

Abstract
Background
Characterizing aggregate genetic risk for alcohol misuse and identifying variants involved in gene-by-environment interaction (G×E) effects has so far been a major challenge. We hypothesized that functional genomic information could be used to enhance detection of polygenic signal underlying alcohol misuse, and to prioritize identification of single nucleotide polymorphisms (SNPs) most likely to exhibit G×E effects.
Methods
We examined these questions in the young adult FinnTwin12 sample (n=1170). We used genome-wide association estimates from an independent sample to derive two types of polygenic scores for alcohol problems in FinnTwin12. Genome-wide polygenic scores included all SNPs surpassing a designated p-value threshold. DNase polygenic scores were a subset of the genome-wide polygenic scores including only variants in DNase I hypersensitive sites (DHSs), which are open chromatin marks likely to index regions with a regulatory function. We conducted parallel analyses using height as a non-psychiatric model phenotype in order to evaluate the consistency of effects. For the G×E analyses, we examined whether SNPs in DHSs were overrepresented among SNPs demonstrating significant G × E effects in an interaction between romantic relationship status and intoxication frequency.
Results
Contrary to our expectations, we found that DNase polygenic scores were not more strongly predictive of alcohol problems than conventional polygenic scores. However, variants in DNase polygenic scores had per-SNP effects that were up to 1.4 times larger than variants in conventional polygenic scores. This same pattern of effects was also observed in supplementary analyses with height. In G×E models, SNPs in DHSs were modestly overrepresented among SNPs with significant interaction effects for intoxication frequency.
Conclusions
These findings highlight the potential utility of integrating functional genomic annotation information in order to increase the signal-to-noise ratio in polygenic scores and identify genetic variants that may be most susceptible to environmental modification.
Keywords: alcohol, functional genomics, gene-environment interplay, polygenic scores
Introduction
Alcohol misuse (i.e., risky drinking and alcohol use disorder) is a top public health problem worldwide (World Health Organization, 2014), and reflects a complex interplay of genetic and environmental influences across development (Pagan et al., 2006). Twin and adoption studies have been critical in demonstrating that genetic influences account for roughly half of the variation in the risk for alcohol use disorder (Verhulst et al., 2015) and other alcohol use behaviors (Dick et al., 2011). Translating findings from family-based research designs of unmeasured genetic variance (i.e., inferred based on resemblance among different types of relatives) into a measured genetic framework to identify the specific variants associated with alcohol outcomes has been challenging (Hart and Kranzler, 2015). Although a few individually important genes and genetic variants have been identified, results from genome-wide association studies (GWAS) of alcohol use disorder underscore its highly polygenic nature (Yang et al., 2014, Hart and Kranzler, 2015, Mbarek et al., 2015). This high level of polygenicity is consistent with emerging findings from GWAS of psychiatric disorders more broadly (Geschwind and Flint, 2015), as well as findings that psychiatric conditions also share much of their polygenic underpinnings (Anttila et al., available online April 2016).
Characterizing Aggregate Genetic Risk for Alcohol Outcomes
In recent years, polygenic approaches have emerged as one method to characterize aggregate measured genetic risk (Wray et al., 2014). These efforts were motivated by the growing recognition that many genes and genetic variants, each of small individual effects, contribute to complex disorders; as well as the practical, clinical goal of being able to accurately predict disease and disorder from genetic information. Most commonly, polygenic scores are created by summing the number of “risk” alleles an individual carries across a selected set of single nucleotide polymorphisms (SNPs), weighted by empirical information from genetic association results obtained from an independent discovery sample. In effect, polygenic scores capture the composite additive effect of these multiple variants. This approach was initially used in the study of schizophrenia (The International Schizophrenia Consortium, 2009) and has since been applied to numerous complex traits (Dudbridge, 2013). As reviewed by Hart & Kranzler (2015), several recent studies have successfully used polygenic score approaches to predict alcohol-related outcomes (Yan et al., 2014, Vrieze et al., 2013, Frank et al., 2012, Kos et al., 2013, Levey et al., 2014).
Polygenic scores can encompass thousands of individual genetic variants spread throughout the genome and include a mixture of true genetic association signal and noise from statistical artifact and stochastic error (Maher, 2015). Conventional polygenic scoring methods have typically accounted for less than 2% of the genetic liability underlying complex traits, although this improves as the discovery sample sizes increases. Simulations indicate that tens of thousands of subjects may still be needed to achieve clinically meaningful prediction with these methods (Dudbridge, 2013). Efforts to amplify true genetic signal and reduce noise could enhance the predictive power of polygenic scores. Although some methods have been developed to improve polygenic scores, as of yet there has been no attempt to use information beyond the discovery GWAS (i.e. p value thresholds for filtering the inclusion of SNPs or linkage disequilibrium structure for weighting SNPs) to further refine the creation of such scores.
The past decade of genomic research has provided a wealth of information about the genetic variants that are being aggregated in these polygenic scores, including information about which variants are more or less likely a priori to have functional consequences on human traits and behaviors (ENCODE Project Consortium, 2012). In the same way that functional genomic information is important for understanding the biological coherence underlying GWAS results, it may also inform better ways to characterize individuals’ aggregate genetic risk for alcohol outcomes. Recent large-scale efforts have established that genetic variants associated with a variety of complex diseases and traits are not randomly distributed throughout the genome, but rather are stratified based on their genomic context (Schork et al., 2013, Finucane et al., 2015). Across many complex diseases and traits, there is modest evidence for an overrepresentation of SNPs with significant GWAS signals in or near protein-coding regions, and even stronger evidence for overrepresentation of SNPs in certain noncoding regions (Hindorff et al., 2009). Once considered “junk DNA”, it is now known that many regions outside of the exons that code for proteins have an indirect biological effect through the regulation of when, in what tissue, and under what circumstances a gene is expressed (ENCODE Project Consortium, 2012). Epigenetic factors near the transcription start site of a gene and in other key regulatory regions can influence gene expression by changing the physical conformation of the DNA, thus changing how accessible the DNA is to the cellular machinery responsible for transcribing genes into proteins.
In particular, GWAS signals are enriched within regions of open chromatin identified by deoxyribonuclease I (DNase I) mapping (Maurano et al., 2012). These so-called DNase I hypersensitive sites (DHS) are regions where DNA is highly accessible (Bell et al., 2011), and likely serves some cis-regulatory function (Thurman et al., 2012). The location of DHS signals overlaps that of many other regulatory markers, indicating that they are a broad, non-specific marker of sites of active regulatory DNA, capturing many different ongoing biological processes affecting gene expression. The enrichment of significant GWAS associations in these regions provides some biological coherence for interpreting the functional impact of variation in these non-coding variants, and also suggest that SNPs located in DHSs (referred to as DHS SNPs) may be more likely to be “true” signals and less likely to be false positives. For this reason, we hypothesized that functional annotation information like DHS location could be used to improve the predictive ability of polygenic scores. Using alcohol problems as our primary outcome, we expected that polygenic scores based on SNPs in regulatory regions (DHSs) would provide stronger predictive power (i.e., account for more variance) compared to conventional, unselected, genome-wide polygenic scores that included a mixture of DHS SNPs and non-DHS SNPs. We focus specifically on localization in DHSs in view of recent evidence that SNPs with lower p values in our discovery sample GWAS were more likely to be in DHS regions (versus non-DHS regions) (Edwards et al., 2015), as well as broader evidence from genomic partitioning analyses that DHS SNPs accounted for the majority (79%) of the heritability across 11 common diseases (Gusev et al., 2014). Additionally, in the absence of existing knowledge about what specific functional annotations would be most advantageous to inform polygenic scores, DHS status provides a non-specific tool for a first look into whether this approach holds promise.
Identifying Genetic Variants Involved in Gene-by-Environment Interaction Effects
Unlike Mendelian disorders such as cystic fibrosis or Huntington’s disease, where a mutation in a single gene is sufficient to cause disorder, the pathway from genotype to phenotype for alcohol outcomes is not necessarily straightforward. Alongside advances in characterizing genetic risk, research has suggested that a number of environmental factors can alter the importance of genetic influences on alcohol outcomes (Young-Wolff et al., 2011), and it has also been suggested that G×E effects may harbor some of the ‘hidden heritability’ for complex behavioral outcomes (Manolio et al., 2009). Despite strong evidence for G×E effects from twin studies (Young-Wolff et al., 2011), the study of G×E using measured genotypes has been controversial (Duncan and Keller, 2011, Dick et al., 2015).
Among the major criticisms is the focus on “usual suspect” candidate genes in the serotonin or dopamine pathways (e.g., SLC6A4 or MAO-A) (Dick et al., 2015). Thus, the field is in need of answers to the question of which SNPs are worth carrying forward into studies of G×E using measured genotypes. One way to answer this question is to examine whether certain types of SNPs (based on genomic information) are overrepresented among SNPs with G×E effects. Thus, in an effort to move away from the candidate gene approach, we tested the exploratory hypothesis that SNPs in regulatory regions would be more likely to have significant G×E effects. We believed DHS SNPs would be enriched for G×E interaction effects given that the DNA variants in DHS regions may be more likely to affect the chromatin structure around a gene that determines whether the DNA is accessible to transcription factors (i.e., the proteins responsible for transcribing DNA to RNA and determining gene expression levels) (Cockerill, 2011). Environmental exposures are known to affect epigenetic processes, and can further drive gene expression or repression via alterations to the availability of transcription factors (Meaney, 2010, Lopez-Maury et al., 2008). For these reasons, we hypothesized that allelic variation in DHS SNPs may be particularly impactful for responsiveness to environmental cues that alter gene expression (Liu et al., 2008).
We examined romantic relationship status as the environmental moderator for these analyses in view of evidence that (1) involvement in a romantic relationship in young adulthood is associated with lower alcohol use (Fleming et al., 2010) and (2) that romantic relationship status changes the degree to which genetic influences are important for alcohol outcomes (Heath et al., 1989, Prescott and Kendler, 2001). Of particular relevance for this exploratory G×E hypothesis, recent analyses of the FinnTwin12 sample indicated that genetic variance for intoxication frequency was attenuated for those in a romantic relationship compared to those not in a romantic relationship (Barr et al., in press). This implies that genetic influences on intoxication frequency are less important for those who are in a relationship, and more important for those who are single. The results from these twin studies suggested that romantic relationship status would be a particularly good “candidate environment” when testing our hypothesis that SNPs in regulatory regions would be enriched for G×E effects. Twin studies of G×E effects using inferred genotypes typically show a fan-shaped pattern of effects, whereby additive genetic factors have more influence in certain environments, and less in others. Detecting a latent G×E effects with inferred genotypes implies that the majority of measured genes are likely to be moderated in the same way, such that the effect of measured genotypic influences on a phenotype varies across levels of the environment.
The Current Study
We examined two research questions related to the incorporation of functional genomic information to understand the genetic and G×E influences on alcohol use outcomes in a population-based sample of young adult Finnish twins (Kaprio, 2013, Kaprio et al., 2002): 1) Do polygenic scores informed by DHS annotation predict lifetime alcohol problems better than conventional polygenic scores that include a mixture of DHS and non-DHS SNPs? And 2) Are DHS variants overrepresented among SNPs with G×E effects for alcohol misuse in a model where romantic relationship status is the environmental moderator? As a set, these questions contribute to efforts to enhance polygenic signal and empirically prioritize variants likely to be involved in G×E effects.
Materials and Method
Sample
Our sample comes from the youngest cohort of the Finnish Twin Cohort Study (FinnTwin12), which was established to examine genetic and environmental factors influencing health-related behaviors (Kaprio, 2013), including the development of alcohol misuse. Participants were recruited from Finland’s Population Registry, permitting comprehensive and unbiased nationwide ascertainment of all twins born across five birth cohorts in Finland from 1983 to 1987. Baseline collection occurred when twins were aged approximately 12 years old, with a sample of some 5600 twins and their families (Kaprio, 2013) and an overall participation rate of 87%. Follow-up surveys occurred at ages 14, 17.5, and 22 years. Of the original epidemiological sample, 1035 families were chosen as part of an intensive subsample, from which 1852 twins (89% participation) completed the adolescent version of the Semi-Structured Assessment for the Genetics of Alcoholism (SSAGA; Bucholz et al., 1994) interview at age 14. Follow up of the intensive subsample when twins were, on average, age 22 (n=1347) included the adult SSAGA. DNA from blood or saliva samples was collected from 1295 twins. Data for the present study is drawn from the psychiatric assessment at age 22 among participants for whom genotypic data were available (n=1170). The sample was 53.6% female (N = 627) and the age range varied from 20–26, with a mean age of 22.42 years (SD=0.72). Participants were fully informed of study procedures and gave written consent to participate. The Helsinki University Central Hospital District’s Ethical Committee and Indiana University’s Institutional Review Board approved the FinnTwin12 study.
Measures
Lifetime alcohol dependence symptoms
The alcohol dependence symptoms (ADsx) measure was the count of the number of lifetime DSM-IV criteria that respondents endorsed from the SSAGA (Bucholz et al., 1994). Responses ranged from 0 to 7. ADsx was natural log-transformed after adding 1 to adjust for the positive skew and to retain participants who endorsed zero symptoms. Individuals who never initiated alcohol use were coded as missing (n = 35).
Frequency of intoxication
For the G×E analyses, we expected that the moderating effect of relationship status would be on a contemporaneous alcohol misuse outcome rather than a cumulative lifetime alcohol misuse outcome, such as ADsx. Accordingly, we used a time-delimited measure of frequency of intoxication for the G×E analyses. Frequency of intoxication was assessed at age 22 by the single item, “How often do you use alcohol in such a way that you get really drunk?” Response options included “never” (0) to “daily” (8). Response categories were transformed to reflect the number of days per month participants were intoxicated and natural log-transformed after adding 1 to adjust for the positive skew and to retain participants who reported “never” (Dick et al., 2001).
Relationship status
Participants were asked, “How long (in years) have you been together with your present partner?” Those who indicated they were not dating were coded as 0. Those who indicated they were in a romantic relationship (dating, married, or living in a common law relationship) of any length were coded as 1.
Genotyping
Genotyping was conducted using the Human670-QuadCustom Illumina BeadChip (Illumina, Inc., San Diego, CA, USA) at the Wellcome Trust Sanger Institute (Kaprio, 2013). Quality control steps included removing SNPs with minor allele frequency < 1%, genotyping success rate < 95%, or Hardy-Weinberg equilibrium p < 1 × 10−6, and removing individuals with genotyping success rate < 95%, a mismatch between phenotypic and genotypic gender, excess relatedness (outside of known families), and heterozygosity outliers. Genotypes were imputed to the 1000 Genomes Phase I (v3) reference panel using ShapeIT (Delaneau et al., 2012) for phasing and IMPUTE2 (Howie et al., 2009) for imputation. Prior analyses indicated a single dimension of ancestry in the sample (Meyers, 2012). Although a single dimension of ancestry does not preclude variation along this dimension, we note that fine-scale population substructure is less of an issue for common variants (versus rare variants), especially in the present sample given the relatively longer LD blocks that make the Finnish population more homogenous than other populations of mixed European ancestry. We also note that in supplementary analyses of the first 10 ancestry principal components and the ADsx and frequency of intoxication measures, we found no substantial evidence of population stratification. Out of 20 possible associations, only a single PC had a p-value of less than 0.10 (PC3 for ADsx, p = 0.04). These converging pieces of evidence suggested that it was not necessary to correct for population stratification, and informed our decision to not include ancestry principal components in our analyses.
Analytic Plan
Polygenic score creation
Summary association statistics used to create the polygenic scores for alcohol problems come from a previously reported GWAS of an alcohol problems factor score conducted in 4,304 Caucasian young adults from the Avon Longitudinal Study of Parents and Children (Edwards et al., 2015); this is the largest GWAS to date of alcohol problems in European young adults. Genotypes in this discovery sample were also imputed to the 1000 Genomes Phase I (v3) reference panel. From this discovery sample, we selected a list of 4,415,289 SNPs also available in FinnTwin12 and with a minor allele frequency > 5% and imputation quality R2 > .90 in both samples, and pruned this SNP set to obtain 212,718 autosomal SNPs (4.8% of the common SNPs) in approximate linkage equilibrium (R2 < .25). This list was further filtered to create two sets of score SNPs with nominal GWAS association p values in the discovery GWAS (thresholds of p < .05 and p < .01, NSNPs = 10,693 and 2,221, respectively), based on preliminary analyses as well as previous work showing these thresholds have the best signal-to-noise ratio/predictive power for polygenic scores (Yang et al., 2014).
Scores were calculated in FinnTwin12 using the score procedure in PLINK version 1.9 (Chang et al., 2015) summing each individual’s total number (imputed dosage) of minor alleles from the score SNPs, with each SNP weighted by the negative log of the GWAS association p value and sign of the association (beta) statistic. As illustrated in Figure 1, identical procedures were used to create a set of DNase I-restricted polygenic scores, except that the final list of LD-pruned SNPs described above was further restricted to SNPs located in DHS sites. The locations of DHSs were based on narrow peak hotspots identified across 53 consolidated epigenomes by the RoadMap Epigenomics Project (http://www.roadmapepigenomics.org). The 53 epigenomes are summarized in the Supplementary Information (Table S1). SNPs were considered to be DHS SNPs if they directly overlapped a DHS or were in perfect linkage disequilibrium (LD) with another SNP overlapping a DHS. Of the 212,718 genome-wide score SNPs, 78,948 (37%) were located in a DHS site, and 3,946 (37%) and 789 (36%) of the DHS score SNPs fell under the GWAS p value thresholds of p < .05 and p < .01, respectively. Hereafter we will use the terms “GW-scores” to refer to polygenic scores created from the genome-wide set of SNPs and “DHS-scores” to refer to polygenic scores from SNPs located in DHS sites only.
Figure 1.
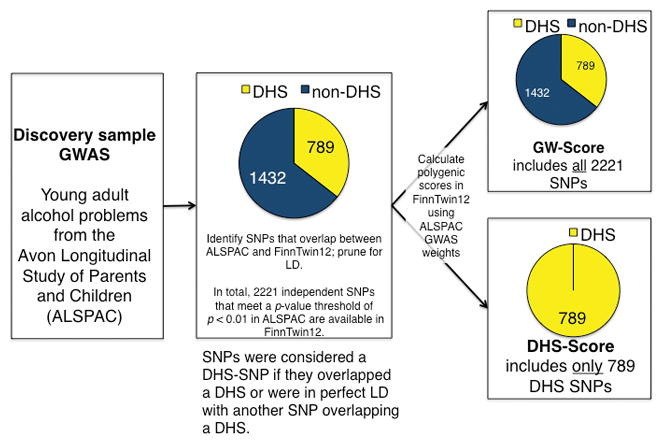
Schematic of DHS- and GW-polygenic score creation in the FinnTwin12 sample using an illustrative p-value of p < 0.01. The GW-score is the weighted linear combination of all SNPs meeting p < 0.01 in the discovery sample (ALSPAC) GWAS (Edwards et al., 2015). The DHS-score is the weighted linear combination of the subset of SNPs meeting p < 0.01 in the discovery sample GWAS that were also located in a DHS site. Abbreviations: DHS = DNase I hypersensitive site; GWAS = Genome-wide association study; LD = linkage disequilibrium; SNP = single nucleotide polymorphism.
Polygenic scores for alcohol phenotypes have had very modest effects in previous studies. Thus, in an attempt to conceptually validate findings coming out of the primary alcohol analyses, we also compared the predictive power of DHS- and GW-scores for height as a secondary outcome. We selected height as a model phenotype given that its molecular genetic etiology is further advanced (relative to alcohol problems) and polygenic scoring methods have already demonstrated substantial success (Wood et al., 2014). We used the same procedure to calculate the polygenic scores for height, with discovery GWAS summary statistics coming from the GIANT Consortium meta-analysis results of ~250,000 adults of European ancestry (Wood et al., 2014; available at http://portals.broadinstitute.org/collaboration/giant). Genotypes from the GIANT study were imputed to the HapMap2 CEU reference population, so the LiftOver tool (http://genome.sph.umich.edu/wiki/LiftOver) was used to harmonize SNP IDs and genomic locations with those of the 1000 Genomes-imputed FinnTwin12 dataset. There were 1,831,837 SNPs in common after filtering, pruned for LD to 193,884 SNPs (31,358 and 15,239 below p thresholds of .05 and .01, respectively). Of these, 76,913 (39.7%) SNPs were located in a DHS, of which 13,593 (43.3% of GW) and 6,918 (45.4% of GW) met p value thresholds of .05 and .01. Height (in centimeters) was measured in the FinnTwin12 sample by a self-report survey item at age 22.
Predictive ability of DHS-scores versus GW-scores
In order to compare the relative strength of the DHS- and GW-scores, we fit a series of separate linear mixed-effects models incorporating each of the GW- and DHS-scores to predict ADsx and height. Each model also included sex (and, for height, age) as covariates. To account for clustering at the family level, we fit mixed models with random intercepts using the lmer function from the lme4 package (version 1.1.11) in R (version 3.2.3). Models were fit with risk scores calculated using SNPs at the p < .01 and p < .05 thresholds from the discovery GWAS. We examined the relative predictive ability of DHS- and GW-scores in two ways. First, we compared the significance of association and the overall variance accounted for (R2) by each score. However, because the number of SNPs included in the polygenic scores differed substantially between the GW-scores and DHS-scores, a direct comparison of the magnitudes of their association statistics may not be meaningful. Thus, as a second approach we calculated an average “per-SNP” effect to facilitate comparisons on the same metric. To do this, we divided the variance accounted for each by each score (R2) by the number of SNPs in that score.
G×E analyses
We examined whether SNPs in regulatory regions were enriched for G×E effects using a chi-square test that compared the proportion of DHS SNPs among the set of SNPs with significant (p < .05) G×E effects relative to the proportion of DHS SNPs in the full genome. For these analyses we used a contemporaneous measure of alcohol misuse, intoxication frequency. We focused on a contemporaneous measure of alcohol misuse in order to ensure that our romantic relationship status environmental moderator and alcohol misuse outcome were temporally matched1. For these analyses, we selected a set of top SNPs in the ALSPAC GWAS (p < 0.005) from the set of 212,718 LD-pruned autosomal SNPs common across the ALSPAC and FinnTwin12 samples, resulting in 1137 SNPs. We focused on the set of the more highly associated SNPs in view of evidence that G×E effects are most likely to be observed for SNPs with smaller p-values (Thomas, 2010). This threshold was arbitrary, but was selected in an attempt to balance testing a large enough number of “top” SNPs with the computational resources required for such tests. For each SNP, we then examined gene-environment interaction effects in a linear model where relationship status was the moderator and frequency of alcohol intoxication was the outcome. G×E models were run using the lme4 package in R in order to account for familial nesting. G×E was tested using a parameterization method that takes into account effects between three gene levels in order to accurately capture interactions that can otherwise be misrepresented when using a single cross-product term (Aliev et al., 2014). The method checks the additive interaction between any two of the three gene levels and corrects for the number of tests. The resulting p-value corresponds to the difference between at least two of the gene levels. Sex and age were included as covariates. Preliminary analyses indicated only a modest association between relationship status and intoxication frequency (r = −.10), and no association between the GW- and DHS-scores and relationship status (range rpb = −0.04 to 0.004, all p > 0.14). The latter null associations, in particular, minimized concerns about gene-environment correlation as a potential confounder when testing G×E effects. Regarding multiple testing concerns, we note that the inferential test of interest for this research question was a chi-square test of the proportion of DHS SNPs among the set of SNPs with a significant (p < .05) G×E effects relative to the overall proportion of DHS SNPs, thus representing a single statistical test for enrichment.
Results
Descriptive Statistics
Table 1 provides an overview of the distributions of the key measures in the FinnTwin12 sample. On average, participants endorsed 1.03 ADsx criteria, and reported being intoxicated 1.52 days per month (SD= 1.79). With respect to relationship status, of the 1,148 nonmissing responses, 58% (n=664) of the twins reported being involved in a relationship, of which 567 (84.8%) had been involved in that relationship for one year or more.
Table 1.
Descriptive statistics for FinnTwin12 sample
| Measure | N / Mean | % / SD | Range | |
|---|---|---|---|---|
| Alcohol dependence symptoms | 1.03 | 1.31 | 0 | 7 |
| Frequency of intoxication (days per month) | 1.52 | 1.79 | 0 | 30 |
| Height (cm) | 172 | 9.38 | 145 | 207 |
| In a Romantic Relationship | 664 | 58.0% | - | - |
| Relationship lasting > 1 year | 567 | 49.4% | - | - |
Note. All percentages based on valid responses.
Do DHS- Scores Predict Outcomes Better than GW-scores?
ADsx
Regression results for GW-scores and DHS-scores predicting ADsx are shown in Table 2. The GW-scores predicted ADsx at both the p<.01 and p<.05 inclusion thresholds (β =.0006, p<.001 and β =.0003, p<.001, respectively). The DHS-scores also predicted ADsx at the p<.01 and p<.05 inclusion thresholds (β =.0008, p=.009 and β =.0004, p=.022, respectively). The overall effect sizes were relatively small, with each GW-score explaining about 1% of the variance in ADsx. Effect sizes for the DHS-scores were also modest, with each explaining ~0.5% of the variation in ADsx. The GW- and DHS-scores included different numbers of SNPs; accordingly, we compared the per-SNP effect sizes for the two types of polygenic scores. Ratios of the DHS to GW per-SNP effect were 1.1 to 1.4 for the per-SNP variance accounted for (R2). This indicates that, on average, each SNP in DHS-scores accounted for 1.1–1.4 times more variance in ADsx compared to each SNP included in the GW-scores.
Table 2.
Linear mixed-effects models for alcohol dependence symptoms (natural log transformed) across two GW and DHS polygenic score thresholds (N = 1,098)
| Threshold | GW scores
|
|||||
|---|---|---|---|---|---|---|
| Beta | 95% CI | P | Pseudo-R2 | SNP Count | Per-SNP R2 | |
| P<.01 | 0.0006 | 0.0003 – 0.0010 | 0.0003 | 0.011 | 2,221 | 4.96E-06 |
| P<.05 | 0.0003 | 0.0001 – 0.0006 | 0.0006 | 0.010 | 10,694 | 9.57E-07 |
| Threshold | DHS scores | |||||
|
| ||||||
| Beta | 95% CI | P | Pseudo-R2 | SNP Count | Per-SNP R2 | |
|
| ||||||
| P<.01 | 0.0008 | 0.0001 – 0.0014 | 0.0094 | 0.005 | 789 | 6.76E-06 |
| P<.05 | 0.0004 | 0.00001 – 0.0008 | 0.0220 | 0.004 | 3,947 | 1.03E-06 |
Notes. All models include sex as a covariate. Pseudo-R2 calculated using the method outlined by Nakagawa et al. (2013). Abbreviations: GW, genome-wide; DHS, DNase I hypersensitive sites.
Height
Regression results for GW-scores and DHS-scores predicting height are shown in Table 3. The GW-scores significantly predicted height at both the p<.01 and p<.05 inclusion thresholds (β =0.0028, p=1×10−47 and β =0.0025, p=3×10−48, respectively). Likewise, the DHS-scores significantly predicted height at the p<.01 and p<.05 inclusion thresholds (β =0.0042, p=1×10−37 and β =0.0040, p=3×10−39, respectively). Compared to polygenic prediction of ADsx, the predictive power of polygenic scores for height was much stronger and the total phenotypic variance accounted for was considerably larger, ranging from 8.6 – 8.9% for GW scores and 6.4 – 6.9% for DHS scores. The per-SNP ratios for DHS to GW effects ranged from 1.6 to 1.8 for R2, indicating that each SNP included in the DHS-scores accounted for, on average, 1.6–1.8 times the variance in height compared to SNPs in GW-scores.
Table 3.
Linear mixed-effects models for height across two GW and DHS polygenic score thresholds (N = 1151)
| Threshold | GW scores
|
|||||
|---|---|---|---|---|---|---|
| Beta | 95% CI | P | Pseudo-R2 | SNP Count | Per-SNP R2 | |
| P<.01 | 0.0028 | 0.0024 – 0.0031 | 1.15E-47 | 0.086 | 15,239 | 5.63E-06 |
| P<.05 | 0.0025 | 0.0022 – 0.0029 | 2.88E-48 | 0.089 | 31,358 | 2.85E-06 |
| Threshold | DHS scores | |||||
|
| ||||||
| Beta | 95% CI | P | Pseudo-R2 | SNP Count | Per-SNP R2 | |
|
| ||||||
| P<.01 | 0.0042 | 0.0036 – 0.0049 | 1.24E-37 | 0.064 | 6,918 | 9.27E-06 |
| P<.05 | 0.0040 | 0.0035 – 0.0046 | 2.98E-39 | 0.069 | 13,593 | 5.05E-06 |
Notes. All models include age and sex as covariates. Pseudo-R2 calculated using the method outlined by Nakagawa et al. (2013). Abbreviations: GW, genome-wide; DHS, DNase I hypersensitive sites.
Are DHS SNPs Enriched for Significant G×E Effects?
Of the top independent 1137 SNPs, 55 (4.8%) showed significant evidence for interaction (p < .05 in the interaction model). In total, 27 of the 409 DHS SNPs showed significant G×E effects (7%) compared to 28 of 728 non-DHS SNPs (4%). A chi-square test of independence indicated that DHS SNPs were overrepresented among significant G×E effects relative to expectation, χ2(1) = 3.92, p = 0.05. This indicates that gene-environment interaction effects for this particular environment were modestly enriched for DHS SNPs.
In supplementary analyses, we also examined whether relationship status moderated the main effect of the aggregate GW- and DHS-scores in predicting alcohol intoxication. None of these interaction effects were significant (all p > 0.05).
Discussion
We tested two hypotheses related to the incorporation of functional genomic information to understand genetic and G ×E effects on alcohol use outcomes. We found that DHS-scores were more parsimonious compared to the GW-scores while capturing the majority of the same signal. The per-SNP effects for variants in the DHS-scores were 1.1 to 1.4 times larger than the per-SNP effect for variants in the GW-scores. We found a similar pattern of effects for a second non-psychiatric phenotype, height. We also found that DHS SNPs were modestly enriched for G×E effects compared to non-DHS SNPs in a model looking at romantic relationship status as the moderator.
These findings add to a growing literature demonstrating that incorporation of functional information about SNPs can advance our understanding of genetic contributions to complex diseases and disorders (Schork et al., 2013, Edwards et al., 2015, Maurano et al., 2012, Finucane et al., 2015). There was minimal loss in predictive power when polygenic scores were limited to variants in DHS regions, which is an encouraging sign that the included variants may be etiologically relevant given their higher a priori probability of having functional consequences. These results also provide some evidence that, like other complex traits (Maurano et al., 2012, Gusev et al., 2014), regulatory mechanisms appear to play a large role in the genetic factors impacting alcohol use outcomes. We should note, however, that the clinical utility of polygenic scoring methods for alcohol problems remains modest: both DHS- and GW- polygenic scores accounted for < 1% of the variance in alcohol dependence symptoms. It was for this reason that we repeated the analyses with height, where there is greater predictive ability associated with polygenic risk scores calculated from large meta-analyses. We were encouraged to find a parallel pattern of results.
Our findings also provide initial evidence that DHS variants are more likely (compared to non-DHS variants) to be involved in gene-by-environment interaction effects. The study of measured gene-by-environment interaction is controversial, in part owing to problems surrounding the selection of the handful of SNPs in “usual suspect” candidate genes commonly examined (Dick et al., 2015, Duncan and Keller, 2011). Our results provide evidence for an empirically based approach that builds on findings from twin studies, GWAS, and functional genomics to select SNPs for studies of measured G×E. Thus, there may be a biologically and empirically justifiable way forward to identify the variants likely to be moderated by environmental factors. Important questions remain about the specific mechanisms underlying these statistical interactions, and where in the pathway from genes to behavior an environmental factor is likely to exert its moderating effect (Moore and Thoemmes, 2016). We speculate that DHS SNPs may be especially responsive to environmental inputs given their involvement in gene regulation (Liu et al., 2008).
Limitations
Our results should be considered in the context of several limitations. First, there was imperfect correspondence between the study populations and alcohol problems measures across the ALSPAC and FinnTwin12 samples. This concern is lessened in view of the genetic overlap evident between multiple dimensions of alcohol use (Dick et al., 2011). Second, the sample sizes of ALSPAC and FinnTwin12 are relatively small given the growing recognition of the large sample sizes needed to precisely estimate small effect sizes. We recognize that there are larger GWAS of alcohol-related behaviors (e.g., Schumann et al., 2016). Several factors guided our choice to use ALSPAC as our discovery sample, including the greater similarity between the ALSPAC and FinnTwin12 sample populations and alcohol problems phenotypes (in contrast to the aging-related cohorts included in the Schumann et al. (2016) study, as well as that study’s focus on an alcohol consumption phenotype). Third, the polygenic scores derived here include only common variants in regions well tagged by the variants in the 1000 Genomes panel. Fourth, there are alternative enrichment (Finucane et al., 2015) and alternative polygenic scoring methods (e.g., LDpred; Vilhjalmsson et al., 2015). Some combination of these may provide additional avenues for optimizing the predictive ability of polygenic scores in the future.
Fifth, we did not take into account the tissue specificity of regulatory markers when delineating DHS SNPs, as all variants located in (or in perfect LD with) a DHS site in any of the RoadMap tissue lines was considered a DHS SNP. Therefore, SNPs that have only a regulatory function in tissues that are not relevant to alcohol use would have been included in the DHS-scores along with true important functional variants, diluting the magnitude of the per-SNP association and the difference in association between SNPs included in the DHS versus non-DHS scores. We performed supplementary analyses using scores that included DHS SNPs limited to brain tissue samples and DHS SNPs present in two or more tissue samples to determine whether SNPs from certain samples were more relevant. Neither of these scores at either p-value threshold predicted ADsx. This may be due to the very small number of markers included in both the brain tissue score (p<.01 = 139; p<.05 = 653) or the two tissue score (p<.01 = 495; p<.05 = 2,571).
Sixth, our environmental moderator (romantic relationship status) captures only one of many relationship features previously implicated in studies of gene-environment interplay for alcohol use and problems (Jarnecke and South, 2014), and our measure of it was rather crude. Although we detect modest evidence that SNPs in DHSs were enriched for G × E effects, we note that these statistical interactions do not in themselves illuminate the biological processes through which these effects occur. Furthermore, as with all studies of G×E with measured genotypes, power is a concern and the results should be interpreted with appropriate caution. We conducted post-hoc power analyses using Quanto (Gauderman, 2002), and when the G×E effect was very small (R2 = 0.0005), we had very low power to detect effects (12%). However, when the G×E effect was somewhat larger (R2 = 0.005) we had 72% power to detect interactions. On a more conceptual level, we note that previous analyses using the FinnTwin12 data have established latent G×E effects for relationship status and intoxication frequency (Barr et al., in press). This gives us more confidence in the G×E results using measured genotypes. Finally, as with other tests of enrichment, the focus of this analysis was not on interpreting the direction of any of the SNP × Relationship Status effects themselves, but rather examining whether there was overall enrichment to inform future studies about “which SNPs and which genes” are worth carrying forward into studies of GxE using measured genotypes.
Conclusions and Future Directions
These findings highlight the potential utility of integrating genomic annotation information in order to increase the signal-to-noise ratio in polygenic scores, and identify genetic variants that may be most susceptible to environmental modification. This work can be expanded in several ways, including extensions to jointly consider multiple annotation categories (Pickrell, 2014) and to make use of alternative weighting schemes to up- and down-weight variants across a range of regulatory marks rather than the blunt filtering tool applied here. Such advancements, in conjunction with ongoing efforts to increase power in gene identification studies, have the potential not only to provide biological insights into the etiology of alcohol misuse and other complex psychiatric disorders, but also to one day provide clinical utility to identify and treat at-risk individuals.
Supplementary Material
Acknowledgments
Funding for this work came from the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health under award numbers R01AA015416, K02AA018755, F31AA024378, and K01AA024152; the National Institute of Mental Health (T32MH20030); the Academy of Finland (grants 100499, 205585, 118555, 141054, 265240, 263278, and 264146); and the Scientific and Technological Research Council of Turkey (TÜBİTAK) under award number 114C117. Genotyping of the FinnTwin12 sample was supported also by the Wellcome Trust Sanger Centre. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the Academy of Finland, or the Scientific and Technological Research Council of Turkey.
Footnotes
Our measure of intoxication frequency was moderately correlated with ADsx (r = .42 p < .001). Comparisons of GW and DHS scores in predicting intoxication frequency showed that GW scores at the p < .001, p < .05, and p < .01 thresholds were significantly associated with intoxication frequency in the expected direction (i.e., higher polygenic score associated with more frequent intoxication). DHS-scores at the p < .001 and p < .01 thresholds were significantly associated with intoxication frequency. Overall, both GW- and DHS-scores predicted intoxication frequency, though less strongly than ADsx (GW-scores: R2 = .000 – .008; DHS-scores: R2 = .000 – .004).
Conflicts of Interest. JK consulted for Pfizer (2012–2014). The authors declare no other conflicts of interest.
References
- Aliev F, Latendresse SJ, Bacanu SA, Neale MC, Dick DM. Testing for measured gene-environment interaction: problems with the use of cross-product terms and a regression model reparameterization solution. Behav Genet. 2014;44:165–81. doi: 10.1007/s10519-014-9642-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttila V, Bulik-Sullivan B, Finucane H, Bras J, Duncan L, Escott-Price V, Falcone G, Gormley P, Malik R, Patsopoulos N, Ripke S, Walters R, Wei Z, Yu D, Lee PH Igap Consortium, Ihgc Consortium, Ilae Consortium on Complex Epilepsies, Imsgc Consortium, Ipdgc Consortium, Metastroke and Intracerebral Hemorrhage Studies of the International Stroke Genetics Consortium, Attention-Deficit Hyperactivity Disorder Working Group of the Psychiatric Genomics Consortium, Anorexia Nervosa Working Group of the Psychiatric Genomics Consortium, Autism Spectrum Disorders Working Group of the Psychiatric Genomics Consortium, Bipolar Disorders Working Group of the Psychiatric Genomics Consortium, Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium, Obsessive Compulsive Disorder and Tourette Syndrome Working Group of the Psychiatric Genomics Consortium, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Breen G, Bulik C, Daly M, Dichgans M, Faraone S, Guerreiro R, Holmans P, Kendler K, Koeleman B, Mathews CA, Scharf JM, Sklar P, Williams J, Wood N, Cotsapas C, Palotie A, Smoller JW, Sullivan P, Rosand J, Corvin A, Neale BM, Brainstorm Consortium. Analysis of shared heritability in common disorders of the brain (available online April 2016) [Google Scholar]
- Barr PB, Salvatore JE, Maes H, Aliev F, Latvala A, Viken RJ, Rose RJ, Kaprio J, Dick DM. Social relationships moderate genetic influences on heavy drinking in young adulthood. J Stud Alcohol Drugs. doi: 10.15288/jsad.2017.78.817. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell O, Tiwari VK, Thoma NH, Schubeler D. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12:554–64. doi: 10.1038/nrg3017. [DOI] [PubMed] [Google Scholar]
- Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JL, Jr, Reich T, Schmidt I, Schuckit MA. A new, semi-structured psychiatric interview for use in genetic linkage studies: A report on the reliability of the SSAGA. J Stud Alcohol. 1994;55:149–158. doi: 10.15288/jsa.1994.55.149. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerill PN. Structure and function of active chromatin and DNase I hypersensitive sites. FEBS J. 2011;278:2182–210. [Google Scholar]
- Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2012;9:179–81. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- Dick DM, Agrawal A, Keller MC, Adkins A, Aliev F, Monroe S, Hewitt JK, Kendler KS, Sher KJ. Candidate gene-environment interaction research: Reflections and recommendations. Perspect Psychol Sci. 2015;10:37–59. doi: 10.1177/1745691614556682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Meyers JL, Rose RJ, Kaprio J, Kendler KS. Measures of current alcohol consumption and problems: two independent twin studies suggest a complex genetic architecture. Alcohol Clin Exp Res. 2011;35:2152–61. doi: 10.1111/j.1530-0277.2011.01564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Rose RJ, Viken RJ, Kaprio J, Koskenvuo M. Exploring gene-environment interactions: Socioregional moderation of alcohol use. J Abnorm Psychol. 2001;110:625–632. doi: 10.1037//0021-843x.110.4.625. [DOI] [PubMed] [Google Scholar]
- Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 2013;9:e1003348. doi: 10.1371/journal.pgen.1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan L, Keller MC. A critical review of the first ten years of measured gene-by-environment interaction research in psychiatry. Am J Psychiat. 2011;168:1041–1049. doi: 10.1176/appi.ajp.2011.11020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AC, Aliev F, Wolen AR, Salvatore JE, Gardner CO, Mcmahon G, Evans DM, Macleod J, Hickman M, Dick DM, Kendler KS. Genomic influences on alcohol problems in a population-based sample of young adults. Addiction. 2015;110:461–470. doi: 10.1111/add.12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh PR, Anttila V, Xu H, Zang C, Farh K, Ripke S, Day FR, Reprogen C, Purcell S, Stahl E, Lindstrom S, Perry JR, Okada Y, Raychaudhuri S, Daly MJ, Patterson N, Neale BM, Price AL Schizophrenia Working Group of the Psychiatric Genomics Consortium, Raci Consortium. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47:1228–35. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming CB, White HR, Catalano RF. Romantic relationships and substance use in early adulthood: An examination of the influences of relationship type, partner substance use, and relationship quality. J Health Soc Behav. 2010;51:153–167. doi: 10.1177/0022146510368930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Cichon S, Treutlein J, Ridinger M, Mattheisen M, Hoffmann P, Herms S, Wodarz N, Soyka M, Zill P, Maier W, Mossner R, Gaebel W, Dahmen N, Scherbaum N, Schmal C, Steffens M, Lucae S, Ising M, Muller-Myhsok B, Nothen MM, Mann K, Kiefer F, Rietschel M. Genome-wide significant association between alcohol dependence and a variant in the ADH gene cluster. Addict Biol. 2012;17:171–80. doi: 10.1111/j.1369-1600.2011.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauderman WJ. Sample size requirements for matched case-control studies of gene-environment interaction. Stat Med. 2002;21:35–50. doi: 10.1002/sim.973. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Flint J. Genetics and genomics of psychiatric disease. Science. 2015;349:1489–1494. doi: 10.1126/science.aaa8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Lee SH, Trynka G, Finucane H, Vilhjalmsson BJ, Xu H, Zang C, Ripke S, Bulik-Sullivan B, Stahl E, Kahler AK, Hultman CM, Purcell SM, Mccarroll SA, Daly M, Pasaniuc B, Sullivan PF, Neale BM, Wray NR, Raychaudhuri S, Price AL Schizophrenia Working Group of the Psychiatric Genomics C Consortium S-S, Schizophrenia Working Group of the Psychiatric Genomics C, Consortium S-S. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet. 2014;95:535–52. doi: 10.1016/j.ajhg.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart AB, Kranzler HR. Alcohol dependence genetics: lessons learned from genome-wide association studies (GWAS) and post-GWAS analyses. Alcohol Clin Exp Res. 2015;39:1312–27. doi: 10.1111/acer.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath AC, Jardine R, Martin NG. Interactive effects of genotype and social-environment on alcohol-consumption in female twins. J Stud Alcohol. 1989;50:38–48. doi: 10.15288/jsa.1989.50.38. [DOI] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarnecke AM, South SC. Genetic and environmental influences on alcohol use problems: Moderation by romantic partner support, but not family or friend support. Alcohol Clin Exp Res. 2014;38:367–375. doi: 10.1111/acer.12263. [DOI] [PubMed] [Google Scholar]
- Kaprio J. The Finnish Twin Cohort Study: an update. Twin Res Hum Genet. 2013;16:157–62. doi: 10.1017/thg.2012.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaprio J, Pulkkinen L, Rose RJ. Genetic and environmental factors in health-related behaviors: Studies on Finnish twins and twin families. Twin Res. 2002;5:366–371. doi: 10.1375/136905202320906101. [DOI] [PubMed] [Google Scholar]
- Kos MZ, Yan J, Dick DM, Agrawal A, Bucholz KK, Rice JP, Johnson EO, Schuckit M, Kuperman S, Kramer J, Goate AM, Tischfield JA, Foroud T, Nurnberger J, Jr, Hesselbrock V, Porjesz B, Bierut LJ, Edenberg HJ, Almasy L. Common biological networks underlie genetic risk for alcoholism in African- and European-American populations. Genes Brain Behav. 2013;12:532–42. doi: 10.1111/gbb.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey DF, Le-Niculescu H, Frank J, Ayalew M, Jain N, Kirlin B, Learman R, Winiger E, Rodd Z, Shekhar A, Schork N, Kiefe F, Wodarz N, Muller-Myhsok B, Dahmen N, Nothen M, Sherva R, Farrer L, Smith AH, Kranzler HR, Rietschel M, Gelernter J, Niculescu AB. Genetic risk prediction and neurobiological understanding of alcoholism. Transl Psychiatry. 2014;4:e391. doi: 10.1038/tp.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li Y, Tollefsbol TO. Gene-environment interactions and epigenetic basis of human diseases. Curr Issues Mol Biol. 2008;10:25–36. [PMC free article] [PubMed] [Google Scholar]
- Lopez-Maury L, Marguerat S, Bahler J. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat Rev Genet. 2008;9:583–93. doi: 10.1038/nrg2398. [DOI] [PubMed] [Google Scholar]
- Maher BS. Polygenic Scores in Epidemiology: Risk Prediction, Etiology, and Clinical Utility. Curr Epidemiol Rep. 2015;2:239–244. doi: 10.1007/s40471-015-0055-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbarek H, Milaneschi Y, Fedko IO, Hottenga JJ, De Moor MH, Jansen R, Gelernter J, Sherva R, Willemsen G, Boomsma DI, Penninx BW, Vink JM. The genetics of alcohol dependence: Twin and SNP-based heritability, and genome-wide association study based on AUDIT scores. Am J Med Genet B Neuropsychiatr Genet. 2015;168:739–48. doi: 10.1002/ajmg.b.32379. [DOI] [PubMed] [Google Scholar]
- Meaney MJ. Epigenetics and the biological definition of gene x environment interactions. Child Dev. 2010;81:41–79. doi: 10.1111/j.1467-8624.2009.01381.x. [DOI] [PubMed] [Google Scholar]
- Meyers J. ProQuest Dissertations & Theses Global Order No. 3523904. 2012. Elucidating genetic and environmental influences on alcohol-related phenotypes. [Google Scholar]
- Moore SR, Thoemmes F. What is the biological reality of gene-environment interaction estimates? An assessment of bias in developmental models. J Child Psychol Psychiatry. 2016;57:1258–1267. doi: 10.1111/jcpp.12579. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Schielzeth H, O’hara RB. A general and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol Evol. 2013;4:133–142. [Google Scholar]
- Pagan JL, Rose RJ, Viken RJ, Pulkkinen L, Kaprio J, Dick DM. Genetic and environmental influences on stages of alcohol use across adolescence and into young adulthood. Behav Genet. 2006;36:483–97. doi: 10.1007/s10519-006-9062-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am J Hum Genet. 2014;94:559–73. doi: 10.1016/j.ajhg.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott CA, Kendler KS. Associations between marital status and alcohol consumption in a longitudinal study of female twins. J Stud Alcohol. 2001;62:589–604. doi: 10.15288/jsa.2001.62.589. [DOI] [PubMed] [Google Scholar]
- Schork AJ, Thompson WK, Pham P, Torkamani A, Roddey JC, Sullivan PF, Kelsoe JR, O’donovan MC, Furberg H, Schork NJ, Andreassen OA, Dale AM Tobacco, Genetics C, Bipolar Disorder Psychiatric Genomics C, Schizophrenia Psychiatric Genomics C. All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet. 2013;9:e1003449. doi: 10.1371/journal.pgen.1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann G, Liu C, O’reilly P, Gao H, Song P, Xu B, Ruggeri B, Amin N, Jia T, Preis S, Segura Lepe M, Akira S, Barbieri C, Baumeister S, Cauchi S, Clarke TK, Enroth S, Fischer K, Hallfors J, Harris SE, Hieber S, Hofer E, Hottenga JJ, Johansson A, Joshi PK, Kaartinen N, Laitinen J, Lemaitre R, Loukola A, Luan J, Lyytikainen LP, Mangino M, Manichaikul A, Mbarek H, Milaneschi Y, Moayyeri A, Mukamal K, Nelson C, Nettleton J, Partinen E, Rawal R, Robino A, Rose L, Sala C, Satoh T, Schmidt R, Schraut K, Scott R, Smith AV, Starr JM, Teumer A, Trompet S, Uitterlinden AG, Venturini C, Vergnaud AC, Verweij N, Vitart V, Vuckovic D, Wedenoja J, Yengo L, Yu B, Zhang W, Zhao JH, Boomsma DI, Chambers J, Chasman DI, Daniela T, De Geus E, Deary I, Eriksson JG, Esko T, Eulenburg V, Franco OH, Froguel P, Gieger C, Grabe HJ, Gudnason V, Gyllensten U, Harris TB, Hartikainen AL, Heath AC, Hocking L, Hofman A, Huth C, Jarvelin MR, Jukema JW, Kaprio J, Kooner JS, Kutalik Z, Lahti J, Langenberg C, Lehtimaki T, Liu Y, Madden PA, Martin N, Morrison A, Penninx B, Pirastu N, Psaty B, Raitakari O, Ridker P, Rose R, Rotter JI, Samani NJ, Schmidt H, Spector TD, Stott D, Strachan D, Tzoulaki I, Van Der Harst P, Van Duijn CM, Marques-Vidal P, Vollenweider P, Wareham NJ, Whitfield JB, Wilson J, Wolffenbuttel B, Bakalkin G, Evangelou E, Liu Y, Rice KM, Desrivieres S, Kliewer SA, Mangelsdorf DJ, Muller CP, Levy D, Elliott P. KLB is associated with alcohol drinking, and its gene product beta-Klotho is necessary for FGF21 regulation of alcohol preference. Proc Natl Acad Sci USA. 2016;113:14372–14377. doi: 10.1073/pnas.1611243113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. Gene-environment-wide association studies: Emerging approaches. Nat Rev Genet. 2010;11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee BK, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song L, Vong S, Weaver M, Yan Y, Zhang Z, Zhang Z, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhulst B, Neale MC, Kendler KS. The heritability of alcohol use disorders: a meta-analysis of twin and adoption studies. Psychol Med. 2015;45:1061–1072. doi: 10.1017/S0033291714002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilhjalmsson BJ, Yang J, Finucane HK, Gusev A, Lindstrom S, Ripke S, Genovese G, Loh PR, Bhatia G, Do R, Hayeck T, Won HH, Kathiresan S, Pato M, Pato C, Tamimi R, Stahl E, Zaitlen N, Pasaniuc B, Belbin G, Kenny EE, Schierup MH, De Jager P, Patsopoulos NA, Mccarroll S, Daly M, Purcell S, Chasman D, Neale B, Goddard M, Visscher PM, Kraft P, Patterson N, Price AL Schizophrenia Working Group of the Psychiatric Genomics Consortium DB, Risk of Inherited Variants in Breast Cancer S. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am J Hum Genet. 2015;97:576–92. doi: 10.1016/j.ajhg.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze SI, Mcgue M, Miller MB, Hicks BM, Iacono WG. Three mutually informative ways to understand the genetic relationships among behavioral disinhibition, alcohol use, drug use, nicotine use/dependence, and their co-occurrence: Twin biometry, GCTA, and genome-wide scoring. Behav Genet. 2013;43:97–107. doi: 10.1007/s10519-013-9584-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, Chu AY, Estrada K, Luan J, Kutalik Z, Amin N, Buchkovich ML, Croteau-Chonka DC, Day FR, Duan Y, Fall T, Fehrmann R, Ferreira T, Jackson AU, Karjalainen J, Lo KS, Locke AE, Magi R, Mihailov E, Porcu E, Randall JC, Scherag A, Vinkhuyzen AA, Westra HJ, Winkler TW, Workalemahu T, Zhao JH, Absher D, Albrecht E, Anderson D, Baron J, Beekman M, Demirkan A, Ehret GB, Feenstra B, Feitosa MF, Fischer K, Fraser RM, Goel A, Gong J, Justice AE, Kanoni S, Kleber ME, Kristiansson K, Lim U, Lotay V, Lui JC, Mangino M, Leach IM, Medina-Gomez C, Nalls MA, Nyholt DR, Palmer CD, Pasko D, Pechlivanis S, Prokopenko I, Ried JS, Ripke S, Shungin D, Stancakova A, Strawbridge RJ, Sung YJ, Tanaka T, Teumer A, Trompet S, Van Der Laan SW, Van Setten J, Van Vliet-Ostaptchouk JV, Wang Z, Yengo L, Zhang W, Afzal U, Arnlov J, Arscott GM, Bandinelli S, Barrett A, Bellis C, Bennett AJ, Berne C, Bluher M, Bolton JL, Bottcher Y, Boyd HA, Bruinenberg M, Buckley BM, Buyske S, Caspersen IH, Chines PS, Clarke R, Claudi-Boehm S, Cooper M, Daw EW, De Jong PA, Deelen J, Delgado G, Denny JC, Dhonukshe-Rutten R, Dimitriou M, Doney AS, Dorr M, Eklund N, Eury E, Folkersen L, Garcia ME, Geller F, Giedraitis V, Go AS, Grallert H, Grammer TB, Grassler J, Gronberg H, De Groot LC, Groves CJ, Haessler J, Hall P, Haller T, Hallmans G, Hannemann A, Hartman CA, Hassinen M, Hayward C, Heard-Costa NL, Helmer Q, Hemani G, Henders AK, Hillege HL, Hlatky MA, Hoffmann W, Hoffmann P, Holmen O, Houwing-Duistermaat JJ, Illig T, Isaacs A, James AL, Jeff J, Johansen B, Johansson A, Jolley J, Juliusdottir T, Junttila J, Kho AN, Kinnunen L, Klopp N, Kocher T, Kratzer W, Lichtner P, Lind L, Lindstrom J, Lobbens S, Lorentzon M, Lu Y, Lyssenko V, Magnusson PK, Mahajan A, Maillard M, Mcardle WL, Mckenzie CA, Mclachlan S, Mclaren PJ, Menni C, Merger S, Milani L, Moayyeri A, Monda KL, Morken MA, Muller G, Muller-Nurasyid M, Musk AW, Narisu N, Nauck M, Nolte IM, Nothen MM, Oozageer L, Pilz S, Rayner NW, Renstrom F, Robertson NR, Rose LM, Roussel R, Sanna S, Scharnagl H, Scholtens S, Schumacher FR, Schunkert H, Scott RA, Sehmi J, Seufferlein T, Shi J, Silventoinen K, Smit JH, Smith AV, Smolonska J, Stanton AV, Stirrups K, Stott DJ, Stringham HM, Sundstrom J, Swertz MA, Syvanen AC, Tayo BO, Thorleifsson G, Tyrer JP, Van Dijk S, Van Schoor NM, Van Der Velde N, Van Heemst D, Van Oort FV, Vermeulen SH, Verweij N, Vonk JM, Waite LL, Waldenberger M, Wennauer R, Wilkens LR, Willenborg C, Wilsgaard T, Wojczynski MK, Wong A, Wright AF, Zhang Q, Arveiler D, Bakker SJ, Beilby J, Bergman RN, Bergmann S, Biffar R, Blangero J, Boomsma DI, Bornstein SR, Bovet P, Brambilla P, Brown MJ, Campbell H, Caulfield MJ, Chakravarti A, Collins R, Collins FS, Crawford DC, Cupples LA, Danesh J, De Faire U, Den Ruijter HM, Erbel R, Erdmann J, Eriksson JG, Farrall M, Ferrannini E, Ferrieres J, Ford I, Forouhi NG, Forrester T, Gansevoort RT, Gejman PV, Gieger C, Golay A, Gottesman O, Gudnason V, Gyllensten U, Haas DW, Hall AS, Harris TB, Hattersley AT, Heath AC, Hengstenberg C, Hicks AA, Hindorff LA, Hingorani AD, Hofman A, Hovingh GK, Humphries SE, Hunt SC, Hypponen E, Jacobs KB, Jarvelin MR, Jousilahti P, Jula AM, Kaprio J, Kastelein JJ, Kayser M, Kee F, Keinanen-Kiukaanniemi SM, Kiemeney LA, Kooner JS, Kooperberg C, Koskinen S, Kovacs P, Kraja AT, Kumari M, Kuusisto J, Lakka TA, Langenberg C, Le Marchand L, Lehtimaki T, Lupoli S, Madden PA, Mannisto S, Manunta P, Marette A, Matise TC, Mcknight B, Meitinger T, Moll FL, Montgomery GW, Morris AD, Morris AP, Murray JC, Nelis M, Ohlsson C, Oldehinkel AJ, Ong KK, Ouwehand WH, Pasterkamp G, Peters A, Pramstaller PP, Price JF, Qi L, Raitakari OT, Rankinen T, Rao DC, Rice TK, Ritchie M, Rudan I, Salomaa V, Samani NJ, Saramies J, Sarzynski MA, Schwarz PE, Sebert S, Sever P, Shuldiner AR, Sinisalo J, Steinthorsdottir V, Stolk RP, Tardif JC, Tonjes A, Tremblay A, Tremoli E, Virtamo J, Vohl MC, Amouyel P, Asselbergs FW, Assimes TL, Bochud M, Boehm BO, Boerwinkle E, Bottinger EP, Bouchard C, Cauchi S, Chambers JC, Chanock SJ, Cooper RS, De Bakker PI, Dedoussis G, Ferrucci L, Franks PW, Froguel P, Groop LC, Haiman CA, Hamsten A, Hayes MG, Hui J, Hunter DJ, Hveem K, Jukema JW, Kaplan RC, Kivimaki M, Kuh D, Laakso M, Liu Y, Martin NG, Marz W, Melbye M, Moebus S, Munroe PB, Njolstad I, Oostra BA, Palmer CN, Pedersen NL, Perola M, Perusse L, Peters U, Powell JE, Power C, Quertermous T, Rauramaa R, Reinmaa E, Ridker PM, Rivadeneira F, Rotter JI, Saaristo TE, Saleheen D, Schlessinger D, Slagboom PE, Snieder H, Spector TD, Strauch K, Stumvoll M, Tuomilehto J, Uusitupa M, Van Der Harst P, Volzke H, Walker M, Wareham NJ, Watkins H, Wichmann HE, Wilson JF, Zanen P, Deloukas P, Heid IM, Lindgren CM, Mohlke KL, Speliotes EK, Thorsteinsdottir U, Barroso I, Fox CS, North KE, Strachan DP, Beckmann JS, Berndt SI, Boehnke M, Borecki IB, Mccarthy MI, Metspalu A, Stefansson K, Uitterlinden AG, Van Duijn CM, Franke L, Willer CJ, Price AL, Lettre G, Loos RJ, Weedon MN, Ingelsson E, O’connell JR, Abecasis GR, Chasman DI, Goddard ME, Visscher PM, Hirschhorn JN, Frayling TM The Electronic Medical Records Genomics Consortium, The Migen Consortium, The Page Consortium, The Lifelines Cohort Study. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Global Status Report on Alcohol and Health. Geneva: World Health Organization; 2014. [Google Scholar]
- Wray NR, Lee SH, Mehta D, Vinkhuyzen AA, Dudbridge F, Middeldorp CM. Research review: Polygenic methods and their application to psychiatric traits. J Child Psychol Psychiatry. 2014;55:1068–87. doi: 10.1111/jcpp.12295. [DOI] [PubMed] [Google Scholar]
- Yan J, Aliev F, Webb BT, Kendler KS, Williamson VS, Edenberg HJ, Agrawal A, Kos MZ, Almasy L, Nurnberger JI, Jr, Schuckit MA, Kramer JR, Rice JP, Kuperman S, Goate AM, Tischfield JA, Porjesz B, Dick DM. Using genetic information from candidate gene and genome-wide association studies in risk prediction for alcohol dependence. Addict Biol. 2014;19:708–21. doi: 10.1111/adb.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Li C, Kranzler HR, Farrer LA, Zhao H, Gelernter J. Exploring the genetic architecture of alcohol dependence in African-Americans via analysis of a genomewide set of common variants. Hum Genet. 2014;133:617–24. doi: 10.1007/s00439-013-1399-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young-Wolff KC, Enoch MA, Prescott CA. The influence of gene-environment interactions on alcohol consumption and alcohol use disorders: A comprehensive review. Clin Psychol Rev. 2011;31:800–816. doi: 10.1016/j.cpr.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.