
Fig. 1.
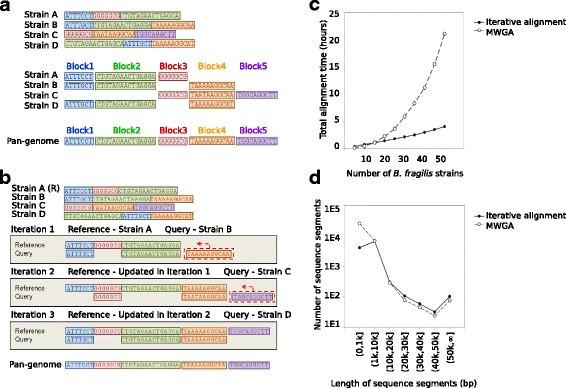
The iterative alignment algorithm. a Conventional multiple whole-genome alignment identifies similar genomic regions (blocks) among genome sequences; the union of all block sequences represents the species pan-genome. b The iterative alignment algorithm is demonstrated with a microbial species having four sequenced strains (strain A–D) where strain A is selected as the representative genome, denoted by (R). In the first alignment iteration, a query genome is selected (strain B) and aligned to the reference (strain A), identifying the genomic region that is present in the query but not in the reference (orange region). The orange region is then appended to the reference genome to generate an updated version. In the next iteration, a new query genome (strain C) is aligned with the updated reference genome. The query-exclusive purple region is identified and appended to the reference genome to generate a new updated version. The iteration continues until all strains are considered. The final reference represents the species pan-genome. c The total alignment time for processing different numbers of B. fragilis strains using multiple whole-genome alignment or iterative alignment. d The distribution of contig sizes that constitute the B. fragilis pan-genome identified using multiple whole-genome alignment or iterative alignment