

Summary
Despite recent advances, many cancers remain refractory to available immunotherapeutic strategies. Emerging evidence indicates that the tolerization of local dendritic cells (DCs) within the tumor microenvironment promotes immune evasion. Here we have described a mechanism by which melanomas establish a site of immune privilege via a paracrine Wnt5a-β-catenin-peroxisome proliferator-activated receptor-γ (PPAR-γ) signaling pathway that drives fatty acid oxidation (FAO) in DCs by upregulating the expression of the carnitine palmitoyltransferase-1A (CPT1A) fatty acid transporter. This FAO shift increased the protoporphyrin IX prosthetic group of indoleamine 2,3-dioxgenase-1 (IDO) while suppressing interleukin-6 (IL-6) and IL-12 cytokine expression, culminating in enhanced IDO activity and the generation of regulatory T cells. We demonstrated that blockade of this pathway augmented anti-melanoma immunity, enhanced the activity of anti-PD-1 antibody immunotherapy, and suppressed disease progression in a transgenic melanoma model. This work implicates a role for tumor-mediated metabolic reprogramming of local DCs in immune evasion and immunotherapy resistance.
Keywords: β-catenin, Wnt5a, dendritic cell tolerization, fatty acid oxidation, indoleamine 2, 3-dioxygenase, regulatory T cells, melanoma microenvironment, immune evasion, immunotherapy resistance, protoporphyrin IX
eTOC Blurb
Previous studies suggest that DC tolerization plays a role in tumor-mediated immune evasion. The mechanism by which cancers promote this process remains poorly understood. Zhao, F. et al. demonstrate that melanomas generate a site of immune privilege by driving DC fatty acid oxidation via a Wnt5a-β-catenin-PPAR-γ signaling pathway that culminates in the induction of IDO enzyme activity. Inhibiting this pathway reverses DC tolerization and enhances anti-PD-1 antibody efficacy in a transgenic model of melanoma.
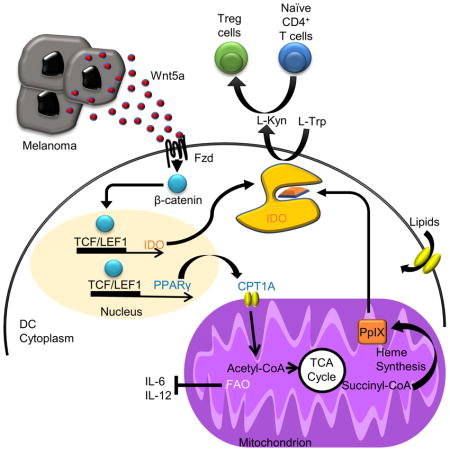
Introduction
While considerable strides have recently been made in cancer immunotherapy, the majority of advanced cancer patients are refractory to this treatment modality. An emerging literature describes active immune evasion mechanisms by which malignancies manipulate their microenvironment to avoid detection and destruction by the adaptive immune response (Mellor and Munn, 2008). The dendritic cell (DC) is now recognized as a key player in the generation of anti-tumor immunity. By processing and presenting antigen, the individual DC induces the activation and modulates the functionality of a large population of naïve T cells (Banchereau and Steinman, 1998). Given the central role of the DC in priming T cells to a developing malignancy, it is reasonable to conclude that cancers may evolve potent mechanisms of immune evasion by targeting DC function. Studies have described DCs within the tumor microenvironment as contributing to tumor pathogenesis, suggesting that these antigen presenting cell populations undergo a tolerization program enabling them to generate an immune privileged microenvironment (Hanks et al., 2013; Scarlett et al., 2012). However, the mechanisms by which cancers induce this DC tolerization program are largely unknown. Recent studies have shown that Batf3+ DCs aid in generating response to immunotherapy (Salmon et al., 2016; Spranger et al., 2017). These studies underscore the importance of understanding DC tolerization as these mechanisms may provide insight into immunotherapy resistance and identify previously unappreciated immunotherapeutic targets.
The immunoregulatory enzyme, indoleamine 2,3-dioxygenase-1 (IDO), plays a vital role in tumor-mediated immune suppression (Munn et al., 2004). By catalyzing the conversion of the essential amino acid tryptophan into the kynurenine metabolic byproducts, IDO supresses the expansion of effector T cells while promoting the differentiation and activation of the immunosuppressive CD4+FoxP3+ regulatory T (Treg) cell population (Fallarino et al., 2006; Sharma et al., 2007). IDO requires the heme prosthetic group, protoporphyrin IX (PpIX), for full enzymatic activity (Shimizu et al., 1978). This is supported by studies demonstrating the inhibition of PpIX synthesis to suppress IDO activity and addition of the heme biosynthetic precursor δ-aminolevulinic acid to augment IDO activity (Thomas et al., 2001). Although IDO is also expressed by some tumors, DC expression of IDO is particularly important for generating an immune privileged site during cancer progression (Munn and Mellor, 2007). Indeed, lymph node DC IDO expression has been correlated with a poor prognosis in melanoma (Munn et al., 2004; Munn et al., 2002). The realization of this critical role of IDO in tumor-mediated immune suppression has led to development of small molecule IDO inhibitors that are now in late phase clinical trials as a strategy to augment anti-tumor immunity (Mullard, 2015). Despite the importance of IDO in tumor-mediated immune tolerance, it is largely unknown how evolving cancers regulate its function.
Previous investigators have described a role for β-catenin signaling in the induction of DC tolerization (Jiang et al., 2007; Manicassamy et al., 2010). Recently, we delineated a paracrine Wnt5a signaling pathway by which melanomas generate an immunotolerant microenvironment (Holtzhausen et al., 2015). By inducing β-catenin activation in nearby DCs, melanoma-derived Wnt5a promotes the transcriptional expression of IDO, culminating in Treg cell generation and establishment of an immune privileged site allowing for melanoma disease progression. While the canonical Wnt ligand, Wnt3a, also induces the expression of IDO by DCs, we have noted that Wnt3a-conditioned DCs fail to induce Treg cell differentiation. These findings suggest that Wnt5a may regulate DC IDO via an unknown post-transcriptional mechanism. A recent RNAseq differential gene expression study correlated Wnt5a with checkpoint inhibitor resistance in advanced melanoma patients, suggesting that this pathway may also be relevant to the clinical management of cancer patients with inhibitors of the PD-1:PD-L1 signaling axis (Hugo et al., 2016).
Prior studies have established that DC maturation, a genetic program that allows for effective DC-dependent effector T cell activation, is accompanied by a glycolytic surge (Krawczyk et al., 2010). Indeed, subsequent work has shown that inhibiting the DC glycolytic pathway diminishes DC-mediated T cell stimulation (Everts et al., 2014). Other studies have demonstrated that Wnt5a modulates cellular metabolism in various systems (Sherwood et al., 2014). Based on these findings, we hypothesized that melanoma-derived Wnt5a metabolically reprograms local DC populations to support IDO enzymatic activity and subsequent Treg cell differentiation and that this represents a mechanism by which cancers hijack an intrinsic switch that shifts the host immune system towards a tolerogenic state. We further propose that this pathway represents a promising pharmacological target for augmenting checkpoint inhibitor immunotherapy.
Results
Melanoma-derived Wnt5a Reprograms DC Energy Metabolism
Toll-like receptor (TLR)-induced DC maturation involves the induction of glycolysis (Krawczyk et al., 2010). We have shown that melanoma-expressed soluble Wnt5a signals via the β-catenin signaling pathway to drive DC tolerization (Holtzhausen et al., 2015). Others have also shown Wnt5a regulates cellular metabolism (Sherwood et al., 2014). Based on these findings, we sought to investigate if Wnt5a alters the metabolism of DCs and whether this may contribute to DC tolerization. Using a biochemical extracellular lactate assay as a surrogate for glycolysis, we found Wnt5a suppresses lactate production by bone marrow-derived DCs (Figures 1A, S1A). Further monitoring of the extracellular acidification rate (ECAR) demonstrated that Wnt5a failed to impact glycolysis within 90 minutes of stimulation, suggesting that Wnt5a may regulate the glycolysis of DCs via a transcriptional mechanism (Figures S1D, E). Indeed, qrt-PCR studies revealed that Wnt5a downregulated the expression of the rate-limiting glycolytic enzymes, hexokinase (HK) and phosphofructokinase-1 (PFK-1) in DCs after 4–8 hrs of stimulation (Figure 1B). Additional ECAR studies demonstrated that Wnt5a pre-treatment suppressed the lipopolysaccharide (LPS)-induced glycolytic surge typically observed during the DC maturation program, indicating that Wnt5a elicits a dominant metabolic effect on DCs (Figures 1C, S1F). Given the observed effect on glycolysis in DCs, we investigated the impact of Wnt5a on DC oxidative phosphorylation (OXPHOS). These studies demonstrated recombinant Wnt5a (rWnt5a) effectively promoted OXPHOS in DCs (Figures 1D, E, S1B, C, G, H). To determine if melanoma-derived Wnt5a was capable of influencing DC metabolism, we analyzed the oxygen consumption rate (OCR) and ECAR of purified DCs stimulated with conditioned media harvested either from a control BrafV600E-Pten−/− melanoma cell line (BrafV600E-Pten−/−-NTC) or a BrafV600E-Pten−/− melanoma cell line genetically silenced for Wnt5a expression (BrafV600E-Pten−/−-Wnt5a-silenced) (Holtzhausen et al., 2015). These studies showed that genetic silencing of Wnt5a diminished the ability of melanomas to promote OXPHOS in DCs, an effect that was partially reversed with the addition of rWnt5a. (Figures 1F, S1I). This data suggests that melanoma-derived Wnt5a promotes OXPHOS in DCs in vitro. However, no changes in either OXPHOS or glycolysis in DCs were observed following Wnt3a treatment (Figures 1D, E, S1G, H, J). To verify that melanoma-derived Wnt5a can modulate DC metabolism in vivo, we purified tumor-infiltrating DCs from both BrafV600E-Pten−/−-NTC and BrafV600E-Pten−/−-Wnt5a-silenced tumors resected from syngeneic mice and measured their real-time OCR. Consistent with our previous findings, this study demonstrates that melanoma-derived Wnt5a promotes DC mitochondrial respiration in situ (Figures 1G, H, S1K). Together, these data reveal that melanoma tissues shift the metabolism of local DC populations from a glycolytic state toward OXPHOS in a Wnt5a-dependent manner.
Figure 1. Melanoma-derived Wnt5a Alters DC Energy Metabolism.

A. Lactate in BMDC culture media from 0–48 hours with rWnt5a treatment. n=6. B. Qrt-PCR analysis of Hk and Pfk expression in DCs following rWnt5a treatment. n=3. C. ECAR (milli-pH units/minute, normalized to 0 min) of untreated (UT) vs. rWnt5a pretreated DCs. Arrow indicates LPS injection. n=6. D. OCR (pico-moles/minute) of DCs pre-treated with rWnt5a or rWnt3a. n=6. Oligo, oligomycin. FCCP, uncoupling agent. Rot, rotenone. E. ECAR of DCs pre-treated with rWnt5a or rWnt3a. n=6. 2DG, 2-deoxyglucose. F. OCR of DCs injected with media alone or concentrated conditioned media (CM) from BrafV600EPten−/−-NTC or -Wnt5aKD cell cultures±rWnt5a. n=6. G. OCR of tumor-infiltrating DCs (TIDCs) isolated from BrafV600EPten−/−-NTC and -Wnt5aKD mice. n=3/group. H. Metabolic parameter calculations based on 1G. n=3/group. KD, knockdown. All data is mean +/- SEM. *P<0.05, **P<0.005. See also Figure S1.
Wnt5a-mediated Metabolic Reprogramming Alters DC Function
Previous studies have demonstrated that inhibition of hexokinase, the initial enzyme in the glycolytic pathway, suppresses DC-induced T cell proliferation while others have found tolerized DCs to exhibit enhanced OXPHOS (Everts et al., 2014; Malinarich et al., 2015). Consistent with these findings and our data showing Wnt5a blocks LPS-induced DC glycolysis, we determined that Wnt5a suppresses LPS-induced DC-mediated antigen-specific T cell proliferation in a manner similar to 2-deoxyglucose (2-DG) (Figures 2A). Notably, this effect was observed in the absence of any alterations in DC surface maturation markers (Figure S2). We have previously shown that Wnt5a promotes DC-mediated Treg cell differentiation both in vitro and in vivo (Holtzhausen et al., 2015). Altogether, these data indicate that inhibition of DC glycolysis and inhibition of DC OXPHOS would have reciprocal effects on Treg cell development. Indeed, co-culturing 2-DG-treated or Wnt5a-treated DCs with naïve CD4+ T cells generated enhanced Treg cell differentiation in vitro while inhibition of DC OXPHOS with oligomycin (oligo) eliminated these Treg cell populations (Figures 2B). Together, these findings imply that Wnt5a drives Treg cell differentiation in the melanoma microenvironment by promoting DC OXPHOS. This is consistent with previous data showing that Wnt3a neither regulates DC metabolism nor promotes DC-mediated Treg cell generation (Figures 1D, E) (Holtzhausen et al., 2015). To examine this question more directly, we purified tumor-infiltrating DCs from BrafV600E-Pten−/−-NTC and BrafV600E-Pten−/−-Wnt5a-silenced tumors and delivered them by intradermal footpad injection into syngeneic Foxp3-mRFP transgenic reporter mice followed by ipsilateral popliteal and inguinal lymph node isolation and Treg cell quantitation by flow cytometry. This confirmed that the BrafV600E-Pten−/−-Wnt5a-silenced tumor-derived DC population previously shown to exhibit diminished OXPHOS (Figures 1G, H) also exhibits suppressed Treg cell differentation in vivo (Figure 2C). In summary, metabolic reprogramming plays a central role in Wnt5a regulation of DC functionality and determines whether a DC drives effector T cell expansion versus Treg cell differentiation (Figure 2D).
Figure 2. DC Function is Regulated by Cellular Metabolism.

A. T cell proliferation assay: DCs loaded with OVA257-264 peptide, pre-treated with rWnt5a or 2DG, stimulated with LPS, and co-incubated with OT-I splenocytes. CD3+CD8+ T cell proliferation measured by CellTrace Violet (CTV) dilution. n=3. right, Representative flow cytometry CTV dilution assay based on 3 independent experiments. Gated on CD3+CD8+ T cells. B. DCs treated with Wnt5a, 2-DG, or Oligo prior to in vitro Treg cell assay measuring DC-induced CD4+FoxP3+ Treg cells. n=3. right, Representative flow cytometry plot of Treg cell analysis based on 3 independent experiments. C. Treg cell analysis of inguinal lymph nodes by flow cytometry. left, Representative of 3 independent experiments. 4 mice/group. D. Schematic illustrating the dynamic spectrum of DC-induced T cell responses based on their metabolic alteration. UT untreated. KD, knockdown. All data are mean +/− SEM. *P<0.05. See also Figure S2.
Wnt5a Induction of DC Fatty Acid Oxidation Promotes Treg Cell Development and Suppresses Effector T Cell Activation
Cancer-associated DCs exhibit higher cytoplasmic lipid content via increased lipid uptake and these elevated lipid stores impair DC antigen processing and presentation (Herber et al., 2010). However, the mechanisms underlying these DC alterations in the tumor microenvironment are unknown. Since our data indicate that melanoma-derived Wnt5a altered DC function, we investigated the impact of Wnt5a and β-catenin on DC lipid content and found this pathway to enhance DC fatty acid uptake and lipid stores (Figures 3A, B).
Figure 3. Wnt5a Promotes Treg Cell Differentiation by Driving Fatty Acid Oxidation in DCs.

A. left, DC uptake of fluorescent dodecanoic acid fatty acid substrate, TF2-C12, measured by flow cytometry after treatment with rWnt5a or vehicle control (UT). n=3. right, DC intracellular lipid content following rWnt5a treatment vs UT. BODIPY, fluorescent lipid probe. n=3. B. left, Fatty acid uptake of DC2.4-β-catKD and DC2.4-NTC cell lines. n=3. right, Microscopic immunofluorescence analysis of BODIPY-stained DC2.4-β-catKD and DC2.4-NTC cell lines (scale bar=1 cm). n=3. green, BODIPY. C. BMDCs pre-treated with rWnt5a vs rWnt5a+ETO prior to OCR analysis. D. Metabolic parameter calculations based on 3C. n=6. E. In vitro Treg cell assay measuring DC-induced CD4+FoxP3+ Treg cells. n=3. below, Representative flow plot based on 3 independent experiments. F. In vivo Treg cell assay measuring DC-induced CD4+FoxP3+ Treg cells following treatment with either rWnt5a or rWnt5a+ETO. n=4/group. right, Representative FoxP3-RFPxCD4-FITC flow plot based on 3 independent experiments. G. BMDCs pulsed with OVA257-264 peptide, treated with ETO or stimulated with LPS, and co-incubated with OT-I splenocytes. CD3+CD8+ T cell proliferation measured by CellTrace Violet dilution. n=3. right, Representative flow cytometry CellTrace Violet dilution assay based on 3 independent experiments. Gated on CD3+CD8+ T cells. H. DC2.4-NTC or DC2.4-CPT1AKD DC lines treated with rWnt5a, injected into foot pads of Foxp3-mRFP mice. Inguinal lymph node was isolated and analyzed by flow cytometry for Treg cells. n=3/group. right, Representative flow plot based on 3 independent experiments. I. DC2.4-NTC or DC2.4-CPT1AKD DC lines were loaded with OVA257-264 peptide, treated with rWnt5a or stimulated with LPS, and co-incubated with OT-I splenocytes. CD8+ T cell proliferation measured by CellTrace Violet (CTV) dilution flow cytometry. n=3. right, Representative flow cytometry CTV dilution assay based on 3 independent experiments. J. BMDCs were transduced with a CPT1A-targeted shRNA-expressing or non-targeting control (NTC) lentivirus, treated in the presence or absence of rWnt5a, pulsed with OVA257-264 peptide, and co-incubated with OT-I splenocytes. Representative flow cytometry CTV dilution assay based on 3 independent experiments. KD, knockdown. All data is mean +/− SEM. *P<0.05. See also Figures S2–S4.
We, therefore, reasoned that Wnt5a may enhance OXPHOS in DCs by promoting fatty acid oxidation (FAO). To determine if Wnt5a regulated FAO in DCs, we analyzed the real-time OCR of DCs treated with rWnt5a in the presence and absence of the carnitine palmitoyl transferase-1 (CPT1) mitochondrial fatty acid transporter inhibitor, etomoxir (ETO). These experiments showed ETO completely ablated Wnt5a induction of both murine and human DC mitochondrial respiration while not impacting DC viability (Figures 3C, D, S3A–B). Since our prior data suggested that OXPHOS in DCs played an important role in DC-mediated Treg cell generation and studies indicated that DC glutaminolysis was not involved in this process (Figure S3C), we investigated the role of FAO in DC-dependent Treg cell generation. This demonstrated ETO treatment potently suppresses the ability of Wnt5a-conditioned DCs to drive Treg cell differentiation in vitro and in vivo following the adoptive transfer of conditioned DCs into Foxp3-mRFP reporter mice (Figures 3E, F). In line with our previous data indicating that inhibition of OXPHOS in DCs promotes antigen-specific T cell proliferation, we found ETO treatment potently induced DC-mediated T cell activation despite a downregulation in DC co-stimulatory receptors based on flow cytometry (Figure 3G, S2). To confirm that off-target effects of ETO did not contribute to this process, we genetically silenced Cpt1a expression in the DC2.4 myeloid DC line and determined the ability of the resulting DC2.4-CPT1A-silenced cell line to induce Treg cell differentiation in vivo as well as to promote effector T cell proliferation in vitro relative to the DC2.4-NTC control cell line (Figure S4). This revealed that genetically targeting CPT1A in the DC2.4 line effectively made these DCs resistant to Wnt5a-induced Treg cell development while promoting their ability to stimulate CD8+ T cell proliferation (Figures 3H, I). To demonstrate that genetic silencing of CPT1A can have similar effects in primary DCs, we engineered a CPT1A-specific shRNA-expressing lentiviral vector and transduced BMDCs before performing OT-I CD8+ T cell proliferation assays (Figures S3D, E). These experiments indeed demonstrated primary CPT1A-silenced DCs induce potent CD8+ T cell proliferation while maintaining resistance to Wnt5a-induced tolerization (Figure 3J).
Overall, these data provide a potential mechanistic explanation for the increased lipid stores previously observed in cancer-associated DCs. In addition, this work implies that Wnt5a shifts DCs from glycolysis towards FAO in the melanoma microenvironment and this metabolic program effectively inhibits effector T cell activation while driving Treg cell differentiation.
The Wnt5a-β-catenin signaling Pathway Regulates DC Fatty Acid Oxidation via the PPAR-γ-CPT1A Axis
Previous investigators have proposed that activation of AMP-activated protein kinase (AMPK) by the AMP analog, AICAR, would antagonize the glycolytic surge required for DC maturation (Krawczyk et al., 2010). Our findings are consistent with this work (Figure S5A). As a result, we hypothesized that Wnt5a shifts DC metabolism from glycolysis to FAO by activating AMPK. However, we found Wnt5a suppressed AMPK activation based on Thr-172 phospho-AMPK immunoblot analysis (Figure S5B). In addition, we detected no impact of Wnt5a on DC Akt Thr-308 phosphorylation, a well characterized promoter of glycolysis in DCs (Figure S5C) (Krawczyk et al., 2010). These results suggest that the Wnt5a-mediated metabolic shift from glycolysis to FAO is independent of AMPK and Akt signaling.
The peroxisome proliferator-activated receptor (PPAR) family of transcription factors regulate the expression of several key factors involved in FAO. Treatment of primary DCs with the β-catenin inhibitor, XAV939, and genetic silencing of β-catenin in the DC2.4 cell line (DC2.4-β-catenin-silenced) promoted glycolysis in DCs, confirming that β-catenin regulates DC metabolism (Figures S5D–G). β-catenin induction of PPAR-γ expression has been previously described (Jansson et al., 2005). Consistent with these data, we found rWnt5a stimulation of primary DCs induced expression of several genes downstream of the PPAR-γ transcription factor previously identified to promote FAO, including CPT1A, using a quantitative polymerase chain reaction (qPCR) array (Figure 4A). We subsequently verified that rWnt5a induces upregulation of Pparg and Cpt1a using quantitative real-time PCR and immunoblot analysis in both murine and human DCs (Figures 4B–D, S5H). To confirm that β-catenin regulates CPT1A expression, we found reduced expression of Cpt1a in the DC2.4-β-catenin-silenced cell line while β-catenin activation of primary DCs via inhibition of the GSK3β enzyme promoted both Pparg and Cpt1a expression (Figures 4E, F, S5E–S5F). Consistent with its inability to alter DC metabolism, Wnt3a also failed to induce expression of both PPAR-γ and CPT1A in DCs (Figure S6A).
Figure 4. Wnt5a Induces DC Fatty Acid Oxidation via the Stimulation of a β-catenin-PPAR Signaling Pathway.

A. BMDCs treated with rWnt5a for 48 hours and analyzed by PCR array. Heatmap gene expression analysis: “F”, fatty acid metabolism genes, “L”, lipid transport genes, “A”, adipogenesis genes. red, high. blue, low. n=3. B. Qrt-PCR analysis of Pparg1 expression by BMDCs treated with rWnt5a. n=3. C. Qrt-PCR analysis of Cpt1a expression by BMDCs treated with rWnt5a. n=3. D. PPARγ and CPT1A Western blot analysis following human monocyte-derived DC treatment with rWnt5a. n=3. Densitometry analysis, S5H. E. Cpt1a qrt-PCR analysis of DC2.4-NTC and DC2.4-β-catKD cell lines. n=3. F. Qrt-PCR analysis of Pparg1 (left) and Cpt1a (right) expression by BMDCs treated with either the GSK3β inhibitor, BIO, vs its control, MeBIO. n=3. G. Western blot analysis of PPARγ following β-catenin immunoprecipitation of Wnt5a-treated BMDCs. n=3. Densitometry analysis, Figure S5I. H. Qrt-PCR and I. Western blot analysis of Ctnnb1, β-catenin and Ido1, IDO in BMDCs isolated from wt and β-catΔDC mice. J. Primary BrafV600E-Pten−/− melanoma progression in wt and β-catΔDC hosts. n=6/group. K. IFN-γ ELISPOT analysis of tumor-infiltrating DCs (TIDCs) TRP2-specific T cells derived from BrafV600EPten−/− melanomas resected from either wt or β-catΔDC mice. n=3/group. Representative of two independent experiments. left, representative wells from ELISPOT plate. KD, knockdown. All data is mean +/− SEM. *P<0.05. See also Figures S4–S6.
PPAR-γ is a transcriptional co-activator complexing with β-catenin to induce genes that drive FAO (Jansson et al., 2005). We therefore performed endogenous co-immunoprecipitation experiments in primary DCs and found PPAR-γ to bind to β-catenin upon Wnt5a stimulation (Figure 4G, S5I). All together, these findings support a mechanism by which Wnt5a signaling promotes PPAR-γ-dependent induction of CPT1A to activate FAO in DCs.
Based on these findings, we directly investigated the impact of this DC signaling pathway on the generation of anti-tumor immunity in vivo. After implanting the BrafV600E-Pten−/− melanoma cell line into syngeneic wild type mice or mice with β-catenin-deleted DCs (β-catΔDC, Figure 4H, I), we monitored primary tumor growth and demonstrated BrafV600E-Pten−/− melanoma growth restriction in β-catΔDC hosts in association with a significant enhancement in melanoma tyrosinase-related protein-2 (TRP2) antigen-specific T cell responses based on IFN-γ ELISPOT (Figures 4J, K). Altogether, these findings suggest that the DC β-catenin-PPAR-γ signaling pathway suppresses the development of T cell-mediated immunity in melanoma.
Wnt5a-induced Fatty Acid Oxidation Regulates the Enzymatic Activity of DC Indoleamine 2,3-dioxygenase
IDO plays a critical role in driving Treg cell development in the tumor microenvironment (Hanks et al., 2013; Holtzhausen et al., 2015; Munn et al., 2004). Despite inducing DC IDO expression, we noted that Wnt3a stimulation failed to condition DCs to promote Treg cell differentiation. In light of the potent impact of FAO on DC-mediated Treg cell generation, we hypothesized that Wnt5a-mediated regulation of FAO was directly modulating the enzymatic activity of IDO. In order to test this hypothesis, we measured production of the IDO byproduct kynurenine in purified DC cultures using high performance liquid chromatography (HPLC). These studies confirmed that rWnt5a promoted DC IDO enzymatic activity and that inhibition of FAO in DCs completely eliminated this effect, suggesting that FAO in DCs regulated the enzymatic activity of IDO (Figure 5A). To demonstrate that this occurs within the melanoma microenvironment, we purified tumor-infiltrating DCs from resected BrafV600E-Pten−/−-NTC and BrafV600E-Pten−/−-Wnt5a-silenced tumors and analyzed kynurenine generation as a surrogate for IDO enzymatic activity. This demonstrated that tumor-infiltrating DCs derived from melanomas lacking Wnt5a expression exhibit lower IDO enzyme activity similar to ETO-treated tumor-infiltrating DCs purified from BrafV600E-Pten−/−-NTC control tumors (Figure 5B). These findings show that Wnt5a-induced FAO plays a critical role in regulating DC IDO enzyme activity within developing melanomas.
Figure 5. Wnt5a-induced FAO in DCs Promotes IDO Enzymatic Activity.

A. Kynurenine HPLC analysis of conditioned media harvested from DCs treated with rWnt5a or rWnt5a+ETO. n=3. B. Kynurenine HPLC analysis of conditioned media harvested from tumor-infiltrating DCs (TIDCs) isolated from BrafV600EPten−/−-NTC and BrafV600EPten−/−-Wnt5aKD melanomas±ETO. n=3/group. C. PpIX flow cytometry analysis of DCs treated with rWnt5a, rWnt5a+ETO, or ETO following δ-aminolevulinic acid (ALA) pre-incubation. n=3. right, Representative flow histogram of PpIX expression based on 3 independent experiments. D. Hemin colorimetric assay of DCs treated with increasing concentrations of rWnt5a ± ETO. n=3. E. Qrt-PCR analysis of Alas1 expression by DCs following the indicated treatments. n=3. F. Qrt-PCR analysis of heme biosynthetic enzymes in DCs treated with rWnt5a. n=3. G. LC-MS analysis of TCA metabolic intermediates derived from DCs following treatment with rWnt5a. arrow, downregulation of α-ketoglutarate. H. Schematic of TCA cycle intermediates (red arrows) and enzymes (blue arrows). Changes in response to Wnt5a shown. I. Qrt-PCR analysis of select TCA cycle enzymes in DCs. n=3. Idh, isocitrate dehydrogenase. Oxoglutarate dyhydrogenase complex: Dld, dihydrolipoyl dehydrogenase. Dlst, dihydrolipoyl succinyltransferase. Oghd, oxoglutarate decarboxylase. J. In vitro Treg cell assay measuring DC-induced CD4+FoxP3+ Treg cells following treatment with either rWnt5a+vehicle control or rWnt5a+SA (Succinylacetone) n=3. KD, knockdown. All data is mean +/− SEM. *P<0.05. See also Figures S4, S6.
Since the IDO apoenzyme requires the heme-derived PpIX prosthetic group for full enzymatic activity and the TCA cycle intermediate, succinyl CoA, serves as the primary substrate for heme synthesis, we conjectured that increased PpIX concentrations may partially explain why FAO in DCs drives IDO function. Thus, we studied the impact of rWnt5a on DC concentrations of PpIX and hemin, the heme breakdown product, using a modified flow cytometry technique and a colorimetric assay, respectively (Hryhorenko et al., 1998). Indeed, this demonstrated that rWnt5a enhanced DC concentrations of the PpIX prosthetic group and its downstream degradation product, hemin, in a FAO-dependent manner (Figure 5C, D). Notably, consistent with its inability to modulate DC metabolism, Wnt3a also failed to enhance PpIX synthesis in DCs (Figure S6B).
Additional studies have determined the PPAR-γ coactivator-1α-dependent and rate-limiting enzyme of heme biosynthesis, aminolevulinic acid synthase-1 (ALAS1), is upregulated in Wnt5a-stimulated DCs (Figures 5E) (Handschin et al., 2005). Indeed, further analysis showed rWnt5a, but not rWnt3a, upregulated expression of Alas1 and several additional enzymes involved in PpIX synthesis, including ALA dehydratase (ALAD), uroporphyrinogen III synthetase, coprophyrinogen III oxidase, and protoporphyrin III oxidase (Figures 5F, S6C).
These data indicate that Wnt5a promotes DC IDO enzymatic activity by enhancing flux through the heme biosynthetic pathway and promoting the synthesis of the PpIX prosthetic group. To elucidate how Wnt5a-induction of FAO in DCs results in increased concentrations of PpIX described above, we utilized high-resolution liquid chromatography-mass spectrometry to measure intermediate TCA metabolites in response to rWnt5a stimulation (Liu et al., 2014). We observed increased quantities of many TCA intermediates except for a decrease in α-ketoglutarate (Figures 5G, H). Further qrt-PCR studies showed rWnt5a stimulated the expression of isocitrate dehydrogenase (Idh1, Idh2) as well as all three components of the downstream α-ketoglutarate dehydrogenase complex (Oghd, Dld, Dlst) which converts α-ketoglutarate to succinyl Co-A (Figures 5H, I). Altogether, these alterations suggest that Wnt5a promotes heme biosynthesis by affecting the TCA cycle and this process contributes to increased quantities of PpIX in DCs (Figure 5H). Finally, we found that inhibition of the enzyme ALAD with succinylacetone also significantly abrogated the ability of Wnt5a stimulated DCs to promote Treg cell generation, demonstrating that modulation of the heme biosynthetic pathway ultimately impacts Treg cell differentiation (Figure 5J).
This work describes a link between cellular metabolism and regulation of immune tolerance via modulation of DC IDO activity and further demonstrates that melanomas manipulate this pathway in a Wnt5a-dependent manner.
Wnt5a-β-catenin Induced Fatty Acid Oxidation is a Key Regulatory Pathway Underlying DC Tolerization
We further demonstrated that FAO inhibition in DCs not only eliminated the impact of Wnt5a on DC-mediated Treg cell differentiation in vitro, it also further suppressed Treg cell differentiation by DCs genetically ablated for IDO (Figure 6A). This data supports the existence of additional mechanisms of DC-mediated Treg cell differentiation beyond IDO that are downstream of the Wnt5a-β-catenin-FAO signaling pathway.
Figure 6. FAO Pathway Modulates DC Cytokine Expression.

A. In vitro Treg cell assay performed using either wild type or Ido1−/− DCs pre-treated with rWnt5a ± ETO and ± anti-IL-6 and anti-IL-12 antagonistic antibodies. n=3. B. Qrt-PCR analysis of Il6, Il12b mRNA in DC2.4-NTC vs. DC2.4-CPT1AKD cells. n=3. C. ELISA analysis of IL-6, IL-12p40 concentrations in the conditioned media of DC2.4-NTC vs. DC2.4-CPT1AKD cell lines. n=3. D. Qrt-PCR analysis of Il6, Il12b mRNA in UT or ETO-treated BMDCs. n=3. E. Qrt-PCR analysis of Il6, Il12b mRNA in BMDCs following treatment with rWnt5a. F. Qrt-PCR analysis of in situ Il6 and Il12b cytokine expression by tumor-infiltrating DCs (TIDCs) purified from BrafV600EPten−/−-NTC or -Wnt5aKD melanomas. n=3/group. G. Schematic diagram showing the proposed β-catenin-dependent pathway regulating IDO and pro-inflammatory cytokine expression via FAO in DCs. KD, knockdown. All data is mean +/− SEM. *P<0.05. See also Figures S4.
Since the local cytokine milieu can influence naïve CD4+ T cell differentiation into Treg cells, we examined the effect of FAO on the DC cytokine expression profile by comparing the expression of several cytokines between the DC2.4-CPT1A-silenced cell line and the DC2.4-NTC control cell line based on qrt-PCR and ELISA. These experiments demonstrated that genetically silencing Cpt1a to inhibit FAO results in significant elevations in expression of the pro-inflammatory cytokines, IL-6 and IL-12, while no significant differences in the expression of IL-10 or TGF-β were noted (Figures 6B, C; data not shown). These alterations in cytokine expression were further recapitulated in primary DC populations exposed to the CPT1A inhibitor, ETO (Figure 6D). Additional studies confirmed Wnt5a suppresses IL-6 and IL-12 expression in primary DCs, implicating the Wnt5a-β-catenin signaling pathway in the regulation of these pro-inflammatory cytokines (Figure 6E). To demonstrate that melanoma-derived Wnt5a induced a similar DC cytokine expression profile in situ, we purified tumor-infiltrating DCs from BrafV600E-Pten−/−-NTC and BrafV600E-Pten−/−-Wnt5a-silenced tumors as above and quantitated both Il6 and Il12b expression by qrt-PCR. These studies supported our previous findings in that BrafV600E-Pten−/− melanomas genetically silenced for Wnt5a were associated with significant elevations in tumor-infiltrating DC IL-6 and IL-12p40 expression (Figure 6F). Together, DC FAO suppresses IL-6 and IL-12 expression in addition to stimulating IDO enzymatic activity, creating an environment that favors Treg cell generation (Figure 6G). Indeed, blocking IL-6 and IL-12 using antagonistic antibodies eliminated the additional suppressive effect of FAO inhibition on Treg cell generation, indicating that the Wnt5a-β-catenin-FAO pathway modulates both IDO and pro-inflammatory cytokine expression in DCs (Figure 6A). Based on previous studies demonstrating that IL-6 promotes the proteosomal degradation of IDO, these data suggest that FAO in DCs may also promote IDO stabilization (Orabona et al., 2008). These dual mechanisms of IDO regulation suggest a central role for the Wnt5a-β-catenin signaling pathway in DC tolerization.
Inhibition of Fatty Acid Oxidation Enhances Anti-PD-1 Antibody Therapy and Suppresses the Progression of an Autochthonous Melanoma Model
The previous results suggest that a paracrine signaling axis mediated by melanoma-expressed Wnt5a induces FAO in local DCs to generate an immunotolerant microenvironment. To assess the impact of melanoma Wnt5a expression on T cell activity in melanoma, we performed IFN-γ ELISPOT assays on tumor-infiltrating lymphocytes (TILs) harvested from BrafV600E-Pten−/−-NTC and BrafV600E-Pten−/−-Wnt5a-silenced tumors. We observed a significant enhancement in IFN-γ-expressing TILs within BrafV600E-Pten−/−-Wnt5a-silenced tumors compared to control tumors, further supporting the immunotolerant role for Wnt5a (Figure 7A). These findings were also associated with elevated Cd274 (PD-L1) expression based on qrt-PCR and immunofluorescence analysis of resected BrafV600E-Pten−/−-Wnt5a-silenced tumor tissues (Figure 7B). Since previous studies have indicated that an inflamed tumor environment characterized by elevated PD-L1 expression is associated with improved responses to anti-PD-1 antibody checkpoint inhibitor therapy, we proposed that pharmacological inhibition of FAO by targeting CPT1A downstream of Wnt5a would augment anti-PD-1 antibody immunotherapy (Spranger et al., 2013). We noted that ETO treatment of BrafV600E-Pten−/− tumor cells has no impact on the intrinsic proliferative capacity of this tumor model (Figure S7). Therefore, any impact of ETO on the efficacy of anti-PD-1 antibody therapy would likely involve the stimulation of anti-tumor immunity. To test this hypothesis, we subcutaneously injected BrafV600E-Pten−/− melanoma cells into syngeneic C57BL/6 mice. Once tumors reached a volume of 80–100 mm3, mice were treated with vehicle, ETO inhibitor, anti-PD-1 antibody, or both. Primary tumor volumes were monitored and melanoma antigen-specific CD8+ T cell responses were quantified by IFN-γ ELISPOT assays. These data showed that ETO-mediated CPT1A inhibition suppresses the progression of BrafV600E-Pten−/− melanoma similar to anti-PD-1 antibody monotherapy while combination anti-PD-1 antibody-ETO therapy resulted in a significant reduction in primary melanoma growth (Figures 7C). Reduced tumor growth correlated with enhanced numbers of CD8+ TILs in combination anti-PD-1 antibody-ETO-treated tumors and a more pronounced induction of TRP2-specific CD8+ T cells suggesting this synergism to be dependent on induction of an effective anti-tumor T cell response (Figures 7D–E). Further work showed ablation of CD8+ T cells in the host eliminated the ability of ETO to suppress BrafV600E-Pten−/− melanoma progression, confirming that FAO inhibition mediates anti-tumor activity in an immune dependent manner (Figure 7F, Figure S7B). These results indicate FAO modulates anti-tumor immunity and is consistent with our previous data suggesting inhibition of the Wnt5a-β-catenin signaling pathway in DCs augments checkpoint inhibitor efficacy (Holtzhausen et al., 2015).
Figure 7. FAO Inhibition Augments the Efficacy of anti-PD-1 Antibody Immunotherapy and Reverses DC Tolerization in a Transgenic Melanoma Model.

A. IFN-γ ELISPOT analysis of tumor-infiltrating T cells derived from BrafV600EPten−/−-NTC or -Wnt5aKD melanomas. n=3/group. left, Wnt5a IHC of resected BrafV600EPten−/−-NTC or -Wnt5aKD melanoma tissues (scale bar=1 cm). right, representative IFN-γ ELISPOT plate based on 3 independent experiments. B. Cd274 qrt-PCR analysis and IF analysis of BrafV600EPten−/−-NTC and BrafV600EPten−/−-Wnt5aKD melanomas (scale bar=1 cm). n=3/group. C. BrafV600EPten−/− melanoma growth in C57BL/6 mice undergoing treatment with vehicle and IgG isotype control, ETO and IgG isotype control, anti-PD-1 ab and vehicle control, or anti-PD-1 ab and ETO. n=6/group. D. CD8+ TIL IHC and IF analysis of BrafV600EPten−/− melanomas resected from mice undergoing the indicated treatment (scale bar=1 cm). BF, brightfield. Tx-R, Texas Red. Representative of 3 tumors/group. E. IFNγ ELISPOT analysis of TRP2-specific tumor-infiltrating T cells isolated from each treatment group from 7C. n=4/group. right, IFN-γ ELISPOT plate. F. ETO treatment of BrafV600EPten−/− melanomas following anti-CD8 ab-mediated T cell depletion or IgG control treatment. n=6/group. G. Investigating the impact of DC-specific FAO on primary melanoma progression in an autochthonous BrafV600EPten−/− model. Pre-treated DCs transferred into the footpad of syngeneic BrafV600EPten−/− mice 3 days prior to tumor induction with 4-HT (4-hydroxytamoxifen) and every 3 days thereafter for 4 weeks (dashed arrows). H. Autochthonous melanoma growth in BrafV600EPten−/− mice undergoing treatment with rWnt5a-treated DCs±ETO following induction of primary melanoma development using 4-HT. n=5/group. I. Tumor-draining lymph node tissue and tumor tissue were analysed for CD4+FoxP3+ Treg cell and CD3+CD8+ T cell (TIL) populations by flow cytometry, respectively. n=4. KD, knockdown. All data is mean +/− SEM. *P<0.05. See also Figure S7.
To investigate the impact of DC-specific FAO inhibition on melanoma progression, primary DCs were treated with Wnt5a with or without ETO prior to their transfer into the draining lymph node bed of developing autochthonous BrafV600E-Pten−/− transgenic melanomas (Figure 7G). Consistently, DC-specific FAO inhibition potently suppressed primary melanoma progression (Figure 7H). Correlative studies showed this effect also coincides with a suppression of Treg cells within local draining LN tissues and enhanced numbers of melanoma-infiltrating CD8+ T cells (Figure 7I). Together, these data suggest the DC Wnt5a-β-catenin-PPAR-γ-CPT1A signaling axis is a pharmacologic target for enhancing the efficacy of cancer immunotherapy.
Discussion
In light of the critical role of DCs in driving effective anti-tumor immunity, we focused on elucidating those tumor-derived mechanisms that impair DC function (Gabrilovich, 2004). Indeed, there is emerging evidence that implicates DC tolerization in tumorigenesis (Hanks et al., 2013; Scarlett et al., 2012). This report demonstrates that melanomas induce local immune tolerance by manipulating the metabolism of DCs within the tumor microenvironment via a paracrine Wnt-β-catenin signaling pathway. Contrary to recently proposed theories that upregulation of IDO strictly represents a negative feedback mechanism of adaptive anti-tumor immunity (Spranger et al., 2013), we define an immune evasion mechanism that has evolved to actively manipulate IDO functionality.
Previous studies have suggested that DC tolerization depends on the β-catenin signaling pathway, however the mechanisms by which tumors control this DC tolerization program and how this pathway ultimately drives immune tolerance has remained unclear (Jiang et al., 2007; Manicassamy et al., 2010). An understanding of these mechanisms could provide pharmacological targets to reverse the immunotolerant microenvironment. We recently demonstrated that melanoma expression of Wnt5a triggers β-catenin-dependent induction of DC IDO expression via a paracrine signaling pathway and this culminates in driving local Treg cell differentiation (Holtzhausen et al., 2015). Others have shown that cellular metabolism regulates DC function as TLR-induced DC maturation is critically dependent upon glycolysis and OXPHOS promotes the development of a pro-tolerogenic state (Everts et al., 2014; Malinarich et al., 2015). Additional studies have described a role for Wnt5a in the regulation of cellular metabolism (Sherwood et al., 2014). Thus, we hypothesized that melanoma expressed Wnt5a metabolically reprograms DCs and this functions as a central mechanism of tumor-mediated immune tolerance.
The data presented here demonstrate that melanoma-derived Wnt5a robustly shifts DCs toward OXPHOS in a manner that is dominant over LPS-induced glycolysis (Everts et al., 2014). Others have suggested that AMPK plays an important role in shifting DC metabolism from glycolysis to an OXPHOS-favored state, however, our data suggests that Wnt5a-mediated metabolic re-programming of DCs is independent of AMPK. After determining that Wnt5a-stimulated DCs were not reliant on glutamine as an energy source for undergoing tolerization and that Wnt5a-stimulated DCs exhibit both enhanced fatty acid uptake and greater lipid stores, we reasoned that DC FAO was critical for driving this phenotype. Notably, a role for FAO in DC tolerance would also be consistent with the metabolic alterations observed in M2 macrophages and myeloid-derived suppressor cells (MDSCs) (Hossain et al., 2015; O’Neill and Pearce, 2016). Indeed, inhibiting FAO pharmacologically or genetically dominantly inhibited DC-mediated Treg cell generation and potently promoted DC-dependent stimulation of CD8+ T cell proliferation. The underlying mechanism of Wnt5a-induced FAO in DCs involve β-catenin-dependent PPAR-γ-mediated expression of CPT1A. We further demonstrate that β-catenin and PPAR-γ form a co-transcriptional activator complex in primary DCs upon Wnt5a exposure and this DC signaling pathway modulates the development of melanoma antigen-specific T cell responses in vivo. These cumulative findings may also explain the induction of FAO in M2 macrophages and MDSCs within the tumor microenvironment.
While the cellular oxidative state has been shown to regulate IDO enzyme activity, to our knowledge a relationship between metabolic regulation and the enzymatic activity of IDO has not been appreciated (Thomas et al., 2001). Here, we have shown that Wnt5a drives heme biosynthesis and the accumulation of the PpIX prosthetic group by both promoting TCA flux and expression of several enzymes involved in this pathway and the heme biosynthetic pathway including the rate limiting enzyme, ALAS1. Since PpIX is a limiting factor of IDO activity (Thomas et al., 2001), we propose that this pathway is a previously unrecognized mechanism of IDO regulation. Although we were unable to measure the metabolic intermediate succinyl CoA due to its relative instability, the diminished quantities of the α-ketoglutarate precursor and the increased expression of each component of the α-ketoglutarate dehydrogenase complex in addition to ALAS1, suggest that Wnt5a-mediated metabolic re-programming promotes heme synthesis by providing increased quantities of substrate while upregulating the expression of key TCA and heme synthesis enzymes.
Our data indicate that FAO in DCs has an impact on DC tolerization that extends beyond IDO. This led us to discover that this metabolic shift potently suppresses two key pro-inflammatory cytokines, IL-6 and IL-12, which contribute to a more favorable mileu for driving Treg cell differentiation. In particular, IL-6 antagonizes Treg cell development in several experimental systems by promoting the proteosomal degradation of IDO (Orabona et al., 2008). These effects on pro-inflammatory cytokine expression by DCs are consistent with previous work showing that Wnt5a suppresses upregulation of these same cytokines in response to LPS (Oderup et al., 2013). Given that FAO in DCs can influence multiple biochemical pathways important for DC tolerization, we speculate that targeting regulators of DC-specific FAO could potently impact the tumor immune microenvironment. Indeed, we have demonstrated that genetic silencing of CPT1A in primary DCs promotes antigen-specific CD8+ T cell activation and adoptive transfer of DCs treated with a pharmacologic FAO inhibitor significantly suppressed melanoma progression in a poorly immunogenic transgenic model of melanoma. Together, these data suggest that targeting the Wnt5a-β-catenin-FAO pathway represents a promising strategy for augmenting checkpoint inhibitor immunotherapy. This is consistent with the robust effect generated by combining a CPT1A-targeted inhibitor with anti-PD-1 antibody therapy in the BrafV600E-Pten−/− melanoma model. Further, since the Wnt5a-β-catenin-FAO pathway regulates several components of DC tolerization that extend beyond IDO, we propose that designing strategies to inhibit this pathway upstream of IDO may be more effective at inducing anti-tumor immunity than strictly targeting the activity of this enzyme.
Melanomas with few tumor-infiltrating lymphocytes (TILs) and a generally non-inflammed microenvironment are poorly responsive to checkpoint inhibitor therapy. In line with our mechanistic DC studies, recent gene expression profiling based on microarray and RNAseq datasets have demonstrated that primary melanomas, as well as other solid tumors, associated with a deficiency in tumor-infiltrating lymphocytes are associated with elevated β-catenin and PPAR-γ signaling (Spranger et al., 2015; Sweis et al., 2016). Despite this finding, a minority of these ‘TIL-poor’ cancers harbor genomic mutations that drive the β-catenin signaling pathway (Luke et al., 2016), suggesting that Wnt-mediated paracrine signaling pathways likely contribute to the elevated β-catenin activation state observed in these non-inflammed tumors. In this work, we have provided functional data indicating that Wnt5a promotes the establishment of an immune privileged, ‘TIL-poor’ melanoma microenvironment by driving FAO in DCs. The importance of Wnt5a in promoting an immune tolerant state is supported by a recent report employing RNAseq differential gene expression analysis demonstrating Wnt5a is one of the most significantly upregulated genes in melanomas refractory to pembrolizumab immunotherapy (Hugo et al., 2016).
Altogether, these findings demonstrate that DC tolerization in the tumor microenvironment contributes to immunotherapy resistance and suggest that Wnt ligand antagonism would be a promising strategy for augmenting anti-PD-1 antibody immunotherapy. Finally, these data further advocate for DC-specific manipulation of the FAO pathway as an approach for designing the next generation of DC-based cancer vaccines.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and fulfilled by the Lead Contact, Dr. Brent A. Hanks (brent.hanks@duke.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In vivo Animal Studies
C57BL/6J (C57, H-2b), BALB/cJ (H-2d), B6.Cg-Braftm1Mmcm Ptentm1Hwu Tg(Tyr-cre/ERT2)13Bos/BosJ (BrafV600EPten−/−, H-2b), C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-1, H-2b), B6.129-Ido1tm1Alm/J (IDO−/−, H-2b) mice were purchased from Jackson Labs. C57BL/6-Foxp3tm1Flv/J (Foxp3-mRFP, H-2b) mice were a gift from H.K. Lyerly (Duke University Medical Center, USA). The CD11c-βcat−/− (βcatΔDC, H-2b) strain was a gift from Santhakumar Manicassamy (Georgia Cancer Center, USA). All experimental groups included randomly chosen littermates of both sexes, ages 6–8 weeks, and of the same strain. Experiments were performed based on a protocol approved by the Institutional Animal Care and Use Committee at Duke University Medical Center.
Cell Lines
Murine bone marrow–derived DCs (BMDCs) were harvested and differentiated using IL-4 and GM-CSF as previously described (Inaba et al., 1992) and purified using CD11c microbeads (Miltenyi Biotec) according to manufacturer’s protocol. DC purity was examined by flow cytometry and consistently found to be >95% CD11c+F4/80− (Figure S3D). BrafV600EPten−/−(male), BrafV600EPten−/−-Wnt5a-silenced (male), and BrafV600EPten−/−-NTC (male) cell lines were generated and cultured as previously described (Holtzhausen et al., 2015). DC2.4, a murine dendritic cell line was kindly provided by Dr. Kenneth L. Rock (University of Massachusetts Medical School), and cultured as previously described (Shen et al., 1997). DC2.4-β-catenin-silenced, DC2.4-CPT1A-silenced, and DC2.4-NTC stable cell lines were generated using a β-catenin-targeted, CPT1A-targeted, or control shRNA-expressing lentivirus (Sigma) followed by 3μg/ml puromycin selection. All cell lines used in this study were tested mycoplasma free by Duke University Cell Culture Facility shared services.
METHOD DETAILS
Dendritic Cell Conditioning
DCs were treated with Wnt3a (100 ng/mL), Wnt5a (200 ng/mL), LPS (1 μg/mL), 1-MT (1 mM), 2DG (1 mM), or ETO (100 μM), 2-DG(2-deoxy-d-glucose, 1 mM), Oligomycin (1 μM), succinylacetone (250 μM), or vehicle control either for 24 or 48 hrs prior to their use in both in vitro and in vivo experiments.
Antibodies, Immunoprecipitation, and Immunoblot Analysis
Primary antibodies including CPT1A (Cell signaling), PPAR-γ (Santa Cruz Biotechnology), β-catenin, β-actin (Millipore), p-AMPK(T172)/AMPK (Cell signaling), p-AKT(T308)/Akt (Cell Signaling) were used at 1:1000. Secondary antibodies including goat anti-rabbit IgG-HRP (Millipore) and goat anti-mouse IgG-HRP (Millipore) were used at 1:5000. Cells were lysed in Laemmli sample buffer after treatment and subjected to SDS-polyacrylamide gel electrophoresis and immunoblot analysis. For immunoprecipitation, cells were lysed in radio immunoprecipitation assay (RIPA) buffer [10 mM sodium phosphate (pH 8.0), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS in the presence of 1 mM DTT, 1 mM phenylmethylsulfonylfluoride, and a protease inhibitor cocktail (Sigma)], precleared with protein A beads, and then incubated with 1 μg of antibody against β-catenin or isotype control IgG with protein A-agarose beads on a rotator overnight at 4°C. After 3 washes with RIPA buffer, immunoprecipitated complexes were eluted in sample buffer by boiling and subjected to immunoblot analysis. Immunoblots were visualized by chemiluminescence substrate (ThermoFisher) and imaged by a ChemiDoc XRSplus system (BioRad).
Flow Cytometry
One million cells were stained with 1 μg per million cells of each fluorochrome conjugated antibodies or commercially available dyes according to the standard protocols and analyzed using a FACSCanto II or LSRII (Becton Dickinson).
RNA Isolation, RT-qPCR, qPCR Array
Total RNA was isolated by RNeasy Plus Mini Kit(Qiagen). RNA(500ng) were used in cDNA Synthesis (iScript, BioRad). Quantification of mRNA for genes involved in PPAR γ signaling was performed using Mouse PrimePCR PPAR Array according to the manufacturer’s protocol (BioRad). Real-time PCR was performed using an ABI7500 Real-Time PCR system (Life Technologies). Data analysis utilized the PrimePCR Analysis Software (BioRad). Conventional qPCR was performed using validated primers (see KEY RESOURCES TABLE), and SsoAdvanced Universal SYBR Green Super mix (BioRad) or Taqman probes (Applied Biosystems) for heme synthesis enzymes.
KEY RESOURCE TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-β-actin antibody, clone:C4 | Santa Cruz Biotechnology | RRID:AB_2714189 |
| Anti-CPT1A Rabbit mAb, clone:D3B3 | Cell Signaling | 12252 |
| Anti-PPAR-γ Antibody,clone:H-100 | Santa Cruz Biotechnology | RRID:AB_654710 |
| Anti-β-catenin, rabbit polyclonal | Millipore Sigma | RRID:AB_310231 |
| Anti-Phospho-AMPKα (Thr172) rabbit mAb, clone:40H9 | Cell Signaling | RRID:AB_331250 |
| Anti-AMPKα Rabbit mAb, clone:D5A2 | Cell Signaling | RRID:AB_10622186 |
| Anti-Phospho-Akt (Thr308) Antibody, rabbit polyclonal | Cell Signaling | RRID:AB_329828 |
| Anti-Akt Antibody, rabbit polyclonal | Cell Signaling | RRID:AB_329827 |
| Anti-Wnt5a Antibody, clone: A-5 | Santa Cruz Biotechnology | RRID:AB_10846090 |
| Anti-PD-L1 Rabbit mAb, Clone: D5V3B | Cell Signaling | 64988 |
| Anti-CD8α XP Rabbit mAb, Clone:D4W2Z | Cell Signaling | 98941 |
| Anti-mouse I-A/I-E(MHCII), Alexa Fluor 488 conjugated, clone:M5/114.15.2 | BD Pharmingen | RRID:AB_11151902 |
| Anti-Mouse CD11c, FITC congugated, clone: HL3 | BD Pharmingen | RRID:AB_395060 |
| Anti-Mouse CD274, PE congugated, clone: MIH5 | BD Pharmingen | RRID:AB_397018 |
| Anti-Mouse CD8a, BV510 congugated, clone: 53-6.7 | BD Pharmingen | RRID:AB_2687548 |
| Anti-Mouse CD3e,PerCP-Cy5.5 congugated, clone: 145-2C11 | BD Pharmingen | RRID:AB_394082 |
| Anti-Mouse CD4, FITC congugated, clone: RM4-5 | BD Pharmingen | RRID:AB_394583 |
| Anti-Mouse Foxp3, PE congugated, clone: MF23 | BD Pharmingen | RRID:AB_1645251 |
| Anti-Mouse CD279, APC congugated, clone: J43 | BD Pharmingen | 562671 |
| Anti-mouse CD40 Pacific Blue conjugated, clone:3/23 | Biolegend | RRID:AB_2561475 |
| Anti-mouse CD80, PerCP/Cy5.5 conjugated, clone: 16-10A1 | Biolegend | RRID:AB_2291392 |
| Anti-mouse CD86, APC conjugated, clone:GL-1 | Biolegend | RRID:AB_493342 |
| Anti-mouse F4/80 Antibody, APC congugated, clone: BM8 | Biolegend | RRID:AB_893481 |
| Anti-mouse IL-6, unconjugated, clone:MP5-20F3 | Biolegend | RRID:AB_315339 |
| Anti-mouse IL-12/IL23 p40, unconjugated, clone:C17.8 | Biolegend | RRID:AB_315375 |
| Goat Anti-Rabbit IgG (H+L)-HRP Conjugate | Bio-Rad | RRID:AB_11125143 |
| Goat Anti-Mouse IgG (H+L)-HRP Conjugate | Bio-Rad | RRID:AB_11125547 |
| InVivoMAB Anti-mouse PD-1, clone: RMP1-14 | BioXCell | RRID:AB_10949053 |
| InVivoMAb rat IgG2a isotype control, clone: 2A3 | BioXCell | RRID:AB_1107769 |
| Anti-mouse CD8 antibody from hybridoma, clone: 53.6.7 | Duke Cell Culture Facility | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human/Mouse Wnt5a Protein | R&D Systems | 645-WN-010 |
| Recombinant Mouse Wnt3a Protein | R&D Systems | 1324-WN-002 |
| Recombinant Mouse IL-4 | BioAbChem | 42-IL4 |
| Recombinant Mouse GM-CSF Protein | R&D systems | 415-ML-010 |
| Glucose | Sigma-Aldrich | G7021-100G |
| Oligomycin | Sigma-Aldrich | O4876-25MG |
| 2-DG (2-Deoxy-D-glucose) | Sigma-Aldrich | D3179-1G |
| FCCP (Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone) | Sigma-Aldrich | C2920-10MG |
| Rotenone | Sigma-Aldrich | 557368-1GM |
| Antimycin A | Sigma-Aldrich | A8674-50MG |
| (+)-Etomoxir sodium salt hydrate | Sigma-Aldrich | E1905-25MG |
| 4-hydroxytamoxifen | Sigma-Aldrich | H6278-10MG |
| L-tryptophan | Sigma-Aldrich | T4196-100G |
| L-kynurenine | Sigma-Aldrich | K8625-25MG |
| BPTES | Sigma-Aldrich | SML0601-5MG |
| AICAR | Sigma-Aldrich | A9978-5MG |
| XAV939 | Sigma-Aldrich | X3004-5MG |
| noble agar | Sigma-Aldrich | A5431-250G |
| NAD+ (β-Nicotinamide adenine dinucleotide) | Sigma-Aldrich | N6522 |
| Polyethylene glycol(PEG)-8000 | Sigma-Aldrich | 89510-250G-F |
| L-Lactic Dehydrogenase from rabbit muscle | Sigma-Aldrich | L2500-10KU |
| Glycine Buffer solution | Sigma-Aldrich | G5418-100ML |
| Sodium L-lactate | Sigma-Aldrich | L7022 |
| LPS (Lipopolysaccharides) | Sigma-Aldrich | L4391-1MG |
| ALA (5-Aminolevulinic acid hydrochloride) | Sigma-Aldrich | A3785-1G |
| Succinylacetone(4,6-Dioxoheptanoic acid) | Sigma-Aldrich | D1415-100MG |
| BODIPY 493/503 | ThermoFisher | D-3922 |
| BIO (GSK-3 Inhibitor IX) | Millipore | 361550-1MG |
| MeBIO(GSK-3 Inhibitor IX, Control) | Millipore | 361556-1MG |
| Critical Commercial Assays | ||
| CD11c MicroBeads UltraPure, mouse | Miltenyi Biotec | 130-108-338 |
| Naive CD4+ T Cell Isolation Kit, mouse | Miltenyi Biotec | 130-104-453 |
| RNeasy Plus Mini Kit | Qiagen | 74134 |
| Fatty Acid Uptake Kit | Sigma-Aldrich | MAK156-1KT |
| Prime PCR PCR Array PPAR targets M96 | Bio-Rad | 10034399 |
| Hemin kit | BioVision | K672 |
| Seahorse Xfe24 FluxPaks | Agilent | 102342-100 |
| Annexin V-FITC Apoptosis Detection Kit | Sigma-Aldrich | APOAF-20TST |
| ATP Determination Kit | ThermoFisher | A22066 |
| Mouse IFN-γ ELISpot PLUS (ALP) | MABTECH | 3321-4APW-2 |
| Mouse IL6 ELISA Kit | ThermoFisher(eBioscience) | 50-112-8863 |
| Mouse IL12(p40) ELISA Set | BD Biosciences | 555165 |
| Warp Red Chromogen Kit | Biocare | WR806 S |
| CellTrace Violet Cell Proliferation Kit | ThermoFisher | C34571 |
| SsoAdvanced Universal SYBR Green Supermix | BIO-Rad | 1725271 |
| iScript Reverse Transcription Supermix | BIO-Rad | 1708841 |
| SsoAdvance Universal Probes Supermix | BIO-Rad | 172-5281 |
| Pierce Gentle Ag/Ab Binding and Elution Buffer Kit | ThermoFisher | 21030 |
| Experimental Models: Cell Lines | ||
| BRAFV600EPTEN−/− melanoma cells | Hanks Lab | PMID: 26041736 |
| BRAFV600EPTEN−/−-NTC | Hanks Lab | PMID: 26041736 |
| BRAFV600EPTEN−/−-Wnt5aKD | Hanks Lab | PMID: 26041736 |
| DC2.4 | Rock Lab | RRID:CVCL_J409 |
| DC2.4-NTC | Hanks Lab | this paper |
| DC2.4-CPT1AKD | Hanks Lab | this paper |
| DC2.4-β-cateninKD | Hanks Lab | this paper |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: BALB/cJ | Jackson Laboratory | RRID:IMSR_JAX:000651 |
| Mouse: B6.Cg-Braftm1Mmcm Ptentm1Hwu Tg(Tyr-cre/ERT2 H-2b)13Bos/BosJ | Jackson Laboratory | RRID:IMSR_JAX:012328 |
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson Laboratory | RRID:IMSR_JAX:003831 |
| Mouse: B6.129-Ido1tm1Alm/J | Jackson Laboratory | RRID:IMSR_JAX:005867 |
| Mouse: C57BL/6-Foxp3tm1Flv/J | Jackson Laboratory | RRID:IMSR_JAX:008374 |
| Mouse: CD11c-bcat−/− | Manicassamy Lab | PMID: 25710911 |
| Oligonucleotides | ||
| Primer: mACTB Forward: GGCTGTATTCCCCTCCATCG | IDT | N/A |
| Primer: mACTB Reverse: CCAGTTGGTAACAATGCCATGT | IDT | N/A |
| Primer: mPPARγ Forward: GCCCTTTGGTGACTTTATGGA | IDT | N/A |
| Primer: mPPARγ Reverse: GCAGCAGGTTGTCTTGGATG | IDT | N/A |
| Primer: mCPT1A Forward: CTCAGTGGGAGCGACTCTTCA | IDT | N/A |
| Primer: mCPT1A Reverse: GGCCTCTGTGGTACACGACAA | IDT | N/A |
| Primer: mCPT1B Forward: TTCAACACTACACGCATCCC | IDT | N/A |
| Primer: mCPT1B Reverse: GCCCTCATAGAGCCAGACC | IDT | N/A |
| Primer: mCPT1C Forward: TCTTCACTGAGTTCCGATGGG | IDT | N/A |
| Primer: mCPT1C Reverse: ACGCCAGAGATGCCTTTTCC | IDT | N/A |
| Primer: mIL6 Forward: TAGTCCTTCCTACCCCAATTTCC | IDT | N/A |
| Primer: mIL6 Reverse: TTGGTCCTTAGCCACTCCTTC | IDT | N/A |
| Primer: mIL10 Forward: GACCAGCTGGACAACATAC | IDT | N/A |
| Primer: mIL10 Reverse: CTGGAGTCCAGCAGACTC | IDT | N/A |
| Primer: mIL12B Forward: GAACACATGCCCACTTGCTG | IDT | N/A |
| Primer: mIL12B Reverse: CGTGCTCATGGCTGGTGCAAAG | IDT | N/A |
| Primer: mTGFβ Forward: GCAACAACGCCATCTATGAG | IDT | N/A |
| Primer: mTGFβ Reverse: TCTTTGCTGTCACAAGAGC | IDT | N/A |
| Primer: mPFK Forward: GGAGGCGAGAACATCAAGCC | IDT | N/A |
| Primer: mPFK Reverse: CGGCCTTCCCTCGTAGTGA | IDT | N/A |
| Primer: mHK3 Forward: TGCTGCCCACATACGTGAG | IDT | N/A |
| Primer: mHK3 Reverse: CCTGTCAGTGTTACCCACAA | IDT | N/A |
| Primer: mCTNNB Forward: TCCCATCCACGCAGTTTGAC | IDT | N/A |
| Primer: mCTNNB Reverse: TCCTCATCGTTTAGCAGTTTTGT | IDT | N/A |
| Primer: mALAS1 Forward: GATGCCAGGCTGTGAAATTTACT | IDT | N/A |
| Primer: mALAS1 Reverse: CTGTTGCGAATCCCTTGGAT | IDT | N/A |
| Primer: mGAPDH Forward: GTCTACATGTTCCAGTATGACTCC | IDT | N/A |
| Primer: mGAPDH Reverse: AGTGAGTTGTCATATTTCTCGTGGT | IDT | N/A |
| Recombinant DNA | ||
| Mission shRNA Plasmid DNA against CPT1A | Sigma-Aldrich | SHCLND-NM_013495 |
| Mission shRNA Plasmid DNA against CTNNB1(β-catenin) | Sigma-Aldrich | SHCLND-NM_001904 |
| Mission shRNA Plasmid DNA against Wnt5a | Sigma-Aldrich | SHCLND-NM_009524 |
| Mission pLKO.1-puro Empty vector Control Plasmid | Sigma-Aldrich | SHC001 |
| Software and Algorithms | ||
| Wave Desktop 2.4 | Agilent | www.agilent.com |
| Graphpad Prism 7 | GraphPad Software | www.graphpad.com |
| ImageJ | NIH | RRID:SCR_003070 |
| FlowJo v10.3 | Flowjo | www.flowjo.com |
| PrimePCR Analysis | Bio-Rad | www.bio-rad.com |
| Image Lab | Bio-Rad | www.bio-rad.com |
| 7500 software v2.3 | Applied Biosystems | www.thermofisher.com |
| ImmunoSpot 5.0 | ImmunoSpot | www.immunospot.com |
| ImmunoCapture6.3.5 | ImmunoSpot | www.immunospot.com |
Murine Cell Isolation
Spleens were diced into 1 mm3 pieces, digested with spleen dissociation buffer (Stemcell Technologies) for 30 minutes at room temperature. Tumors were resected and mechanically disaggregated by gentleMACS (Miltenyi) and digested with RPMI containing collagenase IV (1 mg/ml), hyaluronidase (0.1mg/ml), and deoxyribonuclease (20 U/ml, Sigma) at 37°C for 1 ho ur. A 40-micron filter was used to obtain a single cell suspension for downstream applications. DCs were purified using CD11c microbeads and naïve CD4 T cells were obtained using a naïve T cell isolation kit (Stemcell Technologies). All cell populations were verified for purity by flow cytometry based on a CD45+CD11c+F4/80−IAb/d+ and a CD3+CD4+CD62L+ profile, respectively.
Human Monocyte-derived Dendritic Cells. Human monocyte-derived DCs were generated as previously described (Nair et al., 2012).
ELISPOT
Single cell suspension of tumors were plated at 500,000/well on ELISPOT plate and incubated for 48 hours. Mouse IFNγ ELISPOTPLUS (MABTECH) was performed according to manufacture guidelines. Imaging was conducted using a CTL Immunospot S5 core (Immunospot) and quantified using ImmunoCapture and ImmunoSpot software(Immunospot).
ELISA
Murine IL-6 (eBioscience) and IL-12p40 (Becton Dickinson) ELISAs were performed according to manufacture’s protocol.
Immunohistochemistry/Immunofluorescence
Paraffin-embedded tissues were processed and stained following standard protocols and imaged with a Zeiss CLSM 700 confocal microscope. CD8 (BioLegend) and PD-L1 (Abcam) primary antibodies were utilized where indicated. Warp Red chromogen detection system (BioCare) was used for antigen visualization.
T Cell Proliferation Assay
Splenocytes of OT-1 mice (H-2b) were isolated and stained with CellTrace Violet (ThermoFisher). Preconditioned DCs were loaded with ovalbumin peptide SIINFEKL, and co-cultured at a DC:splenocyte ratio of 40,000:120,000 cells for 72 hrs. CD8+ T cell proliferation was measured by the dilution of Cell Trace Violet dye by flow cytometry.
Treg Cell Assays
In vivo Treg cell assay. DCs (C57, H-2b) were pre-treated for 48 hours and delivered by intradermal injection into the footpad of Foxp3-mRFP mice. Draining inguinal and popliteal lymph nodes were resected 5 days later and analyzed for CD4+Foxp3+ Treg cells. In vitro Treg cell assay, DCs (Balb/c, H-2d) were pre-treated for either 24 or 48 hrs, and re-plated at a 1:3 DC:T-cell ratio with purified allogeneic naïve Foxp3-mRFP(H-2b) CD44loCD62LhiCD4+ Tcells. These co-cultures were incubated for 6 days and quantitated for CD4+FoxP3+ Treg cells by flow cytometry.
BODIPY and Fatty Acid Uptake Assay
Dendritic Cells were stained in 0.5 μg/ml BODIPY 493/503 in PBS for 15 min to determine neutral lipid content (Herber et al., 2010). Fatty acid uptake measurement in DCs were performed using a dodecanoic acid fluorescent TF2-C12 fatty acid (Sigma) according to the manufacturer’s protocol.
Lactate measurement
L-Lactate was measured by lactate dehydrogenase conversion of L-lactate + NAD+ to pyruvate + NADH following treatment with hydralazine (Pesce et al., 1975). Lactate standards and samples were read at NADH specific absorbance 340 nm. For lactate measurement in Wnt5a time course, BMDCs were seeded in a 48-well plate at 1 million cell per well. BMDCs were treated with 100 ng/ml of recombinant Wnt5a from 0 to 48 hours. For extracellular lactate determination, 0.5 ml of supernatant media was collected directly from the culture, deproteinized by polyethylene glycol precipitation (25% w/v PEG-8000, sigma), and clarified by centrifugation at 20,000g for 5 min. For intracellular lactate determination, Cells were washed with ice cold PBS, scraped off in 100 μl Milli-Q water (4°C), freeze (−80 °C) thawed repeatedly 3 times for efficient cell lysis, and then deproteinized as described above. L-lactate was measured by lactate dehydrogenase (LDH, final 2 U/ml, Sigma) conversion of L-lactate + NAD+ (β-Nicotinamide adenine dinucleotide, Sigma) to pyruvate + NADH in Glycine Buffer solution (final concentration Glycine 0.2M, Hydrazine 0.17M to destroy pyruvate allowing reaction to run to complete oxidation of lactate, Chloroform 0.0125%, Sigma). Reaction was incubated at 37°C for 30 minute (without CO2), L-lactate standards and samples were read at NADH specific absorbance 340 nm (Infinite 200 PRO, Tecan).
Cellular Energy Metabolism Analysis
DC energy metabolism was measured using the XFe24 extracellular flux analyzer (Seahorse Bioscience), with the glycolysis stress test kit and the mitochondrial stress test kit as previously described (Everts et al., 2014; Zhao and Klimecki, 2015). For experiments involving LPS injection, DCs were plated in XFe24 plates (200,000/well in 500μl) and treated with LPS for 48 hours prior to XF analysis. DCs were washed and analyzed in XF media (RPMI without sodium bicarbonate, 10mM glucose, 1%FBS, 2mM L-glutamine). ECAR was analyzed in real-time with or without LPS stimulation. For experiments involving BrafV600EPten−/− cell line-derived conditioned media (CM), collected CM was concentrated and desalted with an Amicon Ultra 30K filter according to manufacture’s recommendations (Milipore). Final concentrate was further washed with XF media 3 times, added to DCs immediately prior to OCR analysis. For standard ECAR analysis, XF media (without glucose) was used to wash cells prior to the assay. A final concentration of 10mM glucose, 1μM oligomycin, 50 mM 2-DG(sigma) were injected through XFe24 port A–C. For standard OCR analysis, XF media (with 10mM glucose) was used to wash cells, a final concentration of 1 μM oligomycin, 1.5 μM FCCP (fluoro-carbonyl cyanide phenylhydrazone), 100 nM rotenone, and 1 μM of antimycin-A (Sigma) were injected through XFe24 port A–C.
IDO enzymatic assay and Hemin assay
DC IDO enzyme activity was measured by the conversion of L-tryptophan to L-kynurenine in conditioned media by HPLC (Pallotta et al., 2011). Intracellular hemin was measured using a colorimetric assay kit (BioVision).
PpIX Analysis
Dendritic cells were terminally incubated in the presence of 1mM δ-aminolevulinic acid (ALA) for 4 hrs. Intracellular PpIX was analyzed by flow cytometry as previously described (Hryhorenko et al., 1998).
Metabolomics
Metabolites were extracted from 5 × 106 BMDCs and subjected to LC-MS analysis according to a previously published protocol (Liu et al., 2014a; Liu et al., 2014b). Metabolites were extracted from 5 × 106 BMDCs from each experimental group (3 mice per group), washed with ice cold PBS, and lysed in 80% (v/v) methanol on dry ice. Cell lysates were frozen at −80°C for 15 minutes to disrupt cell membrane and quench enzymatic activity. Samples were then thawed on ice, vortexed rigorously to extract metabolites, and then centrifuged at 20,000g at 4 °C to precipitate proteins and cell debris. Metabolite extracts were then dried using a speed vacuum and subjected to LC-MS analysis (Liu et al., 2014a; Liu et al., 2014b). Data collected from LC-Q exactive MS is processed on Sieve 2.0 (Thermo). For metabolite analysis, theoretical m/z and retention time of 263 known metabolites were used for positive mode, and 197 metabolites were used for negative mode. Data containing detected m/z and relative intensity of different samples were obtained.
Soft Agar Colony Formation Assay
Complete growth media - 0.7% agar was overlaid with complete growth media - 3.5% agar containing 10,000 cells and additional complete growth media. After 2 weeks, colonies were stained with MTT(Sigma) to identify viable colonies and imaged by a ChemiDoc XRSplus system as previously described (Zhao et al., 2014). Images were analyzed with NIH ImageJ to enumerate colony number.
In vivo CD8 Depletion
Hybridoma clone 53-6.7 was expanded at the Duke Cell Culture Facility in hollow fiber cartridges; 10 ml of serum free supernatant was harvested every 2 days. Anti-mouse CD8 antibody was purified by Pierce Gentle Ag/Ab Binding and Elution Buffer Kit according to manufacturer protocols (ThermoFisher). Antibody concentration was determined by BCA protein assay. Anti-CD8 mAb or IgG isotype control was delivered daily for the first three days then every 7 days thereafter by intraperitoneal injection (500 μg/mouse/dose). CD8 depletion was verified by flow cytometry (Figure S7B).
Syngeneic Transplant Tumor Studies
BrafV600EPten−/− cells line were established as previously described (Holtzhausen et al., 2015). 5 × 105 cells were implanted by subcutaneous injection into syngeneic C57BL/6 mice. Tumor growth was monitored by caliper measurement. Etomoxir (Sigma, ETO) was administered daily by oral gavage (25 mg/kg/day) (Collier et al., 1993). Anti-PD-1 rat mAb or rat IgG2a isotype control (BioXCell) was delivered every 3 days by intraperitoneal injection (250 μg/dose).
Autochthonous Tumor Studies
B6.Cg-Braftm1Mmcm Ptentm1Hwu Tg(Tyr-cre/ERT2 H-2b)13Bos/BosJ (BrafV600EPten−/−, H-2b), transgenic mice were subdermally injected with 4-HT (Sigma, 38.75 μg/mouse) to induce primary melanoma development. Three days prior to 4-HT injection, 1 × 106 cells DCs pretreated with Wnt5a +/− ETO were washed and delivered by intra-dermal injection into the hind leg foot pad every 3–4 days until the conclusion of the experiment. Melanoma growth was monitored by orthogonal caliper measurements every 3–4 days between day 15 to day 32.
QUANTIFICATION AND STATISTICAL ANALYSIS
Specific statistical tests are reported in the Figure Legends. GraphPad Prism 7 Windows version was used for all statistical analyses. Unpaired t-test were used to compare mean differences between control and treatment groups. Univariate ANOVA followed by Tukey’s post hoc test were performed to analyze data containing three or more groups. For time lapse extracellular flux analysis repeated measures ANOVA analysis was performed.
Supplementary Material
Highlights.
Melanomas metabolically reprogram local DCs to induce immune tolerance
Melanoma-derived Wnt5a triggers DC FAO via a β-catenin-PPAR-γ pathway
DC FAO drives IDO enzymatic activity by promoting protoporphyrin IX synthesis
Inhibiting the Wnt5a-β-catenin-CPT1A pathway enhances anti-PD-1 antibody activity
Acknowledgments
We would like to thank Dr. Ciriana Orabona (University of Perugia) for assistance with the kynurenine HPLC assay and Dr. Jeff Rathmell (Vanderbilt University) for his assistance with the preparation of the manuscript. B.A.H is supported by start-up funding by the Duke Cancer Institute and the National Cancer Institute of the National Institutes of Health (1K08CA191063-01A1). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors declare no relevant conflicts of interest.
Footnotes
Supplemental Information includes 7 Figures.
Author Contributions.
Conceptualization, FZ, BAH; Methodology, FZ; Investigation, FZ, AH, CX, TT, KSE, ND, JL, XL; Resources, DB, SN, JWL; Formal Analysis, JWL, FZ, BAH; Data Curation: FZ, BAH; Writing, FZ, AH, BAH; Visualization, FZ, AH, BAH; Supervision, BAH; Project Administration, BAH; Funding Acquisition, BAH.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Banchereau J, Steinman R. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1- IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15:323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallarino F, Grohmann U, You S, McGrath BC, Cavener DR, Vacca C, Orabona C, Bianchi R, Belladonna ML, Volpi C, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor {zeta}-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176:6752–6761. doi: 10.4049/jimmunol.176.11.6752. [DOI] [PubMed] [Google Scholar]
- Gabrilovich D. Mechanisms and functional significance of tumor-induced dendritic cell defects. Nature Rev Immunol. 2004;4:941–952. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- Handschin C, Lin J, Rhee J, Peyer AK, Chin S, Wu PH, Meyer UA, Spiegelman BM. Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell. 2005;122:505–515. doi: 10.1016/j.cell.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Hanks BA, Holtzhausen A, Jamieson R, Gimpel P, Campbell O, Sun L, Tewari A, George A, Starr M, Nixon A, et al. Type III TGF-β Receptor Downregulation Generates an Immunotolerant Tumor Microenvironment. J Clin Invest. 2013;123:3925–3940. doi: 10.1172/JCI65745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, Corzo A, Cho HI, Celis E, Lennox B, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16:880–886. doi: 10.1038/nm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzhausen A, Zhao F, Evans K, Tsutsui M, Orabona C, Tyler DS, Hanks BA. Melanoma-derived Wnt5a Promotes Local Dendritic-Cell Expression of IDO and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol Res. 2015;3:1082–1095. doi: 10.1158/2326-6066.CIR-14-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, Del Valle L, Trillo-Tinoco J, Maj T, Zou W, et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol Res. 2015 doi: 10.1158/2326-6066.CIR-15-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryhorenko EA, Rittenhouse-Diakun K, Harvey NS, Morgan J, Stewart CC, Oseroff AR. Characterization of endogenous protoporphyrin IX induced by delta-aminolevulinic acid in resting and activated peripheral blood lymphocytes by four-color flow cytometry. Photochem Photobiol. 1998;67:565–572. [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and Transcriptomic Features of Response to Anti-PD- 1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. The Journal of experimental medicine. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson EA, Are A, Greicius G, Kuo IC, Kelly D, Arulampalam V, Pettersson S. The Wnt/beta-catenin signaling pathway targets PPARgamma activity in colon cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1460–1465. doi: 10.1073/pnas.0405928102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, Jiang S, Whitney JA, Connolly J, Banchereau J, Mellman I. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity. 2007;27:610–624. doi: 10.1016/j.immuni.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ser Z, Locasale JW. Development and quantitative evaluation of a high-resolution metabolomics technology. Anal Chem. 2014;86:2175–2184. doi: 10.1021/ac403845u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke JJ, Bao R, Spranger S, Sweis RF, Lingen MW, Lengyel E, Zha Y, Gajewski TF. Wnt/Beta-catenin pathway activation correlates with immune exclusion across most human cancers. Journal of Clinical Oncology. 2016:34. doi: 10.1158/1078-0432.CCR-18-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, Fairhurst AM, Connolly JE. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J Immunol. 2015;194:5174–5186. doi: 10.4049/jimmunol.1303316. [DOI] [PubMed] [Google Scholar]
- Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, Pulendran B. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science. 2010;329:849–853. doi: 10.1126/science.1188510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. 2008;8:74–80. doi: 10.1038/nri2233. [DOI] [PubMed] [Google Scholar]
- Mullard A. Immunotherapy interest drives IDO deals. Nature reviews Drug discovery. 2015;14:373–373. [Google Scholar]
- Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, Messina JL, Chandler P, Koni PA, Mellor AL. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114:280–290. doi: 10.1172/JCI21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, Marshall B, Chandler P, Antonia SJ, Burgess R, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science. 2002;297:1867–1870. doi: 10.1126/science.1073514. [DOI] [PubMed] [Google Scholar]
- Nair S, Archer GE, Tedder TF. Current Protocols in Immunology. John Wiley & Sons; 2012. Isolation and generation of human dendritic cells; pp. 32.31–32.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213:15–23. doi: 10.1084/jem.20151570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oderup C, LaJevic M, Butcher EC. Canonical and noncanonical Wnt proteins program dendritic cell responses for tolerance. J Immunol. 2013;190:6126–6134. doi: 10.4049/jimmunol.1203002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orabona C, Pallotta MT, Volpi C, Fallarino F, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Grohmann U, Puccetti P. SOCS3 drives proteasomal degradation of indoleamine 2,3- dioxygenase (IDO) and antagonizes IDO-dependent tolerogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20828–20833. doi: 10.1073/pnas.0810278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, Servillo G, Brunacci C, Calvitti M, Bicciato S, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12:870–878. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016;44:924–938. doi: 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, Cubillos-Ruiz JR, Jacobs AC, Gonzalez JL, Weaver J, et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med. 2012;209:495–506. doi: 10.1084/jem.20111413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MD, Baban B, Chandler PR, Hou DY, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, Munn DH. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine-2,3-dioxygenase. J Clin Invest. 2007;117:2570–2582. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158:2723–2730. [PubMed] [Google Scholar]
- Sherwood V, Chaurasiya SK, Ekstrom EJ, Guilmain W, Liu Q, Koeck T, Brown K, Hansson K, Agnarsdottir M, Bergqvist M, et al. WNT5A-mediated beta-catenin-independent signalling is a novel regulator of cancer cell metabolism. Carcinogenesis. 2014;35:784–794. doi: 10.1093/carcin/bgt390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Nomiyama S, Hirata F, Hayaishi O. Indoleamine 2,3-dioxygenase. Purification and some properties. J Biol Chem. 1978;253:4700–4706. [PubMed] [Google Scholar]
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents antitumour immunity. Nature. 2015 doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31:711–723. e714. doi: 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Upregulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Science translational medicine. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweis RF, Spranger S, Bao R, Paner GP, Stadler WM, Steinberg G, Gajewski TF. Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol Res. 2016 doi: 10.1158/2326-6066.CIR-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SR, Salahifar H, Mashima R, Hunt NH, Richardson DR, Stocker R. Antioxidants inhibit indoleamine 2,3-dioxygenase in IFN-gamma-activated human macrophages: posttranslational regulation by pyrrolidine dithiocarbamate. J Immunol. 2001;166:6332–6340. doi: 10.4049/jimmunol.166.10.6332. [DOI] [PubMed] [Google Scholar]
- Zhao F, Klimecki WT. Culture conditions profoundly impact phenotype in BEAS-2B, a human pulmonary epithelial model. Journal of applied toxicology: JAT. 2015;35:945–951. doi: 10.1002/jat.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Malm SW, Hinchman AN, Li H, Beeks CG, Klimecki WT. Arsenite-induced pseudo-hypoxia results in loss of anchorage-dependent growth in BEAS-2B pulmonary epithelial cells. PloS one. 2014;9:e114549. doi: 10.1371/journal.pone.0114549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.