomyelinase gene), W07E6.3 (a putative sphingomyelin synthase gene), ksdr-1 (3-ketosphinganine reductase gene), degs-2 (a putative dihydroceramide desaturase gene), and cgt-2 (1 of 3 orthologs of the glucosylceramide synthase gene), also moderately or weakly suppressed the multivulva phenotype. Only four genes in the test set had no detectable effect on the activated LET-60 phenotype: cgt-1 (1 of 3 orthologues of the glucosylceramide synthase gene), cerk-1 (ceramide kinase gene), spl-1 (sphingosine 1-P lyase gene), and hyl-1 (another orthologue of the CER synthase gene). None of the genes compromised organismal viability when silenced.
We next determined if inhibiting the enzymes in the Cer-SM biosynthetic pathway with pharmacological agents would also revert the multivulva phenotype in C. elegans. We tested myriocin (inhibitor of serine palmitoyl transferase), fuminosin B1 (inhibitor of ceramide synthase), GT11 (dihydroceramide desaturase inhibitor), K1 (ceramide kinase inhibitor), 2-hydroxyoleic acid (2-OHOA; activator of SM synthase), D609 (inhibitor of SM synthase), SKI-II (sphingosine kinase I inhibitor), and l-threo-dihydrosphingosine (l-threo-DHS; a sphingosine kinase I and II inhibitor) (39). While fumonisin B1, K1, and GT11 strongly suppressed the multivulva phenotype, the rest of the drugs only produced a moderate effect. The negative-control nifedipine had no effect (Fig. 5C).
In order to confirm that suppression of the multivulva phenotype occurred at the level of Ras rather than at downstream effector pathways, we examined the ability of the most potent pharmacological and short interfering RNA (siRNA) inhibitors of the let-60 multivulva phenotype to inhibit the lin-1 multivulva phenotype. Lin-1 is a transcription factor that functions as a repressor of vulval development and whose activity is inhibited by phosphorylation by MPK-1 mitogen-activated protein (MAP) kinase downstream of a Ras/MAP kinase pathway. Lin-1 null mutant worms display a multivulva phenotype similar to that induced by constitutively activated let-60; thus, the induction of the lin-1 null phenotype is downstream and independent of let-60 signaling (40). Fendiline, fumonisin B1, K1, and GT11, drugs that most potently reverted the multivulva phenotype in let-60 worms, had no effect on the multivulva phenotype of the lin-1 worms (Fig. 5D). Similarly, knocking down the expression of cgt3, sphk-1, asm-1, and hyl-2, which caused strong reversion of the multivulva phenotype in the let-60 worms, had no effect on the lin-1 worms (Fig. 5E). Taken together, these data strongly suggest that multiple perturbations for SM metabolism block activated let-60 signaling at the level of Ras in C. elegans.
Perturbation of Cer-SM biosynthetic pathway in mammalian cells alters K-Ras localization and function.
We next tested pharmacological agents that target the SM-Cer biosynthetic pathway in mammalian cells. We used quantitative lipidomics to measure SM and Cer content in whole-cell lysates. Interestingly, expression of oncogenic K-Ras increased the cellular content of both lipid species (Fig. 6A). Treatment of cells with fendiline did not alter the overall levels of SM and Cer levels in the WT cells compared to the vehicle-treated controls, although we have shown previously that it significantly alters the subcellular localization of SM by causing its accumulation in endolysosomal vesicles (25). Amitriptyline, SKI-II, and l-threo-DHS significantly increased the cellular content of both lipid species (Fig. 6A).
FIG 6.

Pharmacological modulators of SM-Cer pathway in mammalian cells differentially alter SM levels and distribution. (A) Whole-cell lysates were prepared from MDCK cells stably expressing mGFP–K-RasG12V or WT MDCK cells grown in the presence of vehicle (DMSO) or the denoted drugs for 48 h, and total SM and Cer levels were measured. (B) Representative confocal images of wild-type BHK cells treated with vehicle (DMSO), 20 μM fumonisin B1, 50 nM myriocin, 100 μM D609, 175 μM 2-OHOA, 10 μM SKI-II, 30 μM l-threo-DHS, 30 μM K1, or 10 μM GT11 for 48 h and incubated with 20 μg/ml MBP-GFP-lysenin and DAPI for 15 min at room temperature in serum-free medium. (C) Representative confocal images of wild-type BHK cells treated with vehicle (DMSO), 20 μM fumonisin B1, 50 nM myriocin, 100 μM D609, 175 μM 2-OHOA, 10 μM SKI-II, 30 μM l-threo-DHS, 30 μM K1, or 10 μM GT11 for 48 h permeabilized and stained with GFP-lysenin and DAPI.
We also measured changes in the subcellular distribution of SM after treatment with the pharmacological modulators of the Cer-SM biosynthetic pathway. Although all compounds altered SM localization, there were clear differences in the subcellular localization and abundance of SM between drug-treated cells, as observed with GFP-lysenin staining (Fig. 6B and C). Fumonisin B1, myriocin, GT11, and K1, all of which inhibit enzymes early in the Cer-SM de novo biosynthetic pathway, reduced the cellular content of SM and additionally caused internalization of SM in both live (Fig. 6B) and fixed, permeabilized cells (Fig. 6C). All other compounds increased the cellular content of SM; however, while the SK inhibitors SKI-II and l-threo-DHS increased both PM and internal abundance of SM, 2-OHOA and D609 increased only the intracellular membrane-associated SM while depleting the PM of SM. Effects of 2-OHOA on SM distribution were very similar to those observed with fendiline. Although D609 is an inhibitor of SM synthase, it caused an increase in SM content.
All compounds tested caused mislocalization of Lact-C2 from the PM, but only 2-OHOA and the SK inhibitors, compounds that caused an increase in the cellular SM content, also caused intracellular cholesterol accumulation (Fig. 7A). All compounds caused a dose-dependent mislocalization of K-RasG12V from the PM, with IC50s in concentration ranges similar to the IC50s/50% effective concentrations for their inhibition or activation of their substrate enzymes (Fig. 7B). Furthermore, all compounds reduced the nanoclustering of K-RasG12V, as demonstrated by the reduced Lmax values from EM immunogold spatial mapping (Fig. 7C). Finally, we tested the ability of SKI-II and l-threo-DHS to suppress the proliferation of a panel of pancreatic cancer cells. As with inhibitors of ASM, the SK inhibitors more potently inhibited the proliferation of pancreatic cancer cell lines expressing oncogenic mutant K-Ras than cell lines expressing WT K-Ras (Fig. 7D).
FIG 7.

Perturbation of Cer-SM biosynthetic pathway in mammalian cells alters PtdSer and K-Ras localization and impairs K-Ras function. (A) Representative confocal images of MDCK cells stably coexpressing mGFP-LactC2 and the cholesterol probe mCherry-D4H treated with vehicle (DMSO), 20 μM fumonisin B1, 50 nM myriocin, 100 μM D609, 175 μM 2-OHOA, 10 μM SKI-II, 30 μM l-threo-DHS, 30 μM K1, or 10 μM GT11 for 48 h. mGFP-LactC2 mislocalization was quantified using ImageJ. Mean Manders coefficients for each treatment are shown above the respective images. (B) MDCK cells stably coexpressing mCherry-CAAX and mGFP–K-RasG12V were treated with vehicle (DMSO) or various concentrations of pharmacological modulators of the Cer-SM biosynthetic pathway for 48 h and imaged by confocal microscopy. K-RasG12V mislocalization was quantified using Manders coefficients as the fraction of mCherry-CAAX colocalizing with mGFP–K-RasG12V. The Manders coefficient for DMSO-treated control was 0.292 ± 0.02. (C) Basal PM sheets prepared from MDCK cells expressing mGFP–K-RasG12V after treatment with vehicle (DMSO), 20 μM fumonisin B1, 50 nM myriocin, 100 μM D609, 175 μM 2-OHOA, 10 μM SKI-II, 30 μM l-threo-DHS, 30 μM K1, or 10 μM GT11 for 24 h were labeled with anti-GFP-conjugated gold particles and visualized by EM. The peak values, Lmax, of the weighted mean K function L(r) − r curves are shown as bar graphs (n = 20). Significant differences were evaluated in bootstrap tests (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (D) A panel of wild-type (BxPC-3) or oncogenic mutant K-Ras-expressing (MiaPaCa-2, MOH, MPanc96) pancreatic tumor cells were seeded in 96-well plates and treated for 72 h with vehicle (DMSO) or various concentrations of SKI-II. The number of viable cells was quantified using the CyQuant cell proliferation assay kit (Molecular Probes).
DISCUSSION
We have shown that a wide variety of ASM inhibitors, including tricyclic antidepressants, deplete PM PtdSer, mislocalize K-Ras from the PM, reduce oncogenic K-Ras signaling, and potently inhibit proliferation of pancreatic cancer cells that express oncogenic mutant K-Ras. Using genetic knockdown of genes encoding enzymes in the SM biosynthetic pathway in C. elegans and validated by pharmacological inhibitors of SM biosynthetic enzymes in mammalian cells, we identified multiple enzymes whose inhibition disrupted K-Ras localization and function. All of these perturbations of the SM biosynthetic pathway caused depletion of PM PtdSer that in turn resulted in loss of K-Ras from the PM and reduced nanoclustering of K-Ras that remained PM bound. These changes to K-Ras cellular localization and spatial organization on the PM result in reduced K-Ras signal output, evidenced here by reduced K-Ras MAPK signaling in biochemical assays, reduced proliferation of K-Ras-transformed cancer cells, and reduced growth of K-Ras-driven tumors in nude mice.
Together, our findings show that the maintenance of cellular levels of SM as well as its correct distribution between PM and endomembranes is critical for K-Ras localization and function. For example, all ASM inhibitors caused aberrant accumulation of SM in endolysosomal vesicles. Interestingly, absolute levels of SM and Cer measured by lipidomics revealed no change in the levels of these lipids with fendiline treatment, perhaps due to feedback mechanisms regulating their synthesis. Regardless, our results show that this aberrant distribution of SM is sufficient to induce lysosomal trafficking defects, mimicking genetic deletion of ASM in Niemann-Pick type A and B disease. Concordantly, we showed previously that K-Ras is extensively mislocalized from the PM in patient-derived Niemann-Pick type A and type B cell lines but not in control human fibroblasts (25). Niemann-Pick type A cells express no ASM protein, whereas Niemann-Pick type B cells express ∼2% ASM activity compared to wild-type cells.
C. elegans genetics confirmed by inhibitor studies in mammalian cells showed that multiple enzymes in the spatially distributed metabolic pathways that synthesize and turn over SM when inhibited can block K-Ras localization and function (Fig. 8). Inhibiting these enzymes had variable effects on total SM levels, but all resulted in aberrant SM cellular distributions. Fumonisin B1, myriocin, and GT11, which inhibit enzymes catalyzing the initial reactions in the de novo biosynthesis of SM, decreased cellular levels of SM, as observed by lysenin staining as well as lipidomics, and caused mislocalization of SM from the PM to endolysosomal vesicles. In contrast, sphingosine kinase inhibitors increased total levels of SM, both on the PM and intracellularly, mimicking the phenotype of staurosporine (36), which increases SM synthesis by suppressing ORMDL3 expression and thereby enhancing SPT activity. SK1 has a role in the regulation of ceramide biosynthesis through its ability to divert the precursor of ceramide biosynthesis, dihydrosphingosine phosphate, from the biosynthetic route. Therefore, SK inhibitors alleviate this negative regulation and thereby increase the de novo synthesis of Cer/SM (41–43). Interestingly, 2-OHOA, an activator of SM synthase, and D609, an inhibitor of SM synthase, produced very similar aberrant localization of SM, with a strikingly intense endolysosomal accumulation of SM observed in cells treated with D609. In addition to inhibiting SM synthase, D609 also has been shown to increase cellular Cer levels through activation of SPT as well as to inhibit ASM through off-target effects (44). It seems likely that this combination of effects results in the intracellular accumulation of SM we observed with D609 treatment.
FIG 8.

Model showing the spatial distribution of the de novo and salvage pathways of SM biosynthesis. Genes knocked down in C. elegans and corresponding enzymes encoded are the following: asm-1 (acid sphingomyelinase), cerk (ceramide kinase), cgt-1, cgt-2, and cgt-3 (three orthologues of glucosylceramide synthase), degs-1 (dihydroceramide desaturase), degs-2 (a putative dihydroceramide desaturase), hyl-1 and hyl-2 (orthologues of CER synthase), ksdr-1 (3-ketosphinganine reductase), lagr-1 (ortholog of CER synthase), sms-1 (sphingomyelin synthase 1), asah-1 (acid ceramidase), sphk-1 (sphingosine kinase), spl-1 (sphingosine 1-P lyase), smpd-2 (neutral sphingomyelinase), sptl-1 (serine palmitoyl transferase), and w07e6-3 (a putative sphingomyelin synthase). Color coding indicates the degree of reversion of multivulva phenotype observed with knockdown as indicated in the key.
In C. elegans, RNAi knockdown of ASM and inhibition of ASM with fendiline both strongly suppressed the multivulva phenotype. However, we did not observe a strong correlation in the degree of suppression between RNAi knockdown of all genes and pharmacological inhibition of the cognate enzyme. There are several reasons for this incomplete concordance. For example, in some cases, the efficacy of the knockdown may not have been efficient, as the uptake of RNAi can vary in this organism depending on the route of treatment (45). In addition, the bioavailability of drugs in the worm is unknown, as is the efficacy of the compounds to inhibit the C. elegans orthologs of the mammalian enzymes for which they were selected. More important, however, was the complete concordance between the genetic results in C. elegans and the cognate pharmacology in mammalian cells in their ability to suppress K-Ras function. Importantly, the observations that fendiline, fumonisin B1, K1, and GT11, which potently reverted the Muv phenotype in let-60 worms and knock down expression of cgt3, sphk-1, asm-1, and hyl-2 (causing strong reversion of the Muv phenotype in the let-60 worms), caused no reversion of the Muv phenotype in lin-1 worms strongly support our contention that the suppressive effect of perturbing SM metabolism is operating at the level of Ras in a whole organism.
PtdSer is an anionic lipid that is essential for K-Ras PM binding and nanoclustering (25, 35, 46–48). Indeed, recent work has shown that K-Ras membrane anchor lipid binding specificity is highly selective for PtdSer and that this preference extends beyond the head group to PtdSer species with asymmetric acyl chains (46). Our results show that irrespective of the perturbations induced by different pharmacological modulators of the SM biosynthetic pathway, a unifying effect is an inability of cells treated with these agents to maintain PtdSer on the PM and, hence, K-Ras function. There are several mechanisms that may be relevant. A previous study showed that an increase in SM in the exofacial leaflet of the PM causes internalization of PtdSer (36). Other studies have similarly suggested a role for SM in the maintenance of phospholipid asymmetry on the PM (49, 50). More broadly, SM and Cer are important constituents of cellular membranes and are essential for lipid trafficking (51); indeed, genetic depletion of ASM leads to the lysosomal storage diseases Niemann-Pick A and B, characterized by accumulation of cholesterol and other lipids in the endolysosome (52). Taking these results together with our current data, we posit that SM must be correctly arrayed across cellular membranes at appropriate concentrations to maintain PtdSer on the inner leaflet of the PM. In consequence, SM metabolism is critical for PM lipidomic structure, which is essential for K-Ras membrane binding and function. This proposed mechanism is consistent with the delayed response to ASM and Cer add-back in terms of correcting K-Ras and PtdSer distributions after pharmacological perturbations of SM metabolism. ASM is taken up by endocytosis and macropinocytosis; it is therefore delivered directly to the lumen of the endosomal compartment where it operates. Once in the late endosomal compartment, the ASM first has to correct SM levels followed by redistribution of PtdSer back to the PM. Similarly, Cer has to be recycled through the salvage pathway that includes endosomal-, endoplasmic reticulum-, and Golgi body-localized enzymes (Fig. 8) to reset SM levels and distribution that then in turn allow redistribution of PtdSer back to the PM. Finally, it seems likely that other signaling molecules and complexes operating in lipid-based platforms on the PM also will be modulated by SM metabolism. Therefore, pharmacological tools for modulating sphingolipid pathways may represent novel therapeutic strategies for the treatment of K-Ras-dependent cancer.
MATERIALS AND METHODS
Materials.
R-Fendiline was synthesized by the Translational Chemistry Core Facility at the MD Anderson Cancer Center under good laboratory practice conditions. Structure and purity of fendiline were determined by high-performance liquid chromatography (HPLC), 1H and 13C nuclear magnetic resonance, UV solution, infrared, and high-resolution mass spectra. Staphylococcus aureus ASM and all other drugs were purchased from Sigma-Aldrich and were dissolved in DMSO. Cell culture media were purchased from HyClone. Fetal bovine serum (FBS) was purchased from GIBCO. Penicillin-streptomycin was purchased from Invitrogen. Anti-phospho-p44/42 mitogen-activated protein kinase (MAPK) (ERK1/2) (Thr202/Tyr204) (no. 9101) antibody was purchased from Cell Signaling Technology (Beverly, MA). Mouse anti-GFP (G1546) and anti-β-actin (A1978) antibodies for Western immunoblotting were from Sigma, and rabbit anti-mGFP and anti-mRFP antibodies for immunogold labeling were generated in house. Ceramide (860052P) was purchased from Avanti Polar Lipids.
Cell lines.
BxPC-3, MOH, MiaPaCa-2, and MPanc96 cells were kindly provided by Craig Logsdon, KLE and Hec-1a cells by Karen Lu, and HEC-1b cells by Bryan Hennessey, all at MD Anderson Cancer Center, Houston, TX. All other cell lines were purchased from the American Type Culture Collection (Manassas, VA). MDCK epithelial cells and baby hamster kidney (BHK) cells were grown in Dulbecco's modified Eagle's medium (DMEM)–high-glucose medium supplemented with sodium pyruvate, 2 mM l-glutamine, and 10% FBS or supplemented bovine calf serum, respectively. KLE cells were maintained in DMEM–F-12 medium supplemented with 10% FBS. Hec-1a and Hec-1b cells were grown in McCoy's 5a medium supplemented with 10% FBS. MPanc96 cells were grown in DMEM supplemented with 10% FBS, MiaPaCa-2 cells in DMEM supplemented with 10% FBS and 2.5% horse serum, and SK-CO-1 cells in Eagle's MEM supplemented with 10% FBS. SW948 cells were grown in Leibovitz's L15 medium with 10% FBS. All other cell lines were grown in RPMI 1640 supplemented with 10% FBS. All cell lines were grown at 37°C in 5% CO2.
Lysenin staining.
Two protocols were used to visualize SM in cells labeled with a nontoxic recombinant fragment of lysenin protein tagged with GFP (GFP-Lys), which specifically binds to SM (36, 53). For live cell staining, BHK cells were incubated with 20 μg/ml MBP-GFP-lysenin (kind gift from Gregory Fairn, University of Toronto) and 4′,6-diamidino-2-phenylindole (DAPI) for 15 min at room temperature in serum-free medium. For permeabilized cell staining, BHK cells were fixed with 4% paraformaldehyde (PFA), permeabilized with 0.05% saponin, and then incubated with 20 μg/ml MBP-GFP-lysenin and DAPI for 15 min at room temperature.
ASM add-back.
MDCK cells (1.75 × 105) stably coexpressing mGFP–K-RasG12V and mCherry-CAAX were seeded on a glass coverslip in a 12-well plate and grown with or without fendiline for 48 h. Medium was replaced with fresh medium with or without fendiline containing 2 U/ml ASM, and the incubation was continued for 120 min (25).
Ceramide add-back.
Ceramide dissolved in chloroform at 5 mg/ml was dried under a vacuum in a glass vial to remove the solvent, redissolved in complete growth medium by sonication, and diluted to a final concentration of 10 μM. For confocal microscopic experiments, 1.75 × 105 MDCK cells stably coexpressing mGFP–K-RasG12V and mCherry-CAAX were seeded on a glass coverslip in a 12-well plate and grown with or without fendiline for 48 h. The medium then was replaced with ceramide-containing media (with or without fendiline), and the incubation was continued for 120 min. This protocol was described previously for PtdSer add-back (35), based on the work of Grinstein and coworkers (54), and recently was further characterized and validated (25, 46, 47).
Western blotting.
Cells treated with vehicle or drugs for 48 h were washed in cold phosphate-buffered saline (PBS) and lysed in buffer containing 50 mM Tris-Cl (pH 7.5), 75 mM NaCl, 25 mM NaF, 5 mM MgCl2, 5 mM EGTA, 1 mM dithiothreitol, 100 μM NaVO4, 1% NP-40 plus protease inhibitors. SDS-PAGE and immunoblotting were performed using lysates containing 20 μg of total protein. Signals were detected by enhanced chemiluminescence (Thermo Fisher Scientific) and imaged using a FluorChemQ imager (Alpha Inotech). Quantification of intensities was performed using FluorChemQ software.
Confocal microscopy.
Cells grown on coverslips were fixed with 4% PFA, followed by 50 mM NH4Cl treatment to quench aldehyde groups. The coverslips were mounted in Mowiol and visualized by confocal microscopy (Nikon A1) using a 60× objective.
EM and spatial mapping.
MDCK cells stably expressing GFP–K-RasG12V or GFP-LactC2 were seeded on gold EM grids precoated with pioloform and poly-l-lysine in 3.5-cm dishes. After drug treatment, basolateral PM sheets attached to the EM grids were fixed and immunolabeled with anti-GFP antibody conjugated to 4.5-nm gold particles as previously described (25, 46, 47, 55). For bivariate analysis, PM sheets were labeled with anti-mRFP antibody conjugated to 2-nm gold particles and anti-GFP antibody conjugated to 6-nm gold particles. Digital images of intact immunogold-labeled PM sheets were obtained via a transmission electron microscope at 100,000 magnification. Intact 1-μm2 areas of the PM sheet were identified using ImageJ, and the x-y coordinates of the gold particles were determined. Univariate K functions were calculated as shown in equations 1 and 2 (56, 57):
| (1) |
| (2) |
where K(r) is the univariate K function of a pattern of n gold particles in the study area A, r is cluster radius, ∣∣ · ∣∣ is Euclidean distance, and 1(.) is the indicator function that takes a value of 1 if ∣∣xi − xj∣∣ ≤ r or a value of 0 if ∣∣xi − xj∣∣ > r. Edge correction is implemented using weights where wij−1 is the fraction of the circumference of a circle with a center at xi and radius ∣∣xi − xj∣∣ that falls within the study area. We evaluated K(r) over a range of 1 nm ≤ r ≤ 240 nm. K(r) is linearized to yield L(r) − r as shown in equation 2. For each condition, at least 15 basolateral PM sheets were imaged and analyzed to generate mean L(r) − r curves that then were standardized on the 99% confidence interval (CI) for a random pattern of the same intensity. The peak L(r) − r value is denoted as the Lmax. Statistical significance for differences between replicated point patterns were evaluated in bootstrap tests as described previously (56, 58). Lmax values greater than 1 indicate statistically significant clustering of the gold particle distribution.
To quantify the extent of coclustering of two populations of gold particles (2 nm and 6 nm), we used bivariate K functions calculated as described in equations 3 to 6:
| (3) |
| (4) |
| (5) |
| (6) |
where Kbs(r) is the bivariate K function of the 6-nm (big, or b) gold nanoparticle distribution with respect to the 2-nm (small, or s) gold nanoparticle distribution, and Ksb(r) is the bivariate K function of the small gold particle distribution with respect to the big gold particle distribution. Kbiv(r) combines Kbs(r) and Ksb(r) into a single estimator, where nb is the number of big gold particles and ns is the number of small gold particles. Other symbols are as described for equations 1 and 2. We evaluated Kbiv(r) over a range of 1 nm ≤ r ≤ 240 nm. Kbiv(r) is linearized to yield Lbiv(r) − r as described in equation 6. For each condition, at least 15 basolateral PM sheets were imaged and analyzed to generate mean Lbiv(r) − r curves that then were standardized on the 95% CI for a random pattern of the same intensities, generated by 1,000 Monte Carlo simulations. Bootstrap tests were constructed to test the statistical significance of differences between replicated bivariate functions as described previously (56, 58). For a simple summary statistic, LBI, the area under the curve for each standardized Lbiv(r) − r function was calculated as equation 7:
| (7) |
LBI has an expected value of <100 for proteins that do not cocluster.
Proliferation assays.
BxPC-3 (4 × 103), MiaPaCa-2 (2 × 103), MOH (1.5 × 103), MPanc96 (2 × 103), KLE (1 × 104), Hec-1a (5 × 103), Hec-1b (5 × 103), NCI 1975 (6 × 103), NCI H23 (1 × 104), NCI H508 (4 × 103), SK-CO-1 (3 × 103), and SW948 (4 × 103) cells were seeded in each well of 96-well plates. After 24 h, fresh growth medium supplemented with 1% vehicle (DMSO) or various concentrations of drug was added, and cells were grown for 72 h. Cell numbers were quantified using the CyQuant proliferation assay (Thermo Fisher Scientific) according to the manufacturer's protocol.
Lipidomics.
WT MDCK cells or MDCK cells stably expressing mGFP–K-RasG12V were treated with various drugs for 48 h. Cells were scraped in Dulbecco's phosphate-buffered saline (DPBS), and 1.5 × 106 cells/ml in 300 μl was analyzed. Lipid extraction and analysis using electron spray ionization and tandem mass spectrometry were performed at Lipotype, GmbH (Dresden, Germany), as described previously (59, 60). Automated processing of acquired mass spectra, identification, and quantification of detected lipid species were done by LipidXplorer software. Only lipid identifications with a signal-to-noise ratio of >5, an absolute abundance of at least 1 pmol, and a signal intensity 5-fold higher than that in corresponding blank samples were considered for further data analysis. The abundance of lipids is presented as picomoles of lipids/cell relative to wild-type cells.
C. elegans vulva quantification assay.
let-60(n1046) and lin-1(sy254) worms were kindly provided by Swathi Arur (MD Anderson Cancer Center, Houston, TX) and David Reiner (Texas A&M University), respectively. For drug treatment studies, strain let-60 or lin-1 L1 larvae were cultured in M9 buffer containing Escherichia coli OP50 in the presence of DMSO or drugs. After 4 to 5 days, when worms reached the adult stage, they were scored for the presence of the multivulva phenotype using a DIC/Nomarski microscope. For RNAi knockdown studies, RNAi was induced by feeding let-60 or lin-1 L1 worms through adult stage with E. coli HT115, producing double-stranded RNA to target genes. The presence of the multivulva phenotype was imaged as described above. All RNAi clones were from the C. elegans RNAi (Ahringer) collection (Source BioScience) and were sequenced. Clones absent from the library were constructed as previously described (61).
Mouse xenografts.
All animal studies were performed under an Institutional Animal Care and Use Committee (IACUC)-approved animal protocol (AWC-15-0101), in accordance with National Institutes of Health guidelines (62). Early-passage BxPC-3 or MIAPaCa-2 cells were harvested, and 3 × 106 cells were implanted into the right flanks of female nu/nu mice. The animals were randomized into control and treated groups (10 mice per group). Tumor volume (V) was measured with an external caliper every 3 to 4 days, and it was calculated as V = 0.52(length × width2). Fendiline treatment was initiated when the tumor sizes reached 100 mm3. Fendiline (12.5 mg/kg) was injected daily intraperitoneally for 5 days, with 2 days of no treatment in between. All treatments were continued until any of the subcutaneous tumors reached 1,500 mm3 in volume, when all the animals were sacrificed and the tumors removed.
Statistical analysis.
Unless otherwise stated, Prism (version 5.0c; GraphPad Software) was used for one-way analysis of variance (ANOVA) and two-tailed Student's t tests.
ACKNOWLEDGMENTS
This work was supported by start-up funds from the UT School of Dentistry and Uterine SPORE funding (P50CA098258) provided to D.V.D.H., start-up funds provided to R.V.D.H. by the UT School of Dentistry, a grant to J.F.H. from the Cancer Research and Prevention Institute of Texas (CPRIT RP170233), and an NIH Pathway to Independence Award (K99-CA188593) to K.-J.C.
We thank Gregory Fairn for providing GFP-lysenin.
REFERENCES
- 1.Prior IA, Lewis PD, Mattos C. 2012. A comprehensive survey of Ras mutations in cancer. Cancer Res 72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hancock JF. 2003. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol 4:373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 3.Mitin N, Rossman KL, Der CJ. 2005. Signaling interplay in Ras superfamily function. Curr Biol 15:R563–R574. doi: 10.1016/j.cub.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 4.Plowman SJ, Muncke C, Parton RG, Hancock JF. 2005. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci U S A 102:15500–15505. doi: 10.1073/pnas.0504114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hancock JF, Magee AI, Childs JE, Marshall CJ. 1989. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57:1167–1177. doi: 10.1016/0092-8674(89)90054-8. [DOI] [PubMed] [Google Scholar]
- 6.Hancock JF, Paterson H, Marshall CJ. 1990. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 63:133–139. doi: 10.1016/0092-8674(90)90294-O. [DOI] [PubMed] [Google Scholar]
- 7.Rowinsky EK. 2006. Lately, it occurs to me what a long, strange trip it's been for the farnesyltransferase inhibitors. J Clin Oncol 24:2981–2984. doi: 10.1200/JCO.2006.05.9808. [DOI] [PubMed] [Google Scholar]
- 8.Sebti SM, Der CJ. 2003. Searching for the elusive targets of farnesyltransferase inhibitors. Nat Rev Cancer 3:945–951. doi: 10.1038/nrc1234. [DOI] [PubMed] [Google Scholar]
- 9.Cox AD, Hisaka MM, Buss JE, Der CJ. 1992. Specific isoprenoid modification is required for function of normal, but not oncogenic, Ras protein. Mol Cell Biol 12:2606–2615. doi: 10.1128/MCB.12.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hancock JF, Cadwallader K, Paterson H, Marshall CJ. 1991. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J 10:4033–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baines AT, Xu D, Der CJ. 2011. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem 3:1787–1808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Downward J. 2003. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 13.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, Seshagiri S, Koeppen H, Belvin M, Friedman LS, Malek S. 2010. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 14.Menard D, Niculescu-Duvaz I, Dijkstra HP, Niculescu-Duvaz D, Suijkerbuijk BM, Zambon A, Nourry A, Roman E, Davies L, Manne HA, Friedlos F, Kirk R, Whittaker S, Gill A, Taylor RD, Marais R, Springer CJ. 2009. Novel potent BRAF inhibitors: toward 1 nM compounds through optimization of the central phenyl ring. J Med Chem 52:3881–3891. doi: 10.1021/jm900242c. [DOI] [PubMed] [Google Scholar]
- 15.Neel NF, Martin TD, Stratford JK, Zand TP, Reiner DJ, Der CJ. 2011. The RalGEF-Ral effector signaling network: the road less traveled for anti-Ras drug discovery. Genes Cancer 2:275–287. doi: 10.1177/1947601911407329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nourry A, Zambon A, Davies L, Niculescu-Duvaz I, Dijkstra HP, Menard D, Gaulon C, Niculescu-Duvaz D, Suijkerbuijk BM, Friedlos F, Manne HA, Kirk R, Whittaker S, Marais R, Springer CJ. 2010. BRAF inhibitors based on an imidazo[4,5]pyridin-2-one scaffold and a meta substituted middle ring. J Med Chem 53:1964–1978. doi: 10.1021/jm901509a. [DOI] [PubMed] [Google Scholar]
- 17.Packer LM, Rana S, Hayward R, O'Hare T, Eide CA, Rebocho A, Heidorn S, Zabriskie MS, Niculescu-Duvaz I, Druker BJ, Springer C, Marais R. 2011. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell 20:715–727. doi: 10.1016/j.ccr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. 2010. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts PJ, Der CJ. 2007. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 20.Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, Westover KD. 2014. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc Natl Acad Sci U S A 111:8895–8900. doi: 10.1073/pnas.1404639111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, Sim T, Janne PA, Meyerson M, Marto JA, Engen JR, Gray NS. 2014. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl 53:199–204. doi: 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR. 1999. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 98:69–80. doi: 10.1016/S0092-8674(00)80607-8. [DOI] [PubMed] [Google Scholar]
- 23.Willumsen BM, Christensen A, Hubbert NL, Papageorge AG, Lowy DR. 1984. The p21 ras C-terminus is required for transformation and membrane association. Nature 310:583–586. doi: 10.1038/310583a0. [DOI] [PubMed] [Google Scholar]
- 24.Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI, Waldmann H. 2013. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 25.Cho KJ, van der Hoeven D, Zhou Y, Maekawa M, Ma X, Chen W, Fairn GD, Hancock JF. 2015. Inhibition of acid sphingomyelinase depletes cellular phosphatidylserine and mislocalizes K-Ras from the plasma membrane. Mol Cell Biol 36:363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Hoeven D, Cho KJ, Ma X, Chigurupati S, Parton RG, Hancock JF. 2013. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol Cell Biol 33:237–251. doi: 10.1128/MCB.00884-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beckmann N, Sharma D, Gulbins E, Becker KA, Edelmann B. 2014. Inhibition of acid sphingomyelinase by tricyclic antidepressants and analogons. Front Physiol 5:331. doi: 10.3389/fphys.2014.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gulbins E, Palmada M, Reichel M, Luth A, Bohmer C, Amato D, Muller CP, Tischbirek CH, Groemer TW, Tabatabai G, Becker KA, Tripal P, Staedtler S, Ackermann TF, van Brederode J, Alzheimer C, Weller M, Lang UE, Kleuser B, Grassme H, Kornhuber J. 2013. Acid sphingomyelinase-ceramide system mediates effects of antidepressant drugs. Nat Med 19:934–938. doi: 10.1038/nm.3214. [DOI] [PubMed] [Google Scholar]
- 29.Kornhuber J, Tripal P, Reichel M, Terfloth L, Bleich S, Wiltfang J, Gulbins E. 2008. Identification of new functional inhibitors of acid sphingomyelinase using a structure-property-activity relation model. J Med Chem 51:219–237. doi: 10.1021/jm070524a. [DOI] [PubMed] [Google Scholar]
- 30.Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. 2008. Membrane phosphatidylserine regulates surface charge and protein localization. Science 319:210–213. doi: 10.1126/science.1152066. [DOI] [PubMed] [Google Scholar]
- 31.Ceol CJ, Stegmeier F, Harrison MM, Horvitz HR. 2006. Identification and classification of genes that act antagonistically to let-60 Ras signaling in Caenorhabditis elegans vulval development. Genetics 173:709–726. doi: 10.1534/genetics.106.056465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reiner DJ, Gonzalez-Perez V, Der CJ, Cox AD. 2008. Use of Caenorhabditis elegans to evaluate inhibitors of Ras function in vivo. Methods Enzymol 439:425–449. doi: 10.1016/S0076-6879(07)00430-2. [DOI] [PubMed] [Google Scholar]
- 33.Beitel GJ, Clark SG, Horvitz HR. 1990. Caenorhabditis elegans ras gene let-60 acts as a switch in the pathway of vulval induction. Nature 348:503–509. doi: 10.1038/348503a0. [DOI] [PubMed] [Google Scholar]
- 34.Han M, Sternberg PW. 1990. let-60, a gene that specifies cell fates during C. elegans vulval induction, encodes a ras protein. Cell 63:921–931. doi: 10.1016/0092-8674(90)90495-Z. [DOI] [PubMed] [Google Scholar]
- 35.Cho KJ, Park JH, Piggott AM, Salim AA, Gorfe AA, Parton RG, Capon RJ, Lacey E, Hancock JF. 2012. Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. J Biol Chem 287:43573–43584. doi: 10.1074/jbc.M112.424457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maekawa M, Lee M, Wei K, Ridgway ND, Fairn GD. 2016. Staurosporines decrease ORMDL proteins and enhance sphingomyelin synthesis resulting in depletion of plasmalemmal phosphatidylserine. Sci Rep 6:35762. doi: 10.1038/srep35762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith MM, Levitan DJ. 2004. The Caenorhabditis elegans homolog of the putative prostate cancer susceptibility gene ELAC2, hoe-1, plays a role in germline proliferation. Dev Biol 266:151–160. doi: 10.1016/j.ydbio.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 38.Weinberg F, Schulze E, Fatouros C, Schmidt E, Baumeister R, Brummer T. 2014. Expression pattern and first functional characterization of riok-1 in Caenorhabditis elegans. Gene Expr Patterns 15:124–134. doi: 10.1016/j.gep.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 39.Delgado A, Casas J, Llebaria A, Abad JL, Fabrias G. 2006. Inhibitors of sphingolipid metabolism enzymes. Biochim Biophys Acta 1758:1957–1977. doi: 10.1016/j.bbamem.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 40.Tiensuu T, Larsen MK, Vernersson E, Tuck S. 2005. lin-1 has both positive and negative functions in specifying multiple cell fates induced by Ras/MAP kinase signaling in C. elegans. Dev Biol 286:338–351. doi: 10.1016/j.ydbio.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 41.Siow D, Sunkara M, Morris A, Wattenberg B. 2015. Regulation of de novo sphingolipid biosynthesis by the ORMDL proteins and sphingosine kinase-1. Adv Biol Regul 57:42–54. doi: 10.1016/j.jbior.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 42.Siow D, Wattenberg B. 2011. The compartmentalization and translocation of the sphingosine kinases: mechanisms and functions in cell signaling and sphingolipid metabolism. Crit Rev Biochem Mol Biol 46:365–375. doi: 10.3109/10409238.2011.580097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siow DL, Anderson CD, Berdyshev EV, Skobeleva A, Natarajan V, Pitson SM, Wattenberg BW. 2011. Sphingosine kinase localization in the control of sphingolipid metabolism. Adv Enzyme Regul 51:229–244. doi: 10.1016/j.advenzreg.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adibhatla RM, Hatcher JF, Gusain A. 2012. Tricyclodecan-9-yl-xanthogenate (D609) mechanism of actions: a mini-review of literature. Neurochem Res 37:671–679. doi: 10.1007/s11064-011-0659-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. 2001. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol 2:RESEARCH0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou Y, Prakash P, Liang H, Cho KJ, Gorfe AA, Hancock JF. 2017. Lipid-sorting specificity encoded in K-Ras membrane anchor regulates signal output. Cell 168:239–251. doi: 10.1016/j.cell.2016.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou Y, Wong CO, Cho KJ, van der Hoeven D, Liang H, Thakur DP, Luo J, Babic M, Zinsmaier KE, Zhu MX, Hu H, Venkatachalam K, Hancock JF. 2015. Signal transduction. Membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science 349:873–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou Y, Hancock JF. 2015. Ras nanoclusters: versatile lipid-based signaling platforms. Biochim Biophys Acta 1853:841–849. doi: 10.1016/j.bbamcr.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 49.Tepper AD, Ruurs P, Wiedmer T, Sims PJ, Borst J, van Blitterswijk WJ. 2000. Sphingomyelin hydrolysis to ceramide during the execution phase of apoptosis results from phospholipid scrambling and alters cell-surface morphology. J Cell Biol 150:155–164. doi: 10.1083/jcb.150.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonomini M, Dottori S, Amoroso L, Arduini A, Sirolli V. 2004. Increased platelet phosphatidylserine exposure and caspase activation in chronic uremia. J Thromb Haemost 2:1275–1281. doi: 10.1111/j.1538-7836.2004.00837.x. [DOI] [PubMed] [Google Scholar]
- 51.Jackson CL, Walch L, Verbavatz JM. 2016. Lipids and their trafficking: an integral part of cellular organization. Dev Cell 39:139–153. doi: 10.1016/j.devcel.2016.09.030. [DOI] [PubMed] [Google Scholar]
- 52.Schuchman EH. 2010. Acid sphingomyelinase, cell membranes and human disease: lessons from Niemann-Pick disease. FEBS Lett 584:1895–1900. doi: 10.1016/j.febslet.2009.11.083. [DOI] [PubMed] [Google Scholar]
- 53.Shogomori H, Kobayashi T. 2008. Lysenin: a sphingomyelin specific pore-forming toxin. Biochim Biophys Acta 1780:612–618. doi: 10.1016/j.bbagen.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 54.Kay JG, Koivusalo M, Ma X, Wohland T, Grinstein S. 2012. Phosphatidylserine dynamics in cellular membranes. Mol Biol Cell 23:2198–2212. doi: 10.1091/mbc.E11-11-0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Y, Liang H, Rodkey T, Ariotti N, Parton RG, Hancock JF. 2014. Signal integration by lipid-mediated spatial cross talk between Ras nanoclusters. Mol Cell Biol 34:862–876. doi: 10.1128/MCB.01227-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diggle PJ, Mateu J, Clough HE. 2000. A comparison between parametric and non-parametric approaches to the analysis of replicated spatial point patterns. Adv Appl Prob 32:331–343. doi: 10.1239/aap/1013540166. [DOI] [Google Scholar]
- 57.Ripley BD. 1977. Modelling spatial patterns. J R Stat Soc B 39:172–192. [Google Scholar]
- 58.Plowman SJ, Hancock JF. 2005. Ras signaling from plasma membrane and endomembrane microdomains. Biochim Biophys Acta 1746:274–283. doi: 10.1016/j.bbamcr.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 59.Gerl MJ, Sampaio JL, Urban S, Kalvodova L, Verbavatz JM, Binnington B, Lindemann D, Lingwood CA, Shevchenko A, Schroeder C, Simons K. 2012. Quantitative analysis of the lipidomes of the influenza virus envelope and MDCK cell apical membrane. J Cell Biol 196:213–221. doi: 10.1083/jcb.201108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sampaio JL, Gerl MJ, Klose C, Ejsing CS, Beug H, Simons K, Shevchenko A. 2011. Membrane lipidome of an epithelial cell line. Proc Natl Acad Sci U S A 108:1903–1907. doi: 10.1073/pnas.1019267108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoeven R, McCallum KC, Cruz MR, Garsin DA. 2011. Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans. PLoS Pathog 7:e1002453. doi: 10.1371/journal.ppat.1002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]