

Abstract
The most studied physiological function of biliary epithelial cells (cholangiocytes) is to regulate bile flow and composition, in particular the hydration and alkalinity of the primary bile secreted by hepatocytes. After almost three decades of studies it is now become clear that cholangiocytes are also involved in epithelial innate immunity, in inflammation, and in the reparative processes in response to liver damage. An increasing number of evidence highlights the ability of cholangiocyte to undergo changes in phenotype and function in response to liver damage. By participating actively to the immune and inflammatory responses, cholangiocytes represent a first defense line against liver injury from different causes. Indeed, cholangiocytes express a number of receptors able to recognize pathogen- or damage-associated molecular patterns (PAMPs/DAMPs), such as Toll-like receptors (TLR), which modulate their pro-inflammatory behavior. Cholangiocytes can be both the targets and the initiators of the inflammatory process. Derangements of the signals controlling these mechanisms are at the basis of the pathogenesis of different cholangiopathies, both hereditary and acquired, such as cystic fibrosis-related liver disease and sclerosing cholangitis.
Keywords: Cholangiocytes, inflammation, cytokines, Toll-like receptor, inflammasome
1. Introduction
“Cholangiopathies” are chronic diseases of the biliary tree that if untreated, may progress to biliary-type cirrhosis, liver decompensation or liver cancer. These conditions are rare individually, but frequent as a group and are the cause of significant morbidity and mortality in the pediatric and young adult population. Pathogenesis and treatment of these conditions are not well known; indeed understanding the cholangiopathies represent one of the major unmet needs in Hepatology (1, 2).
The main cell target in cholangiopathies is the epithelium lining the bile ducts (i.e cholangiocytes). Intrahepatic cholangiocytes, particularly those lining the smallest portions of the biliary tree are the first site of reaction to most kinds of biliary injury. In response to several types of damage and inflammatory insults, cholangiocytes show the property of plasticity and acquire the active phenotype of reactive ductular cells (RDC) characterized by de novo secretion of a number of pro-inflammatory and pro-fibrotic cyto/chemokines. RDC have similarities with liver progenitor cells, but possess a reparative function, rather than regenerative function, and therefore are strongly associated with development of liver fibrosis. Thus, cholangiocytes and the resulting RDC are active players in the inflammatory reaction leading on one hand to liver repair, on the other to liver fibrosis and eventually cancer (2, 3).
Chronic inflammation (even of very low grade) is the pathophysiological engine leading to the main pathological and pathophysiological manifestations of cholangiopathies. This working hypothesis, drafted on a paper napkin in 2011 together with Nick LaRusso at the “ra Stua” Hutte in the Italian Alps, became figure 1 of a review article in Gastroenterology (2004) (1), and remains figure 1 also of this review.
Figure 1. Pathogenetic working model for cholangiopathies.
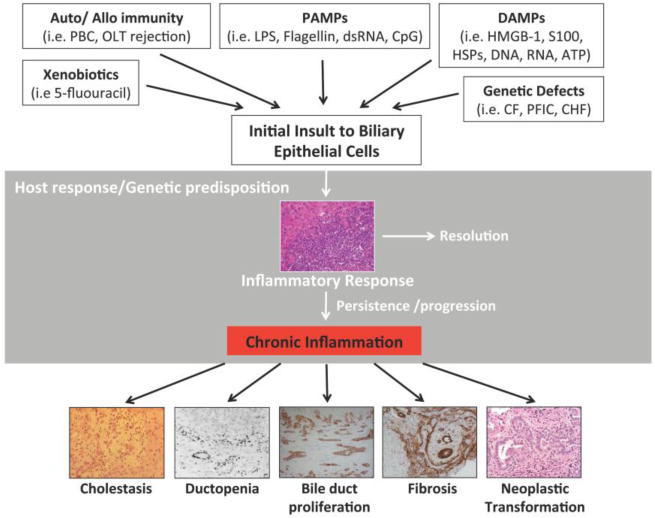
Modified from Lazaridis KN, Strazzabosco M, LaRusso NF. Cholangiopathies: Disorders of biliary epithelia. Gastroenterology 2004; 127: 1565–15771, with permission from Elsevier.
2. Inflammation and cholangiocytes in liver damage
In conditions leading to cholangiopathies, the biliary epithelium is exposed to cytokines and inflammatory mediators produced by infiltrating lymphocytes, macrophages and activated myofibroblasts, as well as to danger associated molecular patterns (or DAMPs) released from nearby damaged liver cells, including hepatocytes and non-parenchymal liver cells. Furthermore, cholangiocytes are exposed to bacterial products (called pathogen associated molecular pattern or PAMPs) originating from the intestine and bloodstream (4).
These pro-inflammatory factors stimulate cholangiocyte proliferative responses, and the secretion of pro-inflammatory cytokines and chemokines. In a first study published in 2001, our group showed that IL1β, IL6, TNFα, and IFNγ alone, but, especially in combination, caused ductular cholestasis by interfering with cAMP-dependent ion transport mechanisms (5). As a result, biliary fluid and bicarbonate secretion were inhibited. This study established an important relationship between inflammation and secretory function of cholangiocytes.
In a follow-up study (6) we also found that exposure to the above cytokines upregulated biliary epithelia expression of nitric oxide synthase 2, inducible (iNOS). Addressing the role of nitric oxide (NO) in biliary inflammation, we observed that upregulation of iNOS generates micromolar concentrations of NO, resulting in the formation of reactive nitrogen oxide species (RNOS) and nytrosylation of important signaling and transport proteins. In particular, we have shown that TNFα, and IFNγ (pro-inflammatory cytokines potentially involved in inflammatory cholangiopathies) stimulate NO production in cultured cholangiocytes and that micromolar concentrations of NO inhibit adenylyl cyclase (AC) activity, cAMP-dependent fluid secretion, as well as cAMP-dependent Cl− and HCO3− transport mediated by CFTR and by AE2, respectively (6). Interestingly, we demonstrated that the same effects were elicited by lipopolisaccaride LPS (see below). Of further interest was that the cholestatic effects of NO and of pro-inflammatory cytokines were prevented by iNOS inhibitors and by agents able to block the formation of RNOS (6). By immunohistochemistry, iNOS was expressed in the bile duct and RDC of a series of chronic liver diseases, indicating that these mechanisms may actually be of pathophysiological relevance in humans, in vivo (6).
Indeed, a strong reactivity for iNOS was reported in cholangiocytes of lipopolysaccharide (LPS)-treated rats, a model of sepsis-associated cholestasis (6). The finding that the inhibitory effects of pro-inflammatory cytokines on cholangiocyte secretion and cAMP production are mediated by iNOS induction and NO production may explains the histological features of the “cholangitis lenta” of sepsis, but also may link chronic inflammation with the cholestatic manifestations in the florid phases of cholangiopathies (1).
Consistent with this interpretation, we found that in PSC, biliary structures, including interlobular and septal ducts, strongly expressed iNOS and nytrotyrosine, suggesting that the bile ducts of PSC are indeed inflamed (6). RNOS, interfering with cAMP-mediated fluid and electrolyte transport, may cause a reduction in bile hydration and alkalinity that would further increase bile duct and hepatocellular damage (this would be now called disruption of the bicarbonate umbrella) (7). Positive nitrotyrosine immunoreactivity in PSC samples confirms the potential role of RNOS and protein nitrosylation in vivo (6). As iNOS expression is regulated by NF-kB, these data represent one of the first indications that NF-kB is upregulated in PSC. This can be a consequence or most likely the cause of the disease (see below). Given the harmful effects of NO on cholangiocyte secretory functions and on DNA repair mechanisms, we suggested that selective inhibition of iNOS expression should be of potential clinical relevance.
Furthermore, these data provided one of the first functional proofs of the presence in cholangiocytes of Toll-like receptors (TLRs) that are activated in response to endotoxins (i.e LPS) (8, 9).
Cytokines release in the setting of inflammation might also affect the barrier function of the biliary epithelium. This function is preserved, in physiologic conditions, by the functional interaction of a cell-cell junctional complex of proteins that includes tight-junctions, adherens junctions and F-actin cytoskeleton (10). Our earlier studies show that the exposure of polarized monolayers of cholangiocytes to cytokines increases the permeability of the epithelium to small dextrans (10KDa) (5). This is consistent with an increase of the paracellular transit possibly mediated by a defective function of the tight junctions. A similar effect was later described in cholangiocytes isolated from CFTR-KO mice, where an aberrant activation of TLR4-NF-kB signaling decreases the epithelial barrier function by destabilizing actin microfilaments and cell-junctional complexes (11).
This pathogenetic mechanism could ultimately contribute to cholestasis since a decrease of the barrier function increases the back-diffusion of toxic bile acids, resulting in further peribiliary inflammation and fibrogenesis.
3. Cholangiocytes and innate immunity
Because of its anatomical localization, and vascular connections with the intestine, the liver is exposed to high amounts of toxic products derived from the microbiota. Human bile under physiological and pathological conditions may contain bacterial components. Cholangiocytes participate to mucosal immune response not only by secreting IgA into the bile (12, 13), but also using a number of innate immune defenses (14, 15). As shown in acute cholangitis from C. parvum (16) the biliary expression of the β-defensin hBD2 is up-regulated in infectious cholangiopathies, dependent the stimulation of toll-like receptors that lead to the activation of NF-kB. Moreover, hBD2 expression on cultured cholangiocytes can be induced by stimulation with TNFα and IL1β, whose release in the portal tract is increased in several chronic cholangiopathies (17).
In physiological conditions, cholangiocytes are involved in protection of the biliary tree against gut-derived pathogens and toxins, mainly by activating Toll-like receptors (TLRs) (16), nuclear receptors (NRs) (18), and producing anti-microbial peptides. The biliary epithelium expresses TLRs (8, 9).
TLRs belong to a family of type I transmembrane proteins that recognize and discriminate between PAMPs (i.e LPS, Flagellin, DNA, RNA) but also endogenous components or DAMPs deriving from necrotic cells. Studies by Takeda et al. and Szabo et al. show that cholangiocytes specifically express TLR 2, 3, 4, 5, and 9 which bind to ligands such as bacterial molecules, double-stranded RNA, gram-negative lipopolysaccharide (endotoxins) and flagellin but also endogenous mediators as hyaluronan and HMGB1 (9, 19). TLR2 is a receptor for the lipoteichoic acid (LTA), a component of the bacterial membrane, TLR3 is a sensor for viral dsRNA and is upregulated in primary biliary cirrhosis and biliary atresia (20, 21). TLR9 is a receptor for CpG DNA, a DNA typical of bacteria, and was reported to be up-regulated in PSC(22).
Signal transduction in response to TLRs results in the recruitment of TIR-domain containing adaptor molecules (MyD88, Mal, TRIF and TRAM) and activation of intracellular signaling pathways that upregulate NF-κB-dependent proinflammatory gene expression. TLR4 signaling is the best described and is the main sensor for LPS. In response to LPS binding, TLR4 dimerizes and activates an intracellular signal cascade that results in NF-kB nuclear import and activation. Once into nucleus, the NF-kB subunit p65 regulates the transcription of a wide range of pro-inflammatory cytokines and chemokines, including tumor necrosis factor (TNFα), IL1, IL6, IL8, IL12, G-CSF and LIX (8, 9, 23).
In addition to bacterial-derived molecules, the biliary epithelium also reacts to DAMPS, also known as alarmins. DAMPs represent a group of molecules normally retained inside the cells, where they participate in different intracellular functions, that upon injury can be released into the extracellular environment and be recognized as endogenous ligands by TLRs (24, 25). Several studies have shown that these “danger signals” are sensed by TLRs of neighboring cells, such as TLR2-4-5-9, and activate mechanisms similar to those classically used by PAMPs (26). This leads to stimulation of the NF-κB-dependent cytokines/chemokines secretion and recruitment of immune cells at the site of injury. Because of their importance in several human diseases, the most studied DAMPs are HMGB-1, S100 proteins (S100A8-9-12) and several Heat-shock proteins (HSPs), together with nuclear DNA complexes and purine nucleotides (ATP) (24).
The liver and in particular the biliary epithelium is continuously exposed to bacterial products coming from the gut microflora. In cholangiocytes, the TLR system is tightly regulated and in physiologic conditions, little or no inflammation occurs, thus avoiding an exaggerated response that could lead to autoimmune or chronic inflammatory disorders (27). The malfunction of one or more of these check points regulating immune tolerance in cholangiocytes may elicit an exaggerated inflammatory responses to pathogens or endotoxins eventually present in the bile. This protective mechanisms that prevent/reduce the damage to the epithelium infact may be altered in a number of cholangiopathies (figure 1).
These findings are even more relevant now, in the light of the hypothesized changes in innate immunity and in gut microbioma in PSC and other liver diseases (28, 29).
Experimental evidence supports the pathophysiological role of these mechanisms in cystic fibrosis related liver disease (CFLD) (11, 30, 31), congenital hepatic fibrosis (CHF) (32) and possibly sclerosing cholangitis. CFLD, was considered to be the prototypic chronic cholangiopathy caused by reduced ductal bile flow generation and reduction in biliary chloride and bicarbonate secretion resulting by the dysfunction of CFTR, but it is now clear that the main pathogenetic event is a lack of tolerance in the innate immune system (30). When CFTR, the apical Cl− channel, is defective, TLR4-dependent inflammatory responses are upregulated. In CFTR-KO mice, experimental colitis induced by dextran sodium sulfate stimulates a brisk inflammatory response centered on the bile ducts that is not observed in WT littermates. This response is characterized by an abundant peribiliary accumulation of CD45+ inflammatory cells including macrophages and neutrophils, and results from a dysregulation of the TLR4/NF-κB axis caused by an aberrant activation of Src tyrosine kinase (30). In normal conditions, Src is sequestered and maintained in an inactive state by a multiprotein complex, composed by CFTR together with the bridge proteins EPB-50, Cbp, and Csk. Absence of CFTR at the apical membrane, as in certain CF patients, leads to the lack of assembly of this complex and a persistent activation of Src that in turn, phosphorylates TLR4, further stimulating the production of pro-inflammatory cytokines in response to PAMPs (11).
This newly described mechanism has great translational potential as it indicates that treatment should not be limited at improving ductal secretion, but at controlling inflammation as well. In a mouse model of CFLD (CFTR-KO mice), our group has shown that PPAR-γ agonists, such as pioglitazione and rosiglitazione, are able to reduce the production of pro-inflammatory cytokine by CFTR-KO cholangiocytes in vitro. In addition, PPAR-γ agonists significantly decreased the inflammatory infiltrate, and biliary damage in CFTR-KO mice treated with DSS (31).
Nuclear receptors (NRs) are a wide group of transcription factors well represented in the liver where they control a range of fundamental physiological functions, from detoxification from bile acids (VDR, FXR), to bile secretion (GR, FXR) (18). Among them, the peroxisome proliferator-activated receptors (PPARs) have been extensively studied and characterized. VDR, LXRs and PPARs, acting as negative regulators, are able to trans-repress the pro-inflammatory signals stimulated by PAMPs. All PPARs isoforms can suppress the pathways activated by the PAMPs-TLRs interaction. PPAR-α, PPAR-β/δ, and PPAR-γ, are all expressed, at different levels, in normal cholangiocytes. Among them, PPAR-γ modulates LPS-induced pro-inflammatory phenotype in CF cholangiocytes, by up-regulating the expression of IkBα, a negative regulator of NF-kB (31).
4. Inflammation, “parainflammation” and autoinflammatory diseases
The biliary epithelium is a preferred target of inflammatory and immune injury to the liver. In inflammatory cholangiopathies, both innate and adaptive immune responses are often sequentially, or simultaneously involved. Stimulation of innate immune responses (see above) by exogenous or endogenous insults (including damage to nearby cells or to hepatocytes) generates an inflammatory reaction that, if protracted, may then self-sustains and perpetuates thanks to the activation of adaptive immune mechanisms (33).
Among the endogenous stimuli able to generate an inflammatory reaction in the biliary tree, cell dysfunction caused by genetic defects plays a distinctive role. Genetically transmitted defects may cause cell/tissue malfunction (stress, altered metabolisms) rather than an overt cell injury. These stress conditions affecting the homeostatic balance of physiological systems are therefore different from those involved in host defense or tissue repair. These conditions do not develop a classic inflammatory response, but rather generate a chronic inflammation of low magnitude (that Ruslan Medzhitov defined as ‘parainflammation’) and it is aimed at restoring a normal cell/tissue homeostasis, rather than defend the host against pathogens (34, 35). As proposed by Medzhitov, parainflammation is an adaptive response to a persistent cell dysfunction shifting the homeostatic set points (35, 36). Unfortunately, in spite of its homeostatic nature, persistent parainflammation becomes maladaptive and may stimulate a fibrotic response.
Diseases caused by genetic mutations of critical proteins expressed by the biliary epithelium in different cell compartments such as the cellular membrane, the primary cilium, and the ER, are paradigmatic of these processes. Again a useful example is CFLD. In this condition, lack of the cystic fibrosis conductance regulator protein (CFTR) at the apical membrane of cholangiocytes impairs the regulation of TLR4-dependent innate immunity response. As result the biliary epithelium loses its endotoxin tolerance, and generates an enhanced inflammatory response, when challenged with TLR4 agonists (several PAMPS and DAMPs). The para-inflammatory process set in motion by the cell dysfunction generates a pro-inflammatory reactive phenotype in the biliary epithelium, the release of cyto/chemokines and the infiltration of the portal spaces with inflammatory cells (30).
Recent studies in mouse models of congenital hepatic fibrosis (CHF) further highlight the mechanisms linking genetically transmitted cholangiocyte dysfunction to biliary fibrosis through parainflammation. CHF is a rare disease, characterized by altered architecture of the biliary tree and extensive biliary fibrosis, leading to severe portal hypertension and its complications(37). CHF is caused by mutations in PKHD1, the gene encoding for fibrocystin (FPC), that is a ciliary protein expressed by ductal epithelial cells (37). The function of FPC is still unknown, and a variety of cellular functions, including proliferation, differentiation, tubulogenesis, planar cell polarity and cell-matrix interaction seem to be influenced by FPC. Biliary cysts, which originate from the abnormal remodeling of the fetal ductal plate structures, progressively enlarge in association with a dense fibrosis and a portal inflammatory infiltrate (38, 39). In this pathogenetic sequence, in sharp contrast with acquired cholangiopathies where portal fibrosis is associated with cell necrosis or apoptosis, an overt necroinflammation of the biliary epithelium is typically absent (39).
Using a mouse model of CHF (Pkhd1del4/del4), we showed that in the early phase of the disease, FPC-defective cholangiocytes recruit inflammatory cells, mostly macrophages, by secreting a variety of chemokines, such as CXCL1, CXCL10, and CXCL12 (32). Chemokines secretion was orchestrated by β-catenin, a signalling molecule whose nuclear translocation and activity are increased in Pkhd1del4/del4 mice following its cAMP/PKA-mediated phosphorylation at the unusual serine-675 residue, which prevents its proteosomal ubiquitination (40). Recruited macrophages secrete TNF-α and TGF-β that up-regulate in cholangiocytes the expression of αvβ6 integrin, an activator of latent TGF-β, an epithelial feature commonly observed in response to duct injury and inflammation (32). In the initial phase of fibrosis, αSMA-positive myofibroblasts are scarce and the peribiliary infiltrate is dominated by classically activated, iNOS expressing M1 macrophages. The number of portal myofibroblasts increases after 6 months of age, along with an increase in alternatively activated M2 macrophages, leading to increased fibrosis and development of portal hypertension. Interestingly, inhibition of macrophage recruitment in vivo by clodronate before the development of portal hypertension, reduced the recruitment of portal myofibroblasts and portal fibrosis (32). Further studies by our group (41) indicate that selective blockade of CXCL10/CXCR3 signaling reduces fibrosis, liver cyst growth and macrophage infiltration in Pkhd1del4/del4 mice indicating that in this parainflammatory context, a major pathogenetic role is played by this specific chemokine axis. The recent finding that increased secretion of CXCL10 is actually under the control of IL-1β (and therefore pSTAT3 and NF-kB), pinpoints CHF as an autoinflammatory disease. Thus, CHF can be considered the archetype of a model of liver fibrogenesis generated by a parainflammatory response to the loss of physiological homeostasis in cholangiocytes.
The maturation of IL-1β and IL-18, produced as an effect of stimulation of innate immunity, requires the activation of inflammasome. These are multi-protein complexes that, by cleaving the inflammatory pro-proteins into their active, secretory forms, promote inflammation (33, 42). Stimulation of inflammasome may also lead to pathological responses as sterile inflammation or autoimmunity, whereas a defective inflammasome response may increase the susceptibility to pathogens. As shown by Maroni et al., the NLP3 inflammasome is increased in reactive cholangiocytes in patients with PSC and in rodent models of biliary damage. The same authors also reported an increase in IL-18 expression following treatment of cultured cholangiocytes with LPS (43).
As previously mentioned, in the pathogenesis of inflammatory diseases, a critical aspect is to distinguish the degree of involvement between the innate and adaptive immune system. The term ‘autoinflammation’ has been introduced in the last few years, to highlight the type of inflammation sustained by abnormal innate immune responses without involvement of the adaptive immune system, which is classically, associated with the production of autoantibodies or activation of autoreactive T cells. In contrast with autoimmunity, in autoinflammation innate immune cells are directly activated by endogenous or exogenous insults, which “light the fire within” (44).
5. Sclerosing cholangitis
Sclerosing Cholangitis is a group of chronic inflammatory diseases of the intrahepatic and/or extrahepatic biliary tract characterized by peribiliary inflammation and obliterative fibrosis leading to strictures and cholestasis, ductopenia, and eventually biliary cirrhosis. As in other chronic inflammatory diseases, development of malignancies (cholangiocarcinoma and eventually colon cancer) is possible. The etiology and pathophysiology of PSC remain unclear and lack of experimental models has hampered this area of research(45–48).
Sclerosing cholangitis is likely a pathological manifestation of a number of different conditions. As shown in table 1, the list of conditions known to be associated with a sclerosing cholangitis is growing and ranges from genetic diseases of channels and transporters in the biliary epithelium or in the hepatocytes, to ischemic damages, infections and autoimmune diseases. Also under the term of Primary Sclerosing Cholangitis there is probably still a number of similar pathophysiological mechanisms, whose etiology is not clear at present. The conditions are usually not associated with inflammatory bowel diseases (IBD).
Table 1.
Conditions associated with sclerosing cholangitis.
| Sclerosing Cholangitis | Cause | Intestinal manifestations |
|---|---|---|
| Primary | Unknown | a ‘syndrome” of concurrent bile duct fibrosis, right-sided colitis and a neoplastic propensity at both these sites |
| Secondary | Cystic Fibrosis | Yes/no |
| ABCB4 deficiency | no | |
| Choledocolithiasis | no | |
| Ischemic cholangitis | no | |
| Portal hypertensive biliopathy | no | |
| AIDS Cholangiopathy | no | |
| Recurrrent pyogenic cholangitis | no | |
| IgG4-associated cholangitis | Yes/no | |
| Mast-cell cholangiopthy | no | |
| Bile duct surgery | no | |
| Abdominal trauma | no |
The association with IBD actually shows strong geographical variation ranging between 80% in Nordic European countries, to 40–50% in Mediterranean countries, to 30% in Japan. Nevertheless, several observations and genetic studies indicate that PSC and colitis is probably a different condition from isolated IBD i.e. “a syndrome of concurrent bile duct fibrosis and right-sided colitis and neoplastic predisposition at both of these sides” that has most likely an autoimmune pathogenesis, as shown by genomics studies (49). However, as only a minority of patients with inflammatory bowel disease (IBD) develop PSC, it is not clear if this syndrome results from a predisposition of the biliary epithelium to inflammatory reactions. It might be that two hits are required: a predisposition to biliary inflammation and a second genetic or most likely environmental factor. It is worth mentioning that diseases of the innate and of the adaptive immunity are actually two extremes in a continuum spectrum of disease conditions and clinical manifestations and there is not a hiatus between inflammatory and autoimmune manifestations (or between innate and adaptive immunity).
The association with IBD indicates that PSC may be caused by aberrant immune responses of the biliary epithelium to PAMPs that enter the portal circulation through a more permeable intestinal mucosa (‘leaky gut’ hypothesis) (50). However, retention of toxic bile acids able to damage the biliary epithelium has been also proposed as an alternative hypothesis (‘toxic bile’ hypothesis) (51). A unified mechanism able to explain the pathophysiology of the disease is lacking. However, these two views may converge if we hypothesize that in PSC, the biliary epithelium possesses an increased reactivity to PAMPs and to DAMPs respectively released by the leaky gut or because of bile acid-induced liver cell damage. This hypothesis implies that a change in cellular homeostasis in cholangiocytes is the cause of PSC, rather than the consequence of the disease.
Consistent with this hypothesis, our preliminary and recently published studies suggest that pro-inflammatory cytokines and endotoxins induce inappropriate innate immune responses in activated cholangiocytes in patients with PSC (52). Preliminary evidence suggests that cholangiocytes isolated from PSC patients show higher NF-κB transcriptional activity and IL-8 secretion when challenged with LPS, providing evidence that IL-8 in cholangiocytes is increased in a cell-autonomous way, and suggesting enhanced innate immune responsiveness to endotoxins. Furthermore, IL-8 was shown increased in bile of patients with PSC (53). Previously published data (54) and our own preliminary data show increased IL-8 secretion and NF-kB transcriptional activity in isolated and cultured PSC cholangiocytes (Strazzabosco unpublished data). Increased expression of NO in bile ducts in PSC suggests activation of the NF-kB pathway (6). Furthermore, Th17 response to pathogen stimulation is increased in patients with PSC (55). Also, Maroni et al. recently reported an increase in Nlrp3 inflammasome expression in the reactive epithelia in biopsies of patients with PSC (43).
Persistent exposure to DAMPs and PAMPs can also trigger cell senescence. This is normally a protective mechanism initiated by the cell to avoid neoplastic transformation. However, if the damage does not resolved, senescent cells can transition to a pathologic reactive phenotype, characterize by hypersecretion of cytokines (e.g IL-6) and chemokines (IL-8), also known as senescence-associated secretory phenotype (SASP). Tabibian et al. has demonstrated the presence in PSC of senescent and SASP cholangiocytes together with Ras activation, a mediator of senescence, suggesting senescence as an additional mechanism contributing to initiation and persistence of the inflammatory response (56).
In aggregate, the above findings are consistent with the hypothesis, that the central pathogenetic mechanism of PSC may be based on aberrant activation of the biliary innate immunity following exposure to PAMPs coming from a leaky intestine or DAMPs released in response to bile acid toxicity.
Genomic studies recently reviewed by Jiang and Karlsen (49) have identified a series non-HLA and more susceptibility loci with notable candidate genes, some of them expressed in the liver or in the gut. Some of them are genes implicated in innate immunity, some implicated with adaptive immunity. The function of these genes for the most part is not understood, and therefore now the ball is in the court of the cell biologist.
6. Conclusions
Consistent with the ability of cholangiocytes to mount a reparative response to many forms of liver damage, the biliary epithelium is heavily involved in inflammation and innate immunity. Reactive cholangiocytes produce a wide range of soluble factors involved in inflammation, such as cytokines, chemokines and growth factors. The imbalance of signals that modulate inflammatory responses in cholangiocytes is emerging as a new pathogenetic mechanism in specific biliary disease settings.
Production of inflammatory mediators in epithelial cells may also be triggered by a genetically determined dysfunction of the epithelial cells CFLD and CHF are two examples relevant for human diseases. In these conditions, a low level inflammatory reaction stimulates secretion of pro-inflammatory cytokines and chemokines that enable cholangiocytes to recruit macrophages and/or neutrophils to the peribiliary region. These observations support the concept that a genetically determined epithelial cell dysfunction may trigger a persistent low level, but long-lasting, inflammatory response, which may ultimately lead to organ scarring, a process called ‘parainflammation’ or auto-inflammation. We hypothesize that this phenomenon may be also relevant in PSC.
This hypothesis will be tested studying the response of human PSC cholangiocytes to TLR agonists. The latest advances in stem cell technology offer the possibility of generating two in vitro models of human cholangiocytes, one derived from induced-pluripotent stem cells (iPSCs) (57–59) and the second from isolated biliary stem cells expanded in vitro as bipotent stem cells in 3D organoids (60, 61). The greatest advantage of iPSCs is that they can be derived from biopsy samples (i.e skin, blood) and they maintain a very high replicative potential that provides an unlimited source of patient-specific cells able to differentiate into the somatic cell of interest and may provide a valuable source to test the efficacy of personalized treatments. On the other hand the organoids have a long-term genetic stability in culture and do not require a transdifferentiation protocol (61).
We believe these recent advances, shed some light on an important pathophysiological aspect of many forms of cholangiopathies, however to achieve a thorough knowledge of the complex immunological functions played by cholangiocytes, both in normal and diseased conditions, further studies are needed.
Highlights.
Cholangiocytes are active players in inflammatory and reparative processes during liver insults.
Chronic inflammation is the pathogenetic factor in cholangiopaties.
Altered adaptive cholangiocytes immunity mechanisms may elicit an exaggerated inflammatory response to pathogens or endotoxins.
Genetic dysfunction in biliary cells may stimulate low-grade inflammatory processes that cause liver damage and fibrosis.
Acknowledgments
Funding: This work was supported by the National Institure of Health (RO1DK096096, RO1DK-079005-07, RO1DK101528, DK034989 Silvio O. Conte Digestive Diseases Research Core Center); by FFC (24/2015); and Partners Seeking a Cure Foundation.
Abbreviations
- RDC
reactive ductular cell
- DAMPs
danger associated molecular patterns
- PAMPs
pathogen associated molecular patterns
- cAMP
cyclic adenosine monophosphate
- NOS
nitric oxide synthase
- RNOS
reactive nitrogen oxide species
- CFTR
cystic fibrosis transmembrane conductance regulator
- AE2
anion exchange protein 2
- LPS
lipopolysaccharide
- PSC
primary sclerosing cholangitis
- TLR
Toll-like receptor
- hBD2
human beta defensing 2
- NR
nuclear receptor
- MyD88
myeloid differentiation primary response gene 88
- Mal
MyD88 adapter like
- TRIF
TIR-domain-containing adapter-inducing interferon-β
- TRAM
TRIF-related adaptor molecule
- G-CSF
Granulocyte-colony stimulating factor
- LIX
Lipopolysaccharide-induced CXC chemokine
- HMGB-1
High mobility group box 1 protein
- ATP
Adenosine triphosphate
- HSP
Heat shock protein
- PPAR-γ
Peroxisome proliferator-activated receptor gamma
- VDR
vitamin D receptor
- FXR
farnesoid X receptor
- GR
glucocorticoid receptor
- CHF
congenital hepatic fibrosis
- PKHD1
Polycystic Kidney And Hepatic Disease 1
- FPC
fibrocystin
- CXCL1
chemokine ligand 1
- CXCL10
C-X-C motif chemokine 10
- CXCL12
C-X-C motif chemokine 12
- IBD
inflammatory bowel disease
- Nlrp3
NLR Family Pyrin Domain Containing 3
- iPSC
induced pluripotent stem cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lazaridis KN, Strazzabosco M, Larusso NF. The cholangiopathies: disorders of biliary epithelia. Gastroenterology. 2004;127:1565–1577. doi: 10.1053/j.gastro.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Strazzabosco M, Fabris L, Spirli C. Pathophysiology of cholangiopathies. J Clin Gastroenterol. 2005;39:S90–S102. doi: 10.1097/01.mcg.0000155549.29643.ad. [DOI] [PubMed] [Google Scholar]
- 3.Fabris L, Strazzabosco M. Epithelial-mesenchymal interactions in biliary diseases. Semin Liver Dis. 2011;31:11–32. doi: 10.1055/s-0031-1272832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harada K, Nakanuma Y. Biliary innate immunity in the pathogenesis of biliary diseases. Inflammation & allergy drug targets. 2010;9:83–90. doi: 10.2174/187152810791292809. [DOI] [PubMed] [Google Scholar]
- 5.Spirli C, Nathanson MH, Fiorotto R, Duner E, Denson LA, Sanz JM, et al. Proinflammatory cytokines inhibit secretion in rat bile duct epithelium. Gastroenterology. 2001;121:156–169. doi: 10.1053/gast.2001.25516. [DOI] [PubMed] [Google Scholar]
- 6.Spirli C, Fabris L, Duner E, Fiorotto R, Ballardini G, Roskams T, et al. Cytokine-stimulated nitric oxide production inhibits adenylyl cyclase and cAMP-dependent secretion in cholangiocytes. Gastroenterology. 2003;124:737–753. doi: 10.1053/gast.2003.50100. [DOI] [PubMed] [Google Scholar]
- 7.Beuers U, Hohenester S, de Buy Wenniger LJ, Kremer AE, Jansen PL, Elferink RP. The biliary HCO(3)(–) umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology. 2010;52:1489–1496. doi: 10.1002/hep.23810. [DOI] [PubMed] [Google Scholar]
- 8.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 9.Szabo G, Dolganiuc A, Mandrekar P. Pattern recognition receptors: a contemporary view on liver diseases. Hepatology. 2006;44:287–298. doi: 10.1002/hep.21308. [DOI] [PubMed] [Google Scholar]
- 10.Meng W, Takeichi M. Adherens junction: molecular architecture and regulation. Cold Spring Harb Perspect Biol. 2009;1:a002899. doi: 10.1101/cshperspect.a002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fiorotto R, Villani A, Kourtidis A, Scirpo R, Amenduni M, Geibel PJ, et al. The cystic fibrosis transmembrane conductance regulator controls biliary epithelial inflammation and permeability by regulating Src tyrosine kinase activity. Hepatology. 2016;64:2118–2134. doi: 10.1002/hep.28817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemaitre-Coelho I, Jackson GD, Vaerman JP. Rat bile as a convenient source of secretory IgA and free secretory component. Eur J Immunol. 1977;7:588–590. doi: 10.1002/eji.1830070818. [DOI] [PubMed] [Google Scholar]
- 13.Nagura H, Smith PD, Nakane PK, Brown WR. IGA in human bile and liver. J Immunol. 1981;126:587–595. [PubMed] [Google Scholar]
- 14.Chuang YH, Lan RY, Gershwin ME. The immunopathology of human biliary cell epithelium. Semin Immunopathol. 2009;31:323–331. doi: 10.1007/s00281-009-0172-5. [DOI] [PubMed] [Google Scholar]
- 15.Syal G, Fausther M, Dranoff JA. Advances in cholangiocyte immunobiology. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1077–1086. doi: 10.1152/ajpgi.00227.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen XM, O’Hara SP, Nelson JB, Splinter PL, Small AJ, Tietz PS, et al. Multiple TLRs are expressed in human cholangiocytes and mediate host epithelial defense responses to Cryptosporidium parvum via activation of NF-kappaB. J Immunol. 2005;175:7447–7456. doi: 10.4049/jimmunol.175.11.7447. [DOI] [PubMed] [Google Scholar]
- 17.Harada K, Ohba K, Ozaki S, Isse K, Hirayama T, Wada A, et al. Peptide antibiotic human beta-defensin-1 and -2 contribute to antimicrobial defense of the intrahepatic biliary tree. Hepatology. 2004;40:925–932. doi: 10.1002/hep.20379. [DOI] [PubMed] [Google Scholar]
- 18.Firrincieli D, Zuniga S, Poupon R, Housset C, Chignard N. Role of nuclear receptors in the biliary epithelium. Dig Dis. 2011;29:52–57. doi: 10.1159/000324129. [DOI] [PubMed] [Google Scholar]
- 19.Takeda K, Akira S. TLR signaling pathways. Seminars in immunology. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Harada K, Sato Y, Itatsu K, Isse K, Ikeda H, Yasoshima M, et al. Innate immune response to double-stranded RNA in biliary epithelial cells is associated with the pathogenesis of biliary atresia. Hepatology. 2007;46:1146–1154. doi: 10.1002/hep.21797. [DOI] [PubMed] [Google Scholar]
- 21.Takii Y, Nakamura M, Ito M, Yokoyama T, Komori A, Shimizu-Yoshida Y, et al. Enhanced expression of type I interferon and toll-like receptor-3 in primary biliary cirrhosis. Lab Invest. 2005;85:908–920. doi: 10.1038/labinvest.3700285. [DOI] [PubMed] [Google Scholar]
- 22.Karrar A, Broome U, Sodergren T, Jaksch M, Bergquist A, Bjornstedt M, et al. Biliary epithelial cell antibodies link adaptive and innate immune responses in primary sclerosing cholangitis. Gastroenterology. 2007;132:1504–1514. doi: 10.1053/j.gastro.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 23.Akira S. Toll-like receptors and innate immunity. Adv Immunol. 2001;78:1–56. doi: 10.1016/s0065-2776(01)78001-7. [DOI] [PubMed] [Google Scholar]
- 24.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pittman K, Kubes P. Damage-Associated Molecular Patterns Control Neutrophil Recruitment. J Innate Immun. 2013 doi: 10.1159/000347132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–519. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- 27.Abreu MT, Fukata M, Arditi M. TLR signaling in the gut in health and disease. J Immunol. 2005;174:4453–4460. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- 28.Mattner J. Impact of Microbes on the Pathogenesis of Primary Biliary Cirrhosis (PBC) and Primary Sclerosing Cholangitis (PSC) Int J Mol Sci. 2016;17 doi: 10.3390/ijms17111864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabibian JH, O’Hara SP, Lindor KD. Primary sclerosing cholangitis and the microbiota: current knowledge and perspectives on etiopathogenesis and emerging therapies. Scand J Gastroenterol. 2014;49:901–908. doi: 10.3109/00365521.2014.913189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiorotto R, Scirpo R, Trauner M, Fabris L, Hoque R, Spirli C, et al. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4-NF-kappaB-mediated inflammatory response in mice. Gastroenterology. 2011;141:1498–1508. doi: 10.1053/j.gastro.2011.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scirpo R, Fiorotto R, Villani A, Amenduni M, Spirli C, Strazzabosco M. Stimulation of nuclear receptor peroxisome proliferator-activated receptor-gamma limits NF-kappaB-dependent inflammation in mouse cystic fibrosis biliary epithelium. Hepatology. 2015;62:1551–1562. doi: 10.1002/hep.28000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Locatelli L, Cadamuro M, Spirli C, Fiorotto R, Lecchi S, Morell CM, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology. 2016;63:965–982. doi: 10.1002/hep.28382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol. 2015;12:387–400. doi: 10.1038/nrgastro.2015.94. [DOI] [PubMed] [Google Scholar]
- 34.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 35.Chovatiya R, Medzhitov R. Stress, inflammation, and defense of homeostasis. Mol Cell. 2014;54:281–288. doi: 10.1016/j.molcel.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–827. doi: 10.1016/j.cell.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321–337. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strazzabosco M, Somlo S. Polycystic liver diseases: congenital disorders of cholangiocyte signaling. Gastroenterology. 2011;140:1855–1859. doi: 10.1053/j.gastro.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strazzabosco M, Fabris L. Development of the bile ducts: essentials for the clinical hepatologist. J Hepatol. 2012;56:1159–1170. doi: 10.1016/j.jhep.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spirli C, Locatelli L, Morell CM, Fiorotto R, Morton SD, Cadamuro M, et al. Protein kinase A-dependent pSer(675) -beta-catenin, a novel signaling defect in a mouse model of congenital hepatic fibrosis. Hepatology. 2013;58:1713–1723. doi: 10.1002/hep.26554. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 41.Kaffe EPF, Mariotti V, Fabris L, Cadamuro M, Amenduni M, Fiorotto R, Strazzabosco M, Spirli C. Inhibition of IL-1b/STAT3/β-catenin-dependent stimulation of the CXCL10/CXCR3 axis prevents macrophage recruitment and progression of the disease in Pkhd1del4/del4 mice, a model of congenital hepatic fibrosis. Journal of Hepatology. 2017;66:S36–S37. [Google Scholar]
- 42.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 43.Maroni L, Agostinelli L, Saccomanno S, Pinto C, Giordano DM, Rychlicki C, et al. Nlrp3 Activation Induces Il-18 Synthesis and Affects the Epithelial Barrier Function in Reactive Cholangiocytes. Am J Pathol. 2017;187:366–376. doi: 10.1016/j.ajpath.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 44.Park H, Bourla AB, Kastner DL, Colbert RA, Siegel RM. Lighting the fires within: the cell biology of autoinflammatory diseases. Nat Rev Immunol. 2012;12:570–580. doi: 10.1038/nri3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lazaridis KN. Sclerosing cholangitis epidemiology and etiology. J Gastrointest Surg. 2008;12:417–419. doi: 10.1007/s11605-007-0344-3. [DOI] [PubMed] [Google Scholar]
- 46.Lee YM, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med. 1995;332:924–933. doi: 10.1056/NEJM199504063321406. [DOI] [PubMed] [Google Scholar]
- 47.O’Mahony CA, Vierling JM. Etiopathogenesis of primary sclerosing cholangitis. Semin Liver Dis. 2006;26:3–21. doi: 10.1055/s-2006-933559. [DOI] [PubMed] [Google Scholar]
- 48.Horsley-Silva JL, Carey EJ, Lindor KD. Advances in primary sclerosing cholangitis. Lancet Gastroenterol Hepatol. 2016;1:68–77. doi: 10.1016/S2468-1253(16)30010-3. [DOI] [PubMed] [Google Scholar]
- 49.Jiang X, Karlsen TH. Genetics of primary sclerosing cholangitis and pathophysiological implications. Nat Rev Gastroenterol Hepatol. 2017;14:279–295. doi: 10.1038/nrgastro.2016.154. [DOI] [PubMed] [Google Scholar]
- 50.Karlsen TH, Schrumpf E, Boberg KM. Update on primary sclerosing cholangitis. Dig Liver Dis. 2010;42:390–400. doi: 10.1016/j.dld.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 51.Karlsen TH, Boberg KM. Update on primary sclerosing cholangitis. J Hepatol. 2013 doi: 10.1016/j.jhep.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 52.Mueller T, Beutler C, Pico AH, Shibolet O, Pratt DS, Pascher A, et al. Enhanced innate immune responsiveness and intolerance to intestinal endotoxins in human biliary epithelial cells contributes to chronic cholangitis. Liver Int. 2011;31:1574–1588. doi: 10.1111/j.1478-3231.2011.02635.x. [DOI] [PubMed] [Google Scholar]
- 53.Zweers SJ, Shiryaev A, Komuta M, Vesterhus M, Hov JR, Perugorria MJ, et al. Elevated interleukin-8 in bile of patients with primary sclerosing cholangitis. Liver Int. 2016 doi: 10.1111/liv.13092. [DOI] [PubMed] [Google Scholar]
- 54.Tabibian JH, Trussoni CE, O’Hara SP, Splinter PL, Heimbach JK, LaRusso NF. Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis. Lab Invest. 2014;94:1126–1133. doi: 10.1038/labinvest.2014.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katt J, Schwinge D, Schoknecht T, Quaas A, Sobottka I, Burandt E, et al. increased Th17 response to pathogen stimulation in patients with primary sclerosing cholangitis. Hepatology. 2013 doi: 10.1002/hep.26447. [DOI] [PubMed] [Google Scholar]
- 56.Tabibian JH, O’Hara SP, Splinter PL, Trussoni CE, LaRusso NF. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology. 2014;59:2263–2275. doi: 10.1002/hep.26993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ogawa M, Ogawa S, Bear CE, Ahmadi S, Chin S, Li B, et al. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nat Biotechnol. 2015;33:853–861. doi: 10.1038/nbt.3294. [DOI] [PubMed] [Google Scholar]
- 58.Sampaziotis F, Cardoso de Brito M, Madrigal P, Bertero A, Saeb-Parsy K, Soares FA, et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat Biotechnol. 2015;33:845–852. doi: 10.1038/nbt.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fiorotto RAM, Cadamuro M, Spirli C, Strazzabosco M. Identification of Src tyrosine kinase as a therapeutic target in cystic fibrosis liver disease using patient specific induced pluripotent stem cells -derived cholangiocytes. Journal of Hepatology. 2017;66:S182. [Google Scholar]
- 60.Broutier L, Andersson-Rolf A, Hindley CJ, Boj SF, Clevers H, Koo BK, et al. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat Protoc. 2016;11:1724–1743. doi: 10.1038/nprot.2016.097. [DOI] [PubMed] [Google Scholar]
- 61.Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 2015;160:299–312. doi: 10.1016/j.cell.2014.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]