

ABSTRACT
The ATP-sensitive potassium channel (KATP) functions as a metabo-electric transducer in regulating insulin secretion from pancreatic β-cells. The pancreatic KATP channel is composed of a pore-forming inwardly-rectifying potassium channel, Kir6.2, and a regulatory subunit, sulphonylurea receptor 1 (SUR1). Loss-of-function mutations in either subunit often lead to the development of persistent hyperinsulinemic hypoglycemia of infancy (PHHI). PHHI is a rare genetic disease and most patients present with immediate onset within the first few days after birth. In this study, we report an unusual form of PHHI, in which the index patient developed hyperinsulinemic hypoglycemia after 1 year of age. The patient failed to respond to routine medication for PHHI and underwent a complete pancreatectomy. Genotyping of the index patient and his immediate family members showed that the patient and other family members with hypoglycemic episodes carried a heterozygous novel mutation in KCNJ11 (C83T), which encodes Kir6.2 (A28V). Electrophysiological and cell biological experiments revealed that A28V hKir6.2 is a dominant-negative, loss-of-function mutation and that KATP channels carrying this mutation failed to reach the cell surface. De novo protein structure prediction indicated that this A28V mutation reoriented the ER retention motif located at the C-terminal of the hKir6.2, and this result may explain the trafficking defect caused by this point mutation. Our study is the first report of a novel form of late-onset PHHI that is caused by a dominant mutation in KCNJ11 and exhibits a defect in proper surface expression of Kir6.2.
KEYWORDS: ATP-Sensitive Potassium Channel (KATP), Inwardly Rectifying Potassium Channel 6.2 (Kir6.2), Persistent Hyperinsulinemic Hypoglycemia of Infancy (PHHI), sulphonylurea Receptor 1 (SUR1)
Introduction
Persistent hyperinsulinemic hypoglycemia of infancy, or PHHI, is the most common cause of severe neonatal hypoglycemia that lasts beyond the first a few hours of life.1 Both sporadic and familial forms of the disease exist, and several genes have been shown to be involved in the pathophysiology. Among these genes, disease-associated alleles for KCNJ11, ABCC8, GLUD1, and GCK are the most common factors that lead to the development of PHHI.2,3 Interestingly, the severity of this disease is highly variable, even among family members that carry the same mutated genes.4
KCNJ11 and ABCC8 genes encode the inwardly-rectifying potassium channel 6.2 (Kir6.2) and the sulphonylurea receptor 1 (SUR1), respectively, and co-assembly of four Kir6.2 with four SUR1 subunits forms a functional ATP-sensitive potassium channel (KATP channel).5 Intracellular ATP blocks the KATP channel via direct interaction with Kir6.2, the pore-forming subunit of the channel. While most excitable cells, including pancreatic β-cells, neurons, cardiac myocytes and skeletal muscles, have Kir6.2 as the KATP channel pore forming subunit, vascular smooth muscle cells have Kir6.1 instead.6 SUR1, on the other hand, is a regulatory protein that confers sensitivity to magnesium nucleotides and drugs, such as sulphonylureas and KATP channel openers.7 At the cellular level, the KATP channel functions as a metabo-electric transducer, since its gating is regulated by the intracellular metabolites such as ATP, long-chain fatty acid-CoA, and phosphatidylinositol-4,5-bisphosphate (PIP2).8 Given that KATP channels are involved in multiple physiological processes, it is not surprising that mutations in either KCNJ11 or ABCC8 can cause a variety of diseases, ranging from diabetes and PHHI to epilepsy, mental retardation (DEND syndrome) and cardiac myopathies.9
Metabolism of glucose by the pancreatic β-cells rapidly increase intracellular ATP ([ATP]i). Elevated [ATP]i closes the KATP channels, which depolarizes the β-cell membrane potentials and subsequently opens the voltage-gated Ca2+ channels. The resulting influx of Ca2+ into the cytosol ultimately triggers insulin secretion via exocytosis.10 Because the KATP maintains the resting membrane potentials of the pancreatic β-cells, loss-of-function in either of the KATP channel subunits may lead to aberrant depolarization of β-cells and excessive insulin release.3 In contrast to mutations in ABCC8, most of the PHHI causing mutations in KCNJ11 are recessive, requiring both parents to be carriers.2,11 In this study, we report a novel dominant form of PHHI, which is caused by a single point mutation (C83T) in KCNJ11 that codes for a Val substitution for Ala at position 28 of the Kir6.2 peptide chain (A28V hKir6.2). The index patient, carrying a maternally inherited A28V hKir6.2, developed diazoxide-nonresponsive, late-onset PHHI requiring a total pancreatectomy. Immediate family members who carry the same heterozygous mutation have also experienced various degrees of hyperinsulinemic hypoglycemic symptomology. Moreover, both the patient and one of his affected siblings developed primary hypopituitarism. Electrophysiological and cell biological studies reveal that the A28V hKir6.2 mutation produces only minuscule KATP currents due to a trafficking defect that prevents surface expression. Our results identify a novel KATP channel defect that causes PHHI and provides additional evidence that the N-terminus of Kir6.2 is involved in KATP channel trafficking.
Results
Clinical findings
The male patient presented at 9 months old with failure-to-thrive, weight loss, and feeding intolerance. Initial evaluations revealed reflux esophagitis and a duodenal ulcer, but persistent symptoms eventually led to diagnoses of hyperinsulinemic hypoglycemia and central hypothyroidism at 16 months. The hypoglycemia was refractory to diazoxide therapy, and after three sequential partial pancreatectomies failed to control the hypoglycemia, removal of the last ∼1% of the pancreas successfully prevented further hypoglycemia and resulted in diabetes mellitus. Histologic examinations of the resected pancreas revealed islet cell hyperplasia. Post pancreatectomy, he developed intractable gastrointestinal bleeding from multiple sites along with hypergastrinemia eventually requiring resection of the stomach and intestine, which also showed endocrine cell hyperplasia.
Notable family history includes asymptomatic hypoglycemia in the mother which did not require treatment. An older sister was diagnosed at 12 months with central hypothyroidism and adrenal insufficiency but a normal pituitary on MRI scan. When she developed repeated severe hypoglycemia despite adequate thyroid, cortisol and growth hormone replacement, she was also diagnosed with hyperinsulinemic hypoglycemia, which also failed to respond to diazoxide.
Commercial sequencing identified a heterozygous variant in KCNJ11 (C83T) that encodes an Ala to Val substitution at amino acid 28 in the Kir6.2 protein in the proband. The patient's mother and sister carry the same A28V Kir6.2 mutation (Fig. 1A).
Figure 1.
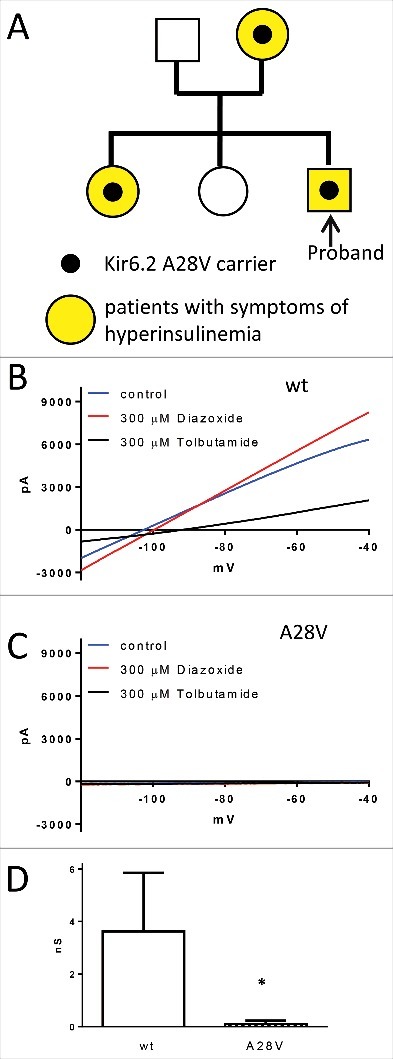
(A) The family pedigree of the index patient. Each family member that carries the A28V hKir6.2 mutation exhibits some degree of PHHI. (B to D) Whole-cell recording of KATP currents in HEK cells. (B) A representative wild-type KATP current (blue trace) was elicited by a voltage ramp pulse (0.5V/s). As predicted, the KATP current was augmented by 300 µM KATP channel opener diazoxide (red trace) and inhibited by 300 µM KATP channel blocker tolbutamide (black trace). (C) In contrast, HEK cells transfected with A28V hKir6.2 exhibited minuscule KATP current (blue trace) and neither 300 µM KATP channel opener diazoxide (red trace) or 300 µM KATP channel blocker tolbutamide (black trace) had an effect on the A28V KATP currents. (D) Summary of wild-type and A28V KATP currents in HEK cells. Wild-type KATP currents (n = 5) were significantly larger than A28V KATP currents (n = 9), as determined by Mann-Whitney U-test (*p < 0.05).
Functional characterization of the hKir6.2 (A28V) mutation
Loss of KATP channel function is one of the most common causes of PHHI.1 We first expressed wild-type or mutant hKir6.2 with hSUR1 in HEK293 cells to examine whether the A28V mutation disrupts KATP channel function. In cells transfected with wild-type hKir6.2 and hSUR1, a weak-inwardly rectifying potassium current was observed, and this current was further augmented by diazoxide, a KATP channel opener, and inhibited by tolbutamide, a KATP channel blocker (Fig. 1B). In contrast, cells transfected with A28V hKir6.2 showed only minuscule potassium currents and adding diazoxide failed to augment the currents (Fig. 1C). These results indicated that A28V hKir6.2 does not form a functional KATP channel (Fig. 1D) in HEK cells.
A28V hKir6.2 impairs KATP channel trafficking
A lack of discernible KATP currents can result from defects in either channel conduction or channel trafficking.3 We next tested whether KATP channels containing A28V hKir6.2 would traffic to the cell membrane by staining KATP channels on the cell surface. To minimize artifacts that caused by manipulating the hKir6.2 subunit, we probed the subcellular location of the KATP channel using a HA-tagged rodent SUR1 (rSUR1) subunit. Prior to the trafficking from the endoplasmic reticulum (ER) to the plasma membrane via the Golgi complex, functional KATP channels must be co-assembled to mask strong ER-retention signals in Kir6.2 and SUR1 subunits.12 Therefore, tracking the HA-tagged SUR1 is a preferred method to assess the cellular localization of fully assembled KATP channels in the presence of Kir6.2 mutants. In the HEK cells transfected with wild-type hKir6.2 and HA-rSUR1, a very strong surface staining pattern was detected (Fig. 2A), as this surface signal clearly outlined the cell boundary (Fig. 2B). This result indicated that the KATP channels containing wild-type hKir6.2 can promote the forward trafficking of HA-tagged rSUR1 by forming functional KATP channels. However, for HEK293 cells transfected with A28V hKir6.2 and HA-rSUR1, most of the HA signal was intracellular (Fig. 2C and D). By normalizing the surface HA signal (green) to total HA signal (red), we found the proportion of HA signal at the cell surface was significantly reduced in cells with KATP channels containing A28V hKir6.2 (Fig. 2E). Based on these results, we conclude that A28V hKir6.2 impairs KATP channel function by suppressing surface expression in HEK cells.
Figure 2.
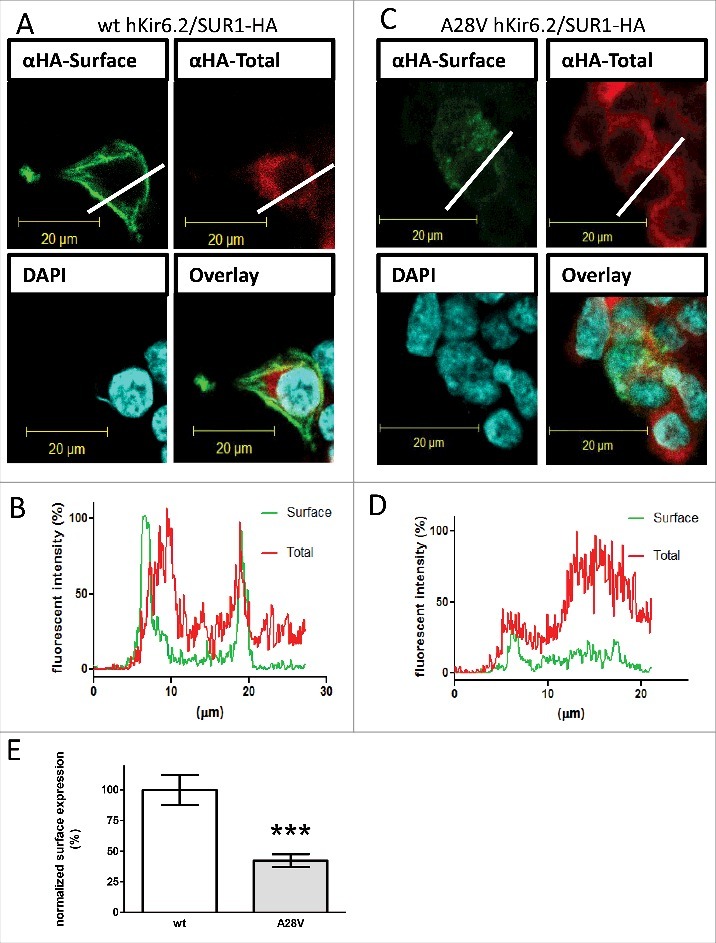
Surface expression of KATP channels in HEK cells. An HA-tag was inserted into the extracellular domain on rSUR1 and then co-transfected with either wild-type hKir6.2 (A) or A28V hKir6.2. As Kir6.2 must be co-assembled with SUR1 for correct trafficking, the subcellular distribution of HA staining should faithfully represent the KATP channel distribution. KATP channels located on the cell surface were labeled green. Total KATP channels were labeled red, and cell nuclei were counterstained with DAPI (blue). The wild-type KATP channels were clearly visible on the cell surface (A), but A28V hKir6.2-containing channels were not readily observed on the cell surface (C). (B&D): Cross-sectional staining intensity profiles of HEK cells expressing wild-type hKir6.2 (C) and A28V hKir6.2 (D). The profiles were determined from cross-sections indicated by the white lines in A and C. Surface staining signals are clearly visible in HEK cells transfected with wild-type hKir6.2, as the green line shows distinct peaks at the cell boundary. By contrast, in HEK cells transfected with A28V hKir6.2, the cell surface boundary is not clearly demarcated (green line, D). (E) Quantitative analysis of KATP channel surface staining signals. KATP channels containing A28V hKir6.2 had greatly reduced surface staining signals compared to wild-type hKir6.2 containing KATP channels (***p < 0.0005, Mann-Whitney U-test n = 11 for each group).
Dominant-negative effect of A28V hKir6.2 on KATP channel trafficking
The patient and the other family members who exhibited hypoglycemia are all heterozygous for the KCNJ11 (C83T) mutation, suggesting that A28V hKir6.2 is a dominant-negative mutation that can suppress normal KATP channel trafficking. To control the expression ratio of wild-type and hKir6.2 (A28V) in the HEK293 cells, we generated a construct to express wide-type and A28V hKir6.2 in tandem, with the two sequences linked by a self-cleaving P2A peptide linker. P2A encodes an RNA skipping signal, so during translation, the ribosome produces a gap in the nascent polypeptide. Thus the two peptide sequences that are linked by the P2A are “self-cleaved” at the P2A sequence and yield two physically separate peptides.13 We generated all four possible arrangements of wild-type and A28V hKir6.2 pairs and examined whether A28V hKir6.2 suppresses KATP channel trafficking to the plasma membrane. Our results showed that cells transfected with the wt-P2A-wt hKir6.2 tandem construct exhibited strong surface signals in HEK293 cells, while those transfected with the A28V-P2A-A28V hKir6.2 construct showed greatly reduced surface signals (Fig. 3A). These results further confirmed the effect of A28V-hKir6.2 in suppressing KATP channel trafficking (Fig. 2A and B). Interestingly, HEK293 cells transfected with either wt-P2A-A28V or A28V-P2A-wt constructs showed diminished surface signals (Fig. 3A), comparable to the HEK293 cells transfected with either A28V monomers (Fig. 2B) or A28V-P2A-A28V homo tandem construct (Fig. 3A and B). These results strongly suggested that A28V hKir6.2 is a dominant-negative mutation in regulating KATP channel trafficking towards plasma membrane.
Figure 3.
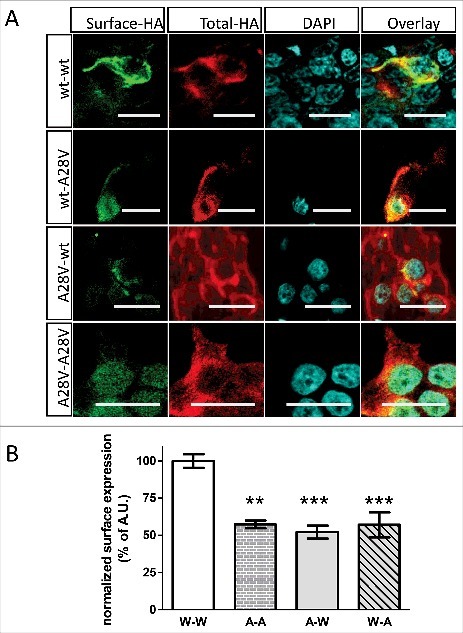
hKir6.2(A28V) has a dominant-negative effect on KATP channel surface expression in HEK cells. (A) An HA-tag was inserted into the extracellular domain of rSUR1 and then co-transfected with one of the hKir6.2 tandem constructs that are linked by a self-cleaving P2A linker. Only cells transfected with the wt-P2A-wt hKir6.2 construct showed strong surface staining (top row). Surface expression was not apparent in HEK cells expressing KATP channels that contain wt-P2A-A28V, A28V-P2A-wt or A28V-P2A-A28V hKir6.2. (B) Quantitative analysis of the surface signal from cells transfected with various hKir6.2 tandem constructs. KATP channels formed from wt-P2A-wt tandem construct expression had much stronger surface staining signals compared to any other hKir6.2 tandem constructs that contain the A28V Kir6.2 mutation. (**p = 0.001, ***p < 0.0005, one-way ANOVA with posthoc multiple comparison test, n = 8 for wt-P2A-wt, 8 for A28V-P2A-A28V, 9 for A28V-P2A-wt, and 14 for wt-P2A-A28V).
Functional characterization of hKir6.2 (A28V) in the Xenopus oocytes
Since the expression of A28V hKir6.2 in HEK cells generated minuscule currents that were too small to characterize the biophysical and pharmacological properties of the mutant channels, we sought to express A28V-hKir6.2 in another cell type that normally shows more permissive membrane trafficking regulation. At lower temperature, the Xenopus oocyte exhibits less stringent trafficking control on membrane protein.14,15 allowing for recording of KATP currents formed by the A28V-hKir6.2 in the Xenopus oocytes. Indeed, when equal mass amounts of RNA were injected into the Xenopus oocytes, the A28V-hKir6.2 containing KATP currents were detectable but significantly smaller than the wild-type KATP currents (Fig. 4A and C). We found that the KATP currents formed by A28V hKir6.2 can be blocked by a specific KATP channel inhibitor, tolbutamide, indicating pharmacological properties are still well-preserved in these mutant channels when trafficked to the plasma membrane (Fig. 4B and D).
Figure 4.
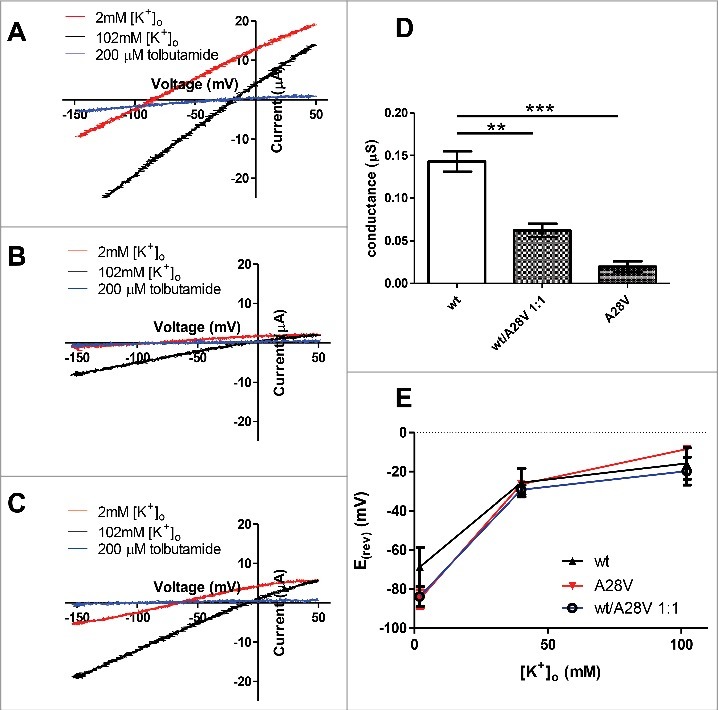
Two-electrode voltage clamp recording of KATP currents in the Xenopus oocytes. (A) A representative wild-type KATP current (red trace) was elicited by a voltage ramp pulse (0.5V/s). As predicted, the KATP current was inhibited by 200 µM KATP channel blocker, tolbutamide (blue trace). Shifting extracellular potassium concentration from 2 mM to 100 mM caused a shift of reversal potential, as predicted by the potassium equilibrium potential. (B) A representative A28V hKir6.2 KATP current (red trace) was elicited by a voltage ramp pulse (0.5V/s). This A28V hKir6.2 KATP current was relatively smaller than the wild-type KATP current, but it could be inhibited by 200 µM tolbutamide (blue trace). The KATP channels containing A28V hKir6.2 were still potassium selective, as the reversal potential followed the potassium equilibrium potential (black trace). (C) A representative recording trace from a Xenopus oocyte injected with an equimolar ratio of wt and A28V hKir6.2 mRNA. KATP current (red trace) was elicited by a voltage ramp pulse (0.5V/s). This wt/A28V KATP current size was in between wild-type and A28V hKir6.2 KATP current, and was inhibited by 200 µM tolbutamide (blue trace). The wt/A28V KATP currents were also potassium selective, as the reversal potential followed the potassium equilibrium potential (black trace). (D) Summary of wt, A28V and wt/A28V KATP currents in the Xenopus oocytes. wt KATP currents (n = 7) were significantly larger than A28V KATP currents (n = 6) and wt/A28V KATP currents (n = 8), as determined by one-way ANOVA with posthoc multiple comparison test, (**p = 0.001, ***p < 0.0005). (E) Summary of the reversal potentials of wt, A28V, and wt/A28V KATP currents. In all three groups, the reversal potentials followed closely with the extracellular potassium concentration, indicating this hKir6.2(A28V) mutant is still potassium selective.
Another plausible mechanism that could contribute to the etiology of PHHI would be if potassium selectivity is impaired in A28V hKir6.2 containing channels. Under this scenario, mutant channels that lose potassium selectivity could cause depolarization rather than hyperpolarization.16 To characterize the ion selectivity in KATP channels, we injected hSUR1, together with either wild-type or A28V hKir6.2, into the Xenopus oocytes and measured the KATP currents in the extracellular ND96 solutions with different potassium concentrations. We found that the reversal potentials of the wild-type and A28V hKir6.2 containing KATP currents closely followed the potassium equilibrium potentials (Fig. 4A, B and E), indicating that the KATP channels formed by A28V hKir6.2 channels remain highly selective for potassium. Finally, we tested whether co-injecting a 1:1 ratio of wild-type and A28V-hKir6.2 would reduce KATP currents in the Xenopus oocytes. Indeed, we observed reduced KATP currents in the oocytes that were co-injected with wild-type and A28V-hKir6.2 RNAs, which further supported the hypothesis that A28V-hKir6.2 is a dominant-negative mutation (Fig. 4B).
A28V suppresses hKir6.2 surface expression via reorienting the ER-retention motif
Since the A28V hKir6.2 mutation dramatically decreased its surface expression, we wondered whether this mutation might alter trafficking signals such as the arginine-lysine-arginine (RKR) ER-retention motif12 or the di-acidic ER-exit motif.17 Although the high-resolution structure of the KATP channel has been solved recently,18,19 the detailed structure of N- and C-terminal regions are yet to be determined. We utilized de novo protein structure prediction method20–23 to construct the N- and C-terminal regions of hKir6.2 according to the existing atomic KATP channel structures (Fig. 5).18 As illustrated in Fig. 5, our model predicted that the N-terminal loop (solid circle) and the C-terminal loop (dashed circle) of the hKir6.2 were surrounded by the ATP-binding pocket at the hKir6.2 and the inter-subunit interface between hKir6.2 and SUR1 (Fig. 5L–N). Alanine to valine mutation at position 28 had little effect on the overall Kir6.2 structure (Fig. 5A–F). However, a superimposition of the wild-type and A28V hKir6.2 structures (Fig. 5G) revealed that the A28V mutation caused a pronounced conformational change at the N-terminal loop (Fig. 5B, E and H) and a counterclockwise rotation and outward shift of the RKR ER-retention motif (Fig. 5 C, F, J and K). On the contrary, A28V mutation only produced a minute change on the ER-exit di-acidic motif (Fig. 5I).
Figure 5.
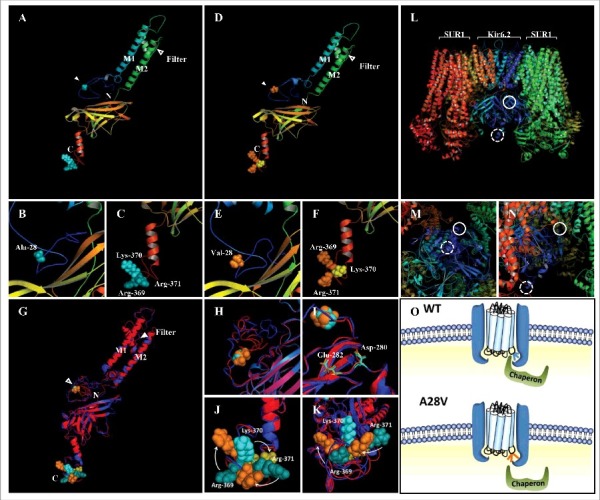
Predicted protein structures of wild-type (A to C) and A28V (D to F) hKir6.2. The first and second transmembrane segments are labeled as M1 and M2, respectively. The selective filter and the position 28 are marked with blank and filled arrowhead, respectively. The N and C represent the N- and C- terminus of the hKir6.2 protein (A and D). (G to K) A superimposed image of the predicted wild-type and A28V hKir6.2. The side chain of position 28 (alanine in wild-type and valine in the mutant) are shown as cyan balls for wild-type and orange balls for the mutant (H and I). Di-acidic motifs of wild-type and A28V hKir6.2 (D280 and E282) are shown as cyan and orange sticks, respectively. (J) The A28V mutation caused a clockwise rearrangement of the RKR motif. (L to N) The fully assembled KATP channel complex (PDB: 5WUA).18 The solid and dashed circles represent the presumptive N-terminal region containing the 28th alanine residue and C- terminal region containing the RKR motif, respectively. (O) A plausible molecular mechanism of the A28V mutation on KATP channel trafficking. In KATP channel formed by the wild-type Kir6.2, the RTR motif is hinged on the neighboring SUR1, and the chaperone may dock onto the KATP channel complex to facilitate the assembled channel complex exiting the ER. In KATP channel formed by the A28V Kir6.2, the C-terminus is distorted and the RKR motif is no longer hidden. The exposed RKR motif may cause a hindrance for the chaperone docking and hence, prevent the forward trafficking of the mutated KATP channel.
Discussion
In this study, we have identified the first dominant-negative mutation in KCNJ11, the gene that encodes the pore-forming subunit of the KATP channels, hKir6.2. Patients that carry one copy of this KCNJ11(C83T) mutation exhibited various symptoms, ranging from a mild PHHI to hypothalamic deficiency.
The atomic structures of the fully assembled KATP channel in the closed conformation provide detailed structural and mechanistic insights into KATP channel assembly and gating.18,19 Nevertheless, the regions that are involved in KATP channel trafficking have yet to be determined, as these trafficking domains are highly dynamic structures. The predicted full-length hKir6.2 structure indicated that the A28 is located on the loosely-packed N-terminal loop; Ala to Val not only re-organizes the N-terminal loop but also distortes the entire C-terminal domain (Fig. 5J and K). We postulate that the distorted C-terminal domain hinders the docking of the chaperones to the RKR ER-retention motif and consequently prevents the KATP channels trafficking to the plasma membrane (Fig. 5O). The trafficking defect may also explain the dominant-negative effect of the A28V mutation, since the forward trafficking of the fully assembled KATP channels can only take place when all the RKR ER-retention motifs on the Kir6.2 and SUR1 are masked.12,24 Exposure of any RKR ER-retention motif traps the channel complex in the ER and reroutes the defected channel to the degradation pathway instead.25
Multiple mutations have been identified in both genes encoding KATP channel subunits (i.e., KCNJ11 and ABCC8) with many of these strongly affect glucose homeostasis.26 Mutations in KCNJ11 can lead to either hyper- or hypo-insulin secretion, depending on their nature.2,27,28 However, mutations in ABCC8 mostly result in hyperinsulinemia.2,3,11 Kir6.2 has evolved to be a high-affinity receptor for [ATP]i.and Kir6.2 mutations located near the ATP binding pocket often render the KATP channel less sensitive to [ATP]i.29–30 As the closure of the KATP channels is a critical link in the pathway by which glucose stimulates insulin secretion, ATP-insensitive KATP channels that remain open keep pancreatic β-cells hyperpolarized even at elevated blood glucose levels. Consequently, glucose fails to stimulate insulin secretion, leading to the development of diabetes.31
Conversely, inactivating mutations in the Kir6.2 gene often cause PHHI, since β-cells without functional KATP channels are depolarized, and continue to secrete insulin at low glucose concentration.3 In contrast, most SUR1 mutations lead to PHHI.3,11,26,32 SUR1 is a high-affinity receptor for intracellular magnesium nucleotides, such as Mg-ATP and Mg-ADP. Binding of the magnesium nucleotides to the SUR1 holds the KATP channels in the open state, even in the presence of physiological [ATP]i that are up to an order of magnitude higher than the normal IC50 of [ATP]i.33 In addition, many mutations in genes encoding Kir6.2 and SUR1 are recessive, thus requiring inheritance of two mutated alleles to exhibit clinical symptoms or signs of disease. The newly identified A28V hKir6.2 mutation is a striking exception to often-observed paradigms in KATP channelopathy: (1) A28V hKir6.2 is a maternally-inherited dominant negative mutation. The index patient and his immediate family members who carry one copy of the same A28V mutation all developed hypoglycemia. (2) KATP channels containing A28V hKir6.2 remain functional if the channel reaches the cell membrane (Fig 4).
In addition to persistent low blood glucose, the index patient and his sister also exhibited some degree of pituitary insufficiency may be secondary to hypothalamic effects. Kir6.2 is highly expressed in many excitable tissues, including pancreas, brain, muscle and heart.34 The KATP channels in neurons and pancreatic β-cells are similar, in that they are formed by Kir6.2 and SUR1. In cardiac muscle, KATP channels are formed by Kir6.2 and SUR2A.7 The differential expression of SUR subunits may also contribute to breadth of pathologies induced by KATP channelopathy. Since Kir6.2 is expressed in the hypothalamus,35,36 we expect the expression of the dominant negative A28V hKir6.2 protein in hypothalamic neurons could impact the KATP channels, disrupt hypothalamic function and thereby effect pituitary function. Previous studies have identified multiple roles for KATP channels in the hypothalamus, ranging from regulation of food intake and glucose homeostasis to hormonal functions.36,37 As the hypothalamus is heavily involved in regulating peripheral glucose homeostasis,38 we also cannot rule out the potential involvement of the hypothalamus in PHHI.39 A similar argument could be made for secretion from the enteroendocrine cells that might explain the gastrointestinal pathology in the index patient.
Most PHHI patients develop clinically evident hypoglycemia within a few days of birth. By contrast, the index patient for the A28V hKir6.2 mutation presented with non-specific symptoms at 9 months and was not diagnosed with PHHI until more than one year after birth, and his older sibling not diagnosed until 12 months of life. One potential explanation for the delayed onset is that the A28V hKir6.2 mutation affects KATP channel trafficking, and the molecular machinery for proper channel trafficking might not be fully developed in infants. In an animal study, mouse β-cells did not exhibit adult-like membrane excitability until postnatal day P3.40
This patient and his affected sister did not respond to the KATP channel opener diazoxide which is often prescribed to manage PHHI symptoms. This failure of diazoxide can be explained by our results. The KATP channels carrying A28V hKir6.2 still had normal pharmacological responses, but the channels did not correctly traffic to the cell surface. This lack of functional KATP channels on the β-cell surface removes the target of activity modulating drugs and makes the affected patients refractory to PHHI medications.
This A28V hKir6.2 mutation affects channel trafficking, and until now, only limited number of therapeutic strategies have been developed to alleviate protein trafficking defects. Lumacaftor, a newly developed small molecule drug and the first medicine to correct CFTR trafficking defects, has been successfully integrated into clinical practice for the treatment of patients with specific CFTR mutations (DelF508).41 Lumacaftor functions as a molecular chaperone and forces the defective channels to travel to the membrane. While it represents a promising start, this drug only benefits ∼4% of CFTR patients that carry this specific CFTR mutation.42 In vitro studies have demonstrated that carbamazepine, an antiepileptic drug, and sulphonylureas such as tolbutamide and glibenclamide may function as molecular chaperones to correct certain KATP channel trafficking defects.43–46 We predict that those molecular chaperones that promote forward trafficking may be beneficial for patients with KATP channels that exhibit trafficking defects.
In summary, we have identified a novel form of late-onset, dominant-negative A28V hKir6.2 mutant that impairs KATP channel trafficking. Patients carrying this mutation developed a novel form of PHHI and hypopituitarism. With the rapid improvement of personalized medicine, we expect novel therapeutic strategies can be developed to treat patients with this KATP channel trafficking defect.
Materials and methods
Whole-cell patch clamp recording in HEK cells
Human Kir6.2 (hKir6.2), SUR1 (hSUR1) (Origene, USA) and rodent (hamster) SUR1 (rSUR1) cDNAs were cloned into the pcDNA3 plasmid. The HA-tagged rSUR1 was generated previously and was kindly provided as a gift from Dr. Lily Jan at the University of California, San Francisco, USA. The HA-epitope was inserted between the sixteenth and seventeenth transmembrane domains of the SUR1 subunit and the sequence of the HA-tag reads as1272LHRELSAGLVYPYDVPDYAHRELSAGLVGLG1284at the site of the epitope insertion.12 Site-directed mutagenesis was performed using Pfu Turbo DNA polymerase (Stratagene, USA) and the A28V mutation was verified by sequencing. HEK293 cells were cultured in DMEM (Thermo Fisher Scientific, USA) containing 10% FBS (Hyclone, USA), 2 mM glutamine, 100 units penicillin and 100 mg/ml streptomycin in a humidified atmosphere of 5% CO2 at 37°C. Cells were plated on poly L-ornithine-coated glass coverslips and transiently transfected with 0.2 μg of the pcDNA3 containing hKir6.2 construct and 0.8 μg of pcDNA3 containing SUR1 construct by using FuGENE 6 (Roche, USA). Cells were used 2–4 days after transfection. KATP currents were recorded using the whole-cell patch-clamp configuration by an Axopatch 700B amplifier (Molecular Device, USA) in a standard extracellular solution containing: 150 mM NaCl, 10 mM HEPES, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2 and pH 7.2, adjusted with NaOH.47 Data were acquired at 10 kHz with pCLAMP software (Molecular Device, USA). Pipettes were pulled from 1.5 mm borosilicate glass capillaries (Sutter Inc, USA). Pipette resistances were 2–4 MΩ when filled with the intracellular solution containing: 135 mM K gluconate, 15 mM KCl, 10 mM HEPES, 5 mM Mg2ATP, 1 mM Na3GTP, 10 mM sodium phosphocreatine, 0.05 mM EGTA and pH 7.2, adjusted with KOH. The access resistances of whole-cell recording ranged between 5 and 20 MΩ and were compensated by ∼80%. All experiments were performed at room temperature (∼25°C). For all studies, all chemicals were purchased from Sigma-Aldrich (USA) if not stated otherwise.
Two electrode recording in Xenopus oocytes
Animal protocols used in this study were approved by the IACUC at Academia Sinica or University of California, San Francisco, and Xenopus oocytes were prepared as described previously.47 Briefly, stage V–VI Xenopus laevis oocytes were harvested, injected with 30 ng of each cRNA, and incubated at 16°C for 2–4 days before recording. hKir6.2 and hSUR1 were cloned into the pGEM vector. hKir6.2 and hSUR1 cDNAs were first linearized by restriction enzyme digestion, and cRNA was then synthesized by using the mMESSAGE mMACHINE SP6 Transcription kit (Thermo-Fisher, USA). Macroscopic currents were recorded from oocytes with two-electrode voltage clamp (GeneClamp 500B, Molecular Device, USA). Electrodes were filled with 3 M KCl and had a resistance between 0.4–1 MΩ. A small chamber with a fast perfusion system (AutoMate Scientific, USA) was used to change extracellular ND96 solution containing: 96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 5 mM HEPES and pH 7.4, adjusted with NaOH (in some cases, the Na+ was replaced with equimolar K+ to keep the osmolarity constant).
Immunofluorescent staining
For non-permeabilized immunostaining of surface KATP channels, HEK cells were first transfected with hKir6.2 and an HA-tagged rSUR1. Three days post transfection, cells were incubated in the blocking solution (5% normal goat serum in PBS; Jackson ImmunoResearch Laboratories, INC. USA) for 10 min at 4°C and then incubated with rat anti-HA antibody (1:200; monoclonal 3F10, Sigma-Aldrich, USA) in the blocking solution for 30 min at 4°C to minimize endocytosis. After washing four times with PBS for 5 min each at 4°C, cells were incubated for 30 min with goat anti-rat Alexa488 secondary antibody (1:1000, Invitrogen, USA) then washed three times with PBS for 5 min each at 4°C. Next, the cells were fixed in the 4% paraformaldehyde/4% sucrose in PBS for 10 min at room temperature then washed three times with PBS for 5 min each. Fixed cells were permeabilized by incubating in blocking solution that contained 0.1% Triton-X-100 for 15 min. Then cells were incubated with the same rat anti-HA antibody (1:200) in the blocking solution for 30 min at room temperature. After washing four times with PBS for 5 min each at room temperature, cells were incubated for 30 min with goat anti-rat Alexa555 secondary antibody (1:1000 Invitrogen, USA), then washed three times with PBS for 5 min each at room temperature. The coverslips were mounted using Fluoromount G mounting medium containing DAPI (Southern Biotech, USA) and images were acquired using a confocal microscope (Zeiss, Germany).
De novo protein structure prediction
The structures of the wild-type and A28V hKir6.2 were predicted by I-TASSER21–23 and the rat Kir6.2 (PDB: 5WUA)18 was used as a template for de novo structure prediction. TM-Align was used to align wild-type and A28V hKir6.2.20 Predicted structures were generated in Pymol.
Data analysis
Imaging data from the immunostained cells were analyzed using ZEN software (Zeiss, Germany). Electrophysiology Data were analyzed with pClamp10 software (Molecular Devices Corp., USA). Results are reported as mean ± SEM. Statistical analysis was performed using Prism 5 (GraphPad, USA), with differences considered significant at p < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001 in all graphs).
Acknowledgements
We thank Dr. Pei-Chun Chen at National Cheng-Kung University, Taiwan for her critical comments and discussions. We thank all members of the Jan laboratory at the UCSF, in particular, Tong Cheng for preparing the constructs, Dr. Huanghe Yang for helping oocyte recording and Dr. Lily Jan for discussions. This study was supported by Academia Sinica and Ministry of Science and Technology, Taiwan (105–2320-B-001 -003 -MY2) and National Institutes of Health grant P30 DK063720.
References
- 1.Goel P, Choudhury SR. Persistent hyperinsulinemic hypoglycemia of infancy: An overview of current concepts. J Indian Assoc Pediatr Surg. 2012;17(3):99–103. doi: 10.4103/0971-9261.98119. PMID:22869973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, Ganapathy K, Bhatti T, Stanley CA, Ganguly A. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. 2013;98(2):E355–363. doi: 10.1210/jc.2012-2169. PMID:23275527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, Ganguly A, Shyng SL, Stanley CA. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest. 2008;118(8):2877–86. doi: 10.1172/JCI35414. PMID:18596924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henwood MJ, Kelly A, Macmullen C, Bhatia P, Ganguly A, Thornton PS, Stanley CA. Genotype-phenotype correlations in children with congenital hyperinsulinism due to recessive mutations of the adenosine triphosphate-sensitive potassium channel genes. J Clin Endocrinol Metab. 2005;90(2):789–94. doi: 10.1210/jc.2004-1604. PMID:15562009 [DOI] [PubMed] [Google Scholar]
- 5.Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev. 1999;20(2):101–35. PMID:10204114 [DOI] [PubMed] [Google Scholar]
- 6.Inagaki N, Gonoi T, Clement JP, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270(5239):1166–70. doi: 10.1126/science.270.5239.1166. PMID:7502040 [DOI] [PubMed] [Google Scholar]
- 7.Inagaki N, Gonoi T, Clement JP, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16(5):1011–1017. doi: 10.1016/S0896-6273(00)80124-5. PMID:8630239 [DOI] [PubMed] [Google Scholar]
- 8.Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, Lazdunski M, Nichols CG, Seino S, Vandenberg CA. International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev. 2005;57(4):509–26. doi: 10.1124/pr.57.4.11. PMID:16382105 [DOI] [PubMed] [Google Scholar]
- 9.McTaggart JS, Clark RH, Ashcroft FM. The role of the KATP channel in glucose homeostasis in health and disease: more than meets the islet. J Physiol. 2010;588(Pt 17):3201–09. doi: 10.1113/jphysiol.2010.191767. PMID:20519313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashcroft FM, Rorsman P. K(ATP) channels and islet hormone secretion: new insights and controversies. Nat Rev Endocrinol. 2013;9(11):660–69. doi: 10.1038/nrendo.2013.166. PMID:24042324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saint-Martin C, Zhou Q, Martin GM, Vaury C, Leroy G, Arnoux JB, de Lonlay P, Shyng SL, Bellanné-Chantelot C. Monoallelic ABCC8 mutations are a common cause of diazoxide-unresponsive diffuse form of congenital hyperinsulinism. Clin Genet. 2015;87(5):448–54. doi: 10.1111/cge.12428. PMID:24814349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zerangue N, Schwappach B, Jan YN, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron. 1999;22(3):537–48. doi: 10.1016/S0896-6273(00)80708-4. PMID:10197533 [DOI] [PubMed] [Google Scholar]
- 13.Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, Kim MK, Shin BA, Choi SY. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One. 2011;6(4):e18556. doi: 10.1371/journal.pone.0018556. PMID:21602908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin-Moshier Y, Marchant JS. The Xenopus oocyte: a single-cell model for studying Ca2+ signaling. Cold Spring Harb Protoc. 2013;(3):185–191. doi: 10.1101/pdb.top066308] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drumm ML, Wilkinson DJ, Smit LS, Worrell RT, Strong TV, Frizzell RA, Dawson DC, Collins FS. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science. 1991;254(5039):1797–99. doi: 10.1126/science.1722350. PMID:1722350 [DOI] [PubMed] [Google Scholar]
- 16.Kanai K, Hirose S, Oguni H, Fukuma G, Shirasaka Y, Miyajima T, Wada K, Iwasa H, Yasumoto S, Matsuo M, et al.. Effect of localization of missense mutations in SCN1A on epilepsy phenotype severity. Neurology. 2004;63(2):329–34. doi: 10.1212/01.WNL.0000129829.31179.5B. PMID:15277629 [DOI] [PubMed] [Google Scholar]
- 17.Taneja TK, Mankouri J, Karnik R, Kannan S, Smith AJ, Munsey T, Christesen HB, Beech DJ, Sivaprasadarao A. Sar1-GTPase-dependent ER exit of KATP channels revealed by a mutation causing congenital hyperinsulinism. Hum Mol Genet. 2009;18(13):2400–13. doi: 10.1093/hmg/ddp179. PMID:19357197 [DOI] [PubMed] [Google Scholar]
- 18.Li N, Wu JX, Ding D, Cheng J, Gao N, Chen L. Structure of a Pancreatic ATP-Sensitive Potassium Channel. Cell. 2017;168(1–2):101–110.e110. doi: 10.1016/j.cell.2016.12.028. PMID:28086082 [DOI] [PubMed] [Google Scholar]
- 19.Martin GM, Yoshioka C, Rex EA, Fay JF, Xie Q, Whorton MR, Chen JZ, Shyng SL. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. Elife. 2017;6:e24149. doi: 10.7554/eLife.24149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Skolnick J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33(7):2302–09. doi: 10.1093/nar/gki524. PMID:15849316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. PMID:18215316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–38. doi: 10.1038/nprot.2010.5. PMID:20360767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8. doi: 10.1038/nmeth.3213. PMID:25549265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwappach B, Zerangue N, Jan YN, Jan LY. Molecular basis for K(ATP) assembly: transmembrane interactions mediate association of a K+ channel with an ABC transporter. Neuron. 2000;26(1):155–67. doi: 10.1016/S0896-6273(00)81146-0. PMID:10798400 [DOI] [PubMed] [Google Scholar]
- 25.Crane A, Aguilar-Bryan L. Assembly, maturation, and turnover of K(ATP) channel subunits. J Biol Chem. 2004;279(10):9080–90. doi: 10.1074/jbc.M311079200. PMID:14699091 [DOI] [PubMed] [Google Scholar]
- 26.Faletra F, Snider K, Shyng SL, Bruno I, Athanasakis E, Gasparini P, Dionisi-Vici C, Ventura A, Zhou Q, Stanley CA, et al.. Co-inheritance of two ABCC8 mutations causing an unresponsive congenital hyperinsulinism: Clinical and functional characterization of two novel ABCC8 mutations. Gene. 2013;516(1):122–5. doi: 10.1016/j.gene.2012.12.055. PMID:23266803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin YW, MacMullen C, Ganguly A, Stanley CA, Shyng SL. A novel KCNJ11 mutation associated with congenital hyperinsulinism reduces the intrinsic open probability of beta-cell ATP-sensitive potassium channels. J Biol Chem. 2006;281(5):3006–12. doi: 10.1074/jbc.M511875200. PMID:16332676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bushman JD, Gay JW, Tewson P, Stanley CA, Shyng SL. Characterization and functional restoration of a potassium channel Kir6.2 pore mutation identified in congenital hyperinsulinism. J Biol Chem. 2010;285(9):6012–23. doi: 10.1074/jbc.M109.085860. PMID:20032456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vedovato N, Cliff E, Proks P, Poovazhagi V, Flanagan SE, Ellard S, Hattersley AT, Ashcroft FM. Neonatal diabetes caused by a homozygous KCNJ11 mutation demonstrates that tiny changes in ATP sensitivity markedly affect diabetes risk. Diabetologia. 2016;59(7):1430–36. doi: 10.1007/s00125-016-3964-x. PMID:27118464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Proks P, Girard C, Baevre H, Njølstad PR, Ashcroft FM. Functional effects of mutations at F35 in the NH2-terminus of Kir6.2 (KCNJ11), causing neonatal diabetes, and response to sulfonylurea therapy. Diabetes. 2006;55(6):1731–37. doi: 10.2337/db05-1420. PMID:16731836 [DOI] [PubMed] [Google Scholar]
- 31.Olson TM, Terzic A. Human K(ATP) channelopathies: diseases of metabolic homeostasis. Pflugers Arch. 2010;460(2):295–306. doi: 10.1007/s00424-009-0771-y. PMID:20033705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shemer R, Avnon Ziv C, Laiba E, Zhou Q, Gay J, Tunovsky-Babaey S, Shyng SL, Glaser B, Zangen DH. Relative expression of a dominant mutated ABCC8 allele determines the clinical manifestation of congenital hyperinsulinism. Diabetes. 2012;61(1):258–63. doi: 10.2337/db11-0984. PMID:22106158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Speier S, Yang SB, Sroka K, Rose T, Rupnik M. KATP-channels in beta-cells in tissue slices are directly modulated by millimolar ATP. Mol Cell Endocrinol. 2005;230(1–2):51–58. doi: 10.1016/j.mce.2004.11.002. PMID:15664451 [DOI] [PubMed] [Google Scholar]
- 34.Inagaki N, Tsuura Y, Namba N, Masuda K, Gonoi T, Horie M, Seino Y, Mizuta M, Seino S. Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heart. J Biol Chem. 1995;270(11):5691–94. doi: 10.1074/jbc.270.11.5691. PMID:7890693 [DOI] [PubMed] [Google Scholar]
- 35.Thomzig A, Laube G, Prüss H, Veh RW. Pore-forming subunits of K-ATP channels, Kir6.1 and Kir6.2, display prominent differences in regional and cellular distribution in the rat brain. J Comp Neurol. 2005;484(3):313–30. doi: 10.1002/cne.20469. PMID:15739238 [DOI] [PubMed] [Google Scholar]
- 36.Yang SB, Tien AC, Boddupalli G, Xu AW, Jan YN, Jan LY. Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron. 2012;75(3):425–36. doi: 10.1016/j.neuron.2012.03.043. PMID:22884327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest. 2008;118(5):1796–1805. doi: 10.1172/JCI32964. PMID:18382766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stanley SA, Kelly L, Latcha KN, Schmidt SF, Yu X, Nectow AR, Sauer J, Dyke JP, Dordick JS, Friedman JM. Bidirectional electromagnetic control of the hypothalamus regulates feeding and metabolism. Nature. 2016;531(7596):647–50. doi: 10.1038/nature17183. PMID:27007848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwartz MW, Seeley RJ, Tschöp MH, Woods SC, Morton GJ, Myers MG, D'Alessio D. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature. 2013;503(7474):59–66. doi: 10.1038/nature12709. PMID:24201279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rozzo A, Meneghel-Rozzo T, Delakorda SL, Yang SB, Rupnik M. Exocytosis of insulin: in vivo maturation of mouse endocrine pancreas. Ann N Y Acad Sci. 2009;1152:53–62. doi: 10.1111/j.1749-6632.2008.04003.x. PMID:19161376 [DOI] [PubMed] [Google Scholar]
- 41.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K Flume PA. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–31. doi: 10.1056/NEJMoa1409547. PMID:25981758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galietta LJ. Managing the underlying cause of cystic fibrosis: A future role for potentiators and correctors. Paediatr Drugs. 2013;15(5):393–402. doi: 10.1007/s40272-013-0035-3. PMID:23757197 [DOI] [PubMed] [Google Scholar]
- 43.Kuo CC, Chen RS, Lu L, Chen RC. Carbamazepine inhibition of neuronal Na+ currents: quantitative distinction from phenytoin and possible therapeutic implications. Mol Pharmacol. 1997;51(6):1077–83. PMID:9187275 [DOI] [PubMed] [Google Scholar]
- 44.Chen PC, Olson EM, Zhou Q, Kryukova Y, Sampson HM, Thomas DY, Shyng SL. Carbamazepine as a novel small molecule corrector of trafficking-impaired ATP-sensitive potassium channels identified in congenital hyperinsulinism. J Biol Chem. 2013;288(29):20942–54. doi: 10.1074/jbc.M113.470948. PMID:23744072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin GM, Chen PC, Devaraneni P, Shyng SL. Pharmacological rescue of trafficking-impaired ATP-sensitive potassium channels. Front Physiol. 2013;4:386. doi: 10.3389/fphys.2013.00386. PMID:24399968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou Q, Chen PC, Devaraneni PK, Martin GM, Olson EM, Shyng SL. Carbamazepine inhibits ATP-sensitive potassium channel activity by disrupting channel response to MgADP. Channels (Austin). 2014;8(4):376–82. doi: 10.4161/chan.29117. PMID:24849284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang SB, Proks P, Ashcroft FM, Rupnik M. Inhibition of ATP-sensitive potassium channels by haloperidol. Br J Pharmacol. 2004;143(8):960–7. doi: 10.1038/sj.bjp.0706017. PMID:15533888 [DOI] [PMC free article] [PubMed] [Google Scholar]