

Abstract
Background and Purpose
Recently, the incidence of Parkinson's disease has shown a tendency to move to a younger population, linked to the constantly increasing stressors of modern society. However, this relationship remains obscure. Here, we have investigated the contribution of stress and the mechanisms underlying this change.
Experimental Approach
Ten‐month‐old α‐synuclein A53T mice, a model of Parkinson's disease (PD), were treated with chronic restraint stress (CRS) to simulate a PD‐sensitive person with constant stress stimulation. PD‐like behavioural tests and pathological changes were evaluated. Differentiated PC12‐A53T cells were treated with corticosterone in vitro. We used Western blot, microRNA expression analysis, immunofluorescence staining, dual luciferase reporter assay and HPLC electrochemical detection to assess cellular and molecular networks after stress treatment. In vivo, stereotaxic injection of shRNA lentivirus was used to confirm our in vitro results.
Key Results
The protein RTP801 is encoded by DNA‐damage‐inducible transcript 4, and it was specifically increased in dopaminergic neurons of the substantia nigra after CRS treatment. RTP801 was post‐transcriptionally inhibited by the down‐regulation of miR‐7. Delayed turnover of RTP801, through the inhibition of proteasome degradation also contributed to its high content. Elevated RTP801 blocked autophagy, thus increasing accumulation of oligomeric α‐synuclein and aggravating endoplasmic reticulum stress. RTP801 inhibition alleviated the symptoms of neurodegeneration during this process.
Conclusions and Implications
RTP801 is a promising target for the treatment of PD, especially for PD‐sensitive patients who live under increased social pressure. Down‐regulation of RTP801 could inhibit the current tendency to an earlier onset of PD.
Abbreviations
- 3‐MA
3‐methyladenine
- BFPT
buried food pellet test
- CHX
cycloheximide
- CRS
chronic restraint stress
- DDIT4
DNA‐damage‐inducible transcript 4
- DOPAC
dihydroxyphenylacetic acid
- ER
endoplasmic reticulum
- PD
Parkinson's disease
- SN
substantia nigra
- SNpc
substantia nigra pars compacta
- VMAT2
vesicular monoamine transporter 2
- αSyn
α‐synuclein
Introduction
Parkinson's disease (PD) is characterized by a loss of dopaminergic neurons in the substantia nigra (SN) and the formation of Lewy bodies with aggregation of α‐synuclein (αSyn). PD has emerged as the second most common neurodegenerative disorder. There are no drugs available to delay or prevent the progression of PD , but studies have identified some factors contributing to deterioration in this condition, including ageing, trauma, and stress. Elucidation of their mechanisms may provide novel clues for the current problems in developing drugs for the treatment of PD.
The increasing stress in modern society means that most people are subjected to long‐term anxiety and emotional disorder. We have focused on the contribution of this increasing stress to the earlier age of onset of PD and to the enhancement of PD morbidity. Mounting evidence indicates that stress enhances the progression of PD. In rodents, stress, such as tail pinch, increased striatal dopamine release and turnover and excitation of striatal dopaminergic nerve terminals, which resulted in cell death (Pei et al., 1990). Subsequent studies demonstrated that stress caused by a unilateral 6‐hydroxydopamine lesion in the nigrostriatal bundle accelerated neural degeneration, which exaggerated motor symptoms in rats (Smith et al., 2008). Chronic stress leads to reduced dopaminergic activity within the ventral tegmental area in rodents and caused increased cortisol levels in PD patients (Djamshidian et al., 2011). One case reported that a 38‐year‐old woman suddenly experienced an early onset of PD symptoms 1 week after learning grievous news, which further aroused attention to the effects of stress on nervous system (Zou et al., 2013). There were some indications of correlations between stress and PD, such as the contribution of reduced T‐lymphocytes to dopaminergic cell loss (Baba et al., 2005; Reynolds et al., 2010), activation of the HPA axis by proinflammatory cytokines and chemokines (Haddad et al., 2002), and a shift of catecholamines into the cytosol to become toxic via auto‐oxidation (Goldstein, 2011). However, there is no strong evidence of the occurrence and deterioration of PD induced by stress.
The protein RTP801 is also known as Dig2 or REDD1, and is encoded by the stress‐responsive gene DNA‐damage‐inducible transcript 4 (DDIT4). RTP801 is a GR target gene, one of the genes activated by glucocorticoids and by stress, such as hypoxia, DNA damage, and nutrient or energy deprivation (Wang et al., 2006; Shimizu et al., 2011). Mice lacking RTP801 (RTP801 KO) are resistant to numerous stress‐induced pathological conditions and may suppress the adverse effects of glucocorticoids (Baida et al., 2014). Abnormalities in RTP801 signalling may also disturb energy homeostasis. A comparison of RTP801 expression in post‐mortem brains from PD and control patients found that RTP801 was highly elevated within neuromelanin‐containing neurons of the SN, but not in cerebellar neurons (Malagelada et al., 2006). Therefore, RTP801 was defined as a stress‐coping regulator but also as a pro‐apoptotic agent in neurodegenerative disorders (Canal et al., 2014). The present study used 10‐month‐old A53T mutant human αSyn transgenic mice as a PD‐sensitive model. In these mice, we identified RTP801 as the reactive factor in stress‐induced PD. These results support the possibility of targeting RTP801, as a means of preventing the tendency to earlier onset of PD which may result from constant social stress.
Methods
Animals
All animal care and experimental procedures complied with the principles outlined in the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Peking Union Medical College and Chinese Academy of Medical Sciences. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
αSyn A53T over‐expressing [B6;C3‐Tg(Prnp‐SNCA*A53T)83Vle/JNju] mice (Giasson et al., 2002; Zhou et al., 2016) and control mice were purchased from the Model Animal Research Center of Nanjing University (MARC, Nanjing, China) and raised in SPF Biotechnology Co., Ltd. (Beijing, China). Experiments were performed in the SPF laboratory animal room of the Institute of Materia Medica & Neuroscience Center, Chinese Academy of Medical Sciences and Peking Union Medical College. All animals were maintained in cages (five animals per cage) under constant temperature and humidity and exposed to a 12:12 h light–dark cycle with unrestricted access to tap water and food, except as described in the experiments.
Animal experiments
First set of experiments
The two mouse genotypes, αSyn A53T (male, aged 10 months, weight = 31 ± 2 g, n = 20) and wild‐type (WT) mice (male, aged 10 months, weight = 32 ± 2 g, n = 20), were randomly divided into four groups: WT, WT + chronic restraint stress (CRS), A53T, and A53T + CRS (n = 10 per group). The animals were subjected to the behaviour test after 4 weeks of CRS treatment. Half of the animals were anaesthetized with pentobarbital sodium, and brains were perfusion‐fixed with 4% paraformaldehyde in 0.01 M PBS. Brains were removed, and midbrain paraffin sections were sliced for immunofluorescence (see below for details) . Half of the animals were killed via cervical dislocation and decapitated to obtain brain tissue for protein or miRNA extraction. Dopamine and dihydroxyphenylacetic acid (DOPAC) were extracted from the striatum and assayed as described later. operators of the behavioural tests were blinded to treatments.
Second set of experiments
αSyn A53T mice (male, aged 10 months, weight = 31 ± 2 g, n = 24) were randomly divided into three groups: A53T + scramble shRNA, A53T + scramble shRNA + CRS, and A53T + RTP801 shRNA + CRS (n = 8 per group). The animals were anaesthetized with isoflurane and stereotaxically injected with RTP801 shRNA lentivirus or control lentivirus into the substantia nigra par compacta (SNpc), as described below. Animals were allowed to recover for 7 days before starting CRS treatment. The rotarod test was performed 2 days after the end of the 4 week CRS treatment. Three animals from each group were anaesthetized and perfusion‐fixed for immunofluorescence. The other five animals in each group were killed by cervical dislocation and used for Western blotting and dopamine assays. Animals were randomized for treatment. The operators were blinded in the behaviour test.
Chronic restraint stress (CRS)
The CRS procedure was modified according to a previously reported procedure (Liu et al., 2013; Huang et al., 2015). Restraint was performed by placing each animal into a 50 mL tube and adjusting it with plaster tape to ensure that the animal was unable to move. There were 3 cm holes in the lid and the far end for breathing. The stress was maintained 4 h each day for 4 weeks (6 days per week). Stress was started at different times each day (between 10:00 and 20:00 h) to minimize predictability. No pain was involved in getting the mice into the tubes. The control littermates were kept in their original cage, but food and water were provided only during the stress interval of the experimental groups to exclude the effect on body weight caused by different times of feeding.
Behavioural tests
All behavioural tests were performed between 9:00 and 14:00 h under normal animal room lighting. The buried food pellet test (BFPT) was performed on day 28 to day 32 in the first set of experiments. The rotarod test was performed on day 34, and the pole test was performed on day 35 (Figure 1A). The rotarod test was performed on day 37 in the second set of experiments (Figure 8A).
Figure 1.
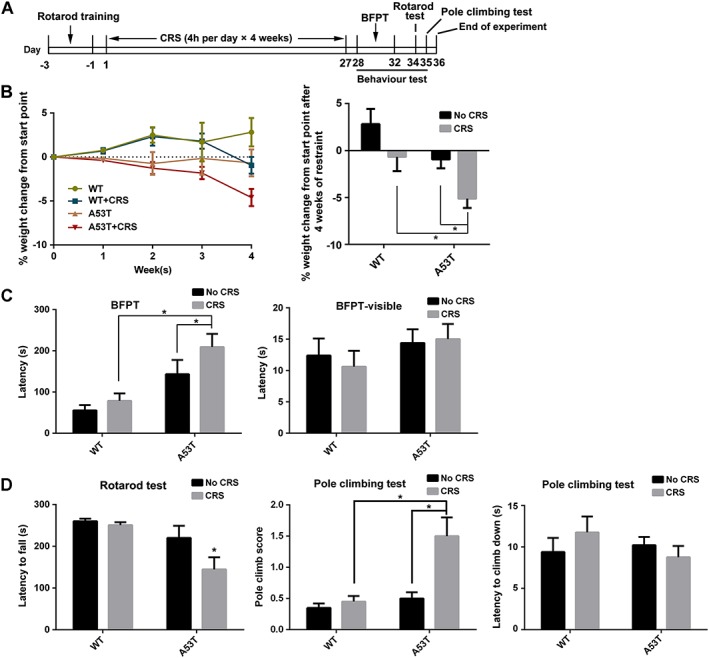
CRS accelerates PD‐like symptoms in A53T mice. (A) Scheme of the experimental procedure. (B) Body weight was monitored for the 4 weeks of CRS treatment and the corepsoding time course is presented in the left‐hand graph. Relative changes in weight were compared after CRS, with the changes after 4 weeks shown in the right‐hand graph. (C) BFPT and BFPT‐visible tests were performed after CRS treatment on Day 28 to Day 32. Latency of the tested mice to find the food pellet below the bedding in BFPT was recorded to test olfactory dysfunction. Latency of the tested mice to find the food pellet on the top of the bedding (BFPT‐visible) was recorded as a control. (D) Rotarod test was performed 2 days after BFPT test on Day 34. Pole climbing test was performed 1 day after the rotarod test. Latency to fall off the rotarod and the score and latency to climb down the pole were recorded. Data shown are means ± SEM, n = 10. *P<0.05, significantly different as indicated; two‐way ANOVA followed by Newman–Keuls test.
Figure 8.
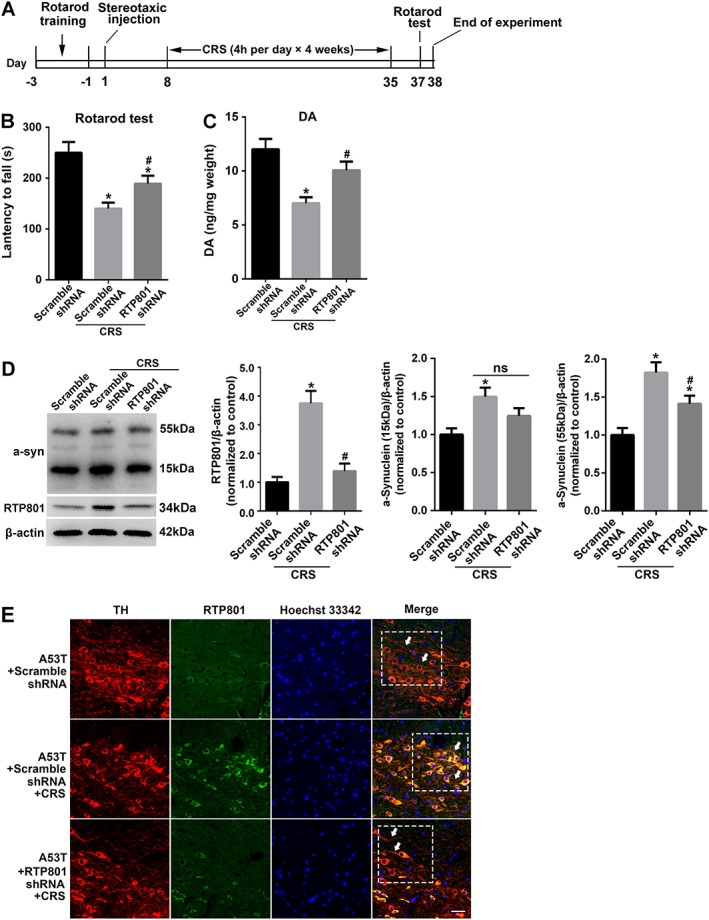
In vivo down‐regulation of RTP801 alleviates the pathological changes in A53T mice with CRS treatment. (A) Scheme of the in vivo experimental procedure. Lentivirus‐mediated RTP801 shRNA or scramble shRNA was stereotaxically injected into SNpc. CRS treatment was performed 7 days after the injection (n = 8). (B) Latency to fall off the rotarod were recorded 2 days after CRS treatment to evaluate motor coordination and balance of the experimental animals (mean ± SEM, n = 8). (C) Dopamine levels in the striatum were evaluated (mean ± SEM, n = 5). (D) The content of RTP801 and the accumulation of αSyn in mesencephalon were examined using Western blots. Data shown are means ± SEM, n = 5. *P<0.05, significantly different from scramble shRNA, # P<0.05, significantly different from scramble shRNA without CRS. (E) Immunofluorescence staining of TH (red) and RTP801 (green) in SNpc. Scale bar = 20 μm (mean ± SEM. n = 3).
Buried food pellet test (BFPT)
Mice were subjected to a food‐restricted diet (0.2 g chow per mouse per 24 h) 2 days before and throughout the three following experimental days. Mice had free access to water and underwent one trial per day for three successive days. The mouse was allowed 5 min to adapt in the cage prior to each trial, and the cage was identical to the real test cage without food pellets. The mouse was placed in the test cage and allowed to search for a food pellet (approximately 1 g), which was randomly buried approximately 0.5 cm below the bedding layer before each trial. The latency time for the mouse to find the food pellet within 5 min was recorded. The mice were allowed to consume the pellet and returned to their cages after the test. A visible pellet test was performed as a control to confirm the specificity of the BFPT, in which the food pellet was placed on the surface of the bedding (BFPT‐visible). Each mouse underwent the control test after the daily trial and consumed the food pellet.
Rotarod test
Motor coordination and balance were assessed using a rotarod apparatus (IITC Life Science, CA, USA) as reported in our previous research (Heng et al., 2016). Briefly, the rotarod was programmed to rotate with speeds that increased from 5 to 30 rpm in 300 s. The mice were placed on the rolling rod and tested three times with an interval time of at least 30 min to test the latency time to fall off the rotarod.
Pole‐climbing test
The pole‐climbing test was performed according to Davies et al. (2011). Briefly, the apparatus was a wooden pole with a height of 50 cm and a diameter of 1 cm wrapped with gauze to prevent slipping. The pole was positioned in the home cage and covered with bedding to protect the mice from injury. The mice were allowed free exploration for 1 min in the cage, and they were placed on top of the wooden pole and trained to climb down the pole. Each mouse was tested after three training sessions, and their latency to climb down the pole and scores for their locomotor activities were recorded. A 0 indicated no significant dysfunction; 0 to 0.5 indicated a helical or sliding step; 0.5–1 indicated several interruptions when climbing down, but the animal still grasped the pole; 1.5 indicated falling down after a sliding step; and 2 indicated an immediate fall from the pole. Each mouse was evaluated in three trials in a single day.
Cell culture and treatment
PC12 cells were cultured and treated with NGF as reported previously (Greene and Tischler, 1976). Briefly, cells were cultured in DMEM supplemented with 1% horse serum, penicillin/streptomycin and 50 ng·mL−1 NGF (Mouse Nerve Growth Factor for Injection, Staidson Biopharmaceuticals Co., Ltd, Beijing, China) for 7 days. Corticosterone was dissolved in 95% ethanol at a concentration of 1 mg·mL−1 and stored at −20°C until use. The stock solution was diluted to working solution using DMEM (Zhou et al., 2010). NGF withdrawal and treatments with corticosterone were performed on day 7 in vitro and assessed 48 h later for various assays, unless otherwise stated.
Cell viability/death assessment
Cell viability was measured by MTT assay. Briefly, cells were seeded in 96‐well plates (5 × 103 cells per well) and cultured as described above. Cells were treated 7 days after NGF withdrawal. Cells were incubated at 37°C for 4 h in a 0.5 mg·mL−1 MTT solution, and the media were carefully removed. DMSO (100 μL) was added to each well to dissolve the violet formazan crystals. The absorbance at 570 nm was measured. All values were normalized to the control group. Two technical replicates were included within each independent experiment for cell culture experiments. n denotes the number of independent assays.
Oligonucleotide synthesis and vector construction
MiR‐7 mimics (Sense: 5′‐UGGAAGACUAGUGAUUUUGUUGU‐3′, anti‐sense: 5′‐AACAAAAUCACUAGUCUUCCAUU‐3), negative control (sense: 5′‐UUCUCCGAACGUGUCACGUUU‐3′ anti‐sense: 5′‐ACGUGACACGUUCGGAGAAUU‐3′), anti‐miR‐7 (5′‐ACAACAAAAUCACUAGUCUUCCA‐3′) and control‐anti‐miR (5′‐CAGUACUUUUGUGUAGUACAA‐3′) were synthesized by GenePharma (Shanghai, China). RTP801 siRNA were prepared by GenePharma based on the following sequences: 5‐AAGACTCCTCATACCTGGATG‐3, which targeting both mouse and rat RTP801 sequence.
RTP801 constructs were generated by PCR cloning (RTP801 forward, 5′‐GAATTCGAACCATGCCTAGCCTTTGGGATCG‐3′; RTP801 reverse, 5′‐CTCGAGTCAACACTCTTCAATGAGCA‐3′) from pCMS‐EGFP‐RTP801, which was a gift from Lloyd Greene & Cristina Malagelada (Addgene plasmid #65057) (Malagelada et al., 2006), then cut with EcoRI and XhoI enzyme sites and ligated to pcDNA3.1 vector. The constructed pcDNA‐RTP801 was verified by DNA sequencing. DDIT4‐3′UTR and DDIT4‐MUT‐3′UTR were constructed into pmirGLO dual‐luciferase miRNA target expression vector (Promega, USA) with cloning sites SacI and XhoI by GenePharma. The above oligonucleotides and vectors were transfected with Lipofectamine 2000 according to manufacturer's instruction. RTP801 shRNA lentivirus vector was constructed and packaged by Obio Technology Corp., Ltd. (Shanghai, China).
RNA extraction and microRNA expression analysis
Total cellular RNA was extracted using Trizol reagent. MiRNA detection by real time analysis involved reverse transcription of cDNA using a small RNA‐specific stem‐loop RT primer as follows: 5‐AGCATTCGTCTCGACACAGCAACAAAATC‐3′. The generated cDNA was amplified with the specific primer for miR‐7 (5′‐TGACTCTGCTGGAAGACTAGTGAT‐3′ and 5′‐TAGAGCATTCGTCTCGACACAG‐3′). ΔCT method was used to calculate the relative expression and normalized to uniformly expressed U6.
Western blot
Western blot analysis was performed as described (Zhang et al., 2016b). Briefly, proteins were separated by electrophoresis on respective concentration of polyacrylamide gels and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 3% BSA and then incubated with primary antibodies at 4°C overnight, then followed by horseradish peroxidase‐conjugated secondary antibody and detected with the enhanced chemiluminescence plus detection system (PPLYGEN, Beijing, China). The signal protein bands were scanned by ImageQuant LAS4000 mini (GE Healthcare, Sweden) and analysed by Gel‐Pro Analyser software (Media Cybernetics, Bethesda, MD, USA).
Dual luciferase reporter assay
Cells were seeded in 96‐well plates and incubated as described and then transfected with various constructs as indicated in the respective experiment. Six hours after transfection, the treatment was performed as described respectively. Twenty‐four hours after transfection, cells were lysed with passive lysis buffer for 20 min. Firefly and Renilla luciferase activities were determined in lysates using the dual luciferase assay system (Promega, USA) in GloMax®‐96 Glomax luminometer (Promega, USA).
Immunofluorescence studies
Mice were anaesthetized, and the brains were perfusion‐fixed with 4% paraformaldehyde in 0.01 M PBS. The brains were removed after perfusion. Paraffin sections (4 μm) of the midbrain were cut for immunofluorescence. After deparaffinization and rehydration of the paraffin sections, antigen retrieval was performed with citrate buffer (0.1 M, pH 6.0) at 95°C for 10 min, then treated with 0.5% Triton X‐100 and blocked with 5% BSA. The sections were incubated with primary antibodies overnight at 4°C, then followed by Alexa Fluor secondary antibody (1:500) with or without Hoechst33342 (1:1000) for 2 h in room temperature. Finally, sections were washed three times with PBS, mounted with prolong anti‐fade reagent and observed with a confocal laser scanning microscope (LSM‐700, Leica, Wetzlar, Germany).
Determination of dopamine and DOPAC
Dopamine and its metabolites, DOPAC, were assayed according to the protocol described by Zhang et al. (2016a). Briefly, the striatum of mice was removed after 4 weeks CRS, then weighed and homogenized (1 mg:10 μL) in ice cold 0.6 M HClO4 solution including internal standard (isoprenaline, 250 ng·mL−1). After centrifugation (20 000× g, 4°C, 20 min), the supernatant (300 μL) was obtained and mixed with another solution (150 μL including 20 mM potassium citrate, 300 mM K2HPO4 and 2 mM Na2EDTA). After 30 min on ice, the mixture was centrifuged at 20 000× g for 20 min. The supernatant was collected and samples injected into the HPLC system (Waters e2695, USA) equipped with Electrochemical Detector (Waters 2465, USA) and C18 column (4.6 mm, 150 mm; Atlantis T3; Waters, USA). The mobile phase consisted of 100 mM sodium acetate, 85 mM citric acid, 0.2 mM Na2EDTA, 0.5 mM sodium 1‐octanesulfonate and 15% methanol (pH 3.68). The flow rate was set at 1 mL·min−1.
Stereotaxic injections
Animals were anaesthetized with 1.5% isoflurane mixed with medical air (20% oxygen, 80% nitrogen) at a flow rate of 0.7 L·min−1 and placed in a stereotaxic apparatus fixed with a mouse adaptor that positioned the skull horizontally between the bregma and lambda. RTP801 shRNA lentivirus or control lentivirus were injected into the SNpc at a flow rate of 0.1 μL·min−1 with a final volume of 0.5 μL. The needle was left in place for 5 min after injection. The stereotaxic coordinates for SNpc were as follows: −3.08 mm from the bregma, 1.5 mm to the midline and 4.5 mm below the subdural matter. The animals were allowed to recover for 7 days after a single injection, before starting any treatment.
Data and statistical analysis
Data and statistical analyses in this study complied with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as the means ± SEM. Statistical analyses were performed using GraphPad Prism 6. A two‐tailed Student's t‐test was used for comparisons between two groups. One‐way ANOVA or two‐way ANOVA analysis followed by the Newman–Keuls post hoc test was used for comparisons between multiple groups. P < 0.05 was considered statistically significant.
Materials
Primary antibodies to αSyn (C‐20), TH (H196), TH (F‐11) and vesicular monoamine transporter 2 (VMAT2) (D4) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Primary antibodies to ATF4 (D4B8), CHOP (L63F7) and GRP78 (BiP, C50B12) were purchased from Cell Signaling Technology (Danvers, MA, USA). Primary antibodies to p62 (ab56416) and DDIT4 (ab191871) were purchased from Abcam (Cambridge, MA, USA). LC3 (L8918) rabbit polyclonal antibody, corticosterone, 3‐methyladenine (3‐MA) and salubrinal were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Cycloheximide (CHX) was purchased from Beyotime Institute of Biotechnology (Nantong, Jiangsu Province, China). MG‐132 was purchased from Selleck Chemicals. Alexa Fluor 546 Donkey Anti‐Rabbit IgG (Invitrogen™), Alexa Fluor 488 Donkey Anti‐Rabbit IgG (Invitrogen), Alexa Fluor 546 Donkey Anti‐Mouse IgG (Invitrogen) and Alexa Fluor 488 Donkey Anti‐Mouse IgG (Invitrogen) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Horseradish peroxidase‐coupled secondary antibodies were purchased from KPL (Gaithersburg, MD, USA).
DMEM and reduced serum medium Opti‐MEM were purchased from Gibco (Carlsbad, CA, USA). Lipofectamine 2000 were purchased from Thermo Fisher Scientific (Waltham, MA, USA). FBS and horse serum were obtained from Hyclone (Rockford, IL, USA). PVDF membranes were obtained from Millipore (Bedford, MA, USA). Prolong anti‐fade kit was purchased from Molecular Probes (Carlsbad, CA, USA). Other materials were obtained from standard suppliers or as indicated in the text.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2015 a,b).
Results
CRS accelerates the PD‐like disease process in A53T mice
Ten‐month‐old A53T mutant human αSyn transgenic mice were used as a PD‐sensitive model to investigate the effect of CRS on PD‐like symptoms. The times and the procedures used in this model are summarised in Figure 1A. Body weight is an indirect parameter of HPA axis activation, and it was monitored for 4 weeks. The results demonstrated that CRS significantly affected gains in body weight in A53T mice. The change in body weight in A53T mice was more sensitive to the CRS treatment than the WT mice after 4 weeks of CRS treatment (Figure 1B). The BFPT was performed to evaluate olfactory dysfunction, which is a typical early symptom of PD patients. CRS clearly prolonged the latency to find the pellet compared to control mice. Figure 1C shows that there were no significant motor differences between groups, as manifested by the similar latencies in the BFPT‐visible test. These results demonstrated that CRS aggravated the olfactory disorder in A53T mice. The rotarod and pole climb tests evaluated motor coordination. A53T mice treated with CRS exhibited an apparently poor performance in the rotarod test compared to the untreated A53T group (Figure 1D). The pole climb tests of CRS‐treated A53T mice were accompanied by slipping in a helical route or even a fall, which indicated pole‐grasping failure in these mice. The scores of CRS‐treated A53T mice were notably higher than the other groups, which demonstrates bradykinesia (Figure 1D). Most of the PD‐like animals could not hold firmly and even quickly fell from the pole, but the latency to climb down was not significantly different.
CRS aggravates the pathological process of PD in A53T mice
Pathological accumulation of αSyn in neuronal cells and synapses are characteristic of PD. A rabbit anti‐αSyn (C‐20) antibody was used to detect the most abundant bands at approximately 15 and 55 kDa, which are the monomeric and oligomeric forms of αSyn, respectively, to examine the effect of CRS on the accumulation of αSyn (Rockenstein et al., 2014). CRS exposure induced a robust increase in monomeric and oligomeric αSyn in the mesencephalon compared to the untreated group (Figure 2A). Fluorescence immunostaining of tyrosine hydroxylase (TH) and the vesicular transporter VMAT2, which are essential for the survival of dopaminergic neurons (Lohr et al., 2014), exhibited a significant loss of TH‐ and VMAT2‐positive neurons in SNpc after CRS treatment, demonstrating damage to dopaminergic neurons in the pathological site of PD (Figure 2B). Western blot analysis of TH also confirmed the loss of TH‐positive dopaminergic neurons (Figure 2C). The levels of dopamine and its metabolite 3,4‐DOPAC in striatum were analysed by HPLC electrochemical detection, and both were significantly decreased in CRS‐treated A53T mice (Figure 2D). Collectively, these results demonstrated that CRS significantly exacerbated the pathological markers of PD.
Figure 2.
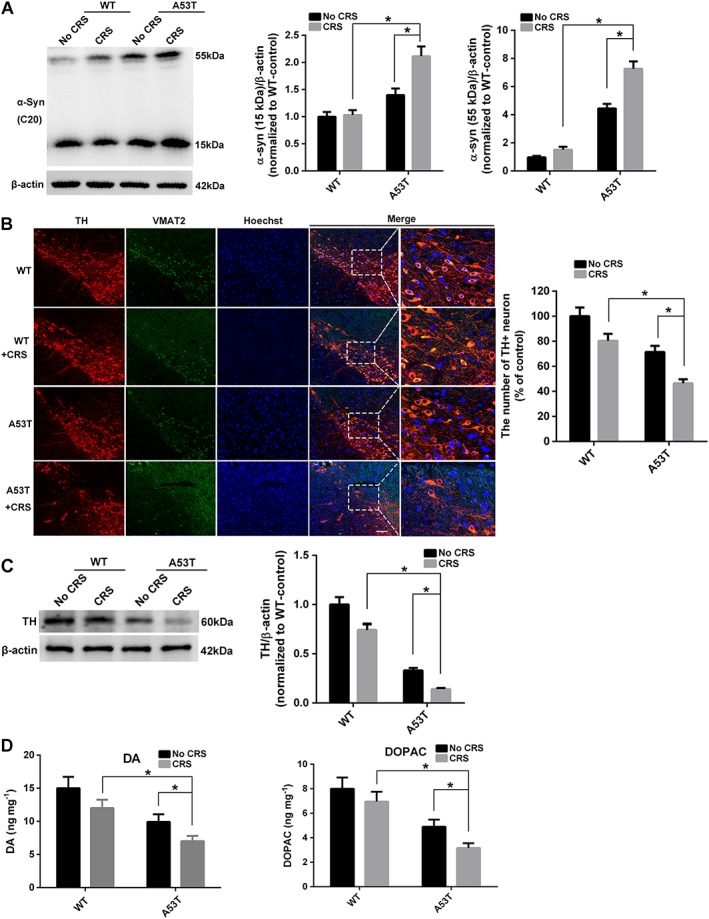
CRS deteriorates the pathological process in A53T mice. (A) Western blot was used to examine the accumulation of αSyn 15 and 55 kDa in mesencephalon (n = 5). (B) Immunofluorescence staining of TH (red) and VMAT2 (green) in SNpc. Bar = 20 μm. The number of TH+ neuron in SNpc was calculated (mean ± SEM, n = 5). (C) Western blot to examine TH expression in mesencephalon (n = 5). (D) Dopamine (DA) and DOPAC levels in the striatum were assayed by HPLC electrochemical detection. Data shown are means ± SEM, n = 5. *P<0.05, significantly different as indicated.
RTP801 is relevant to neurodegeneration after CRS treatment
Immunofluorescence of RTP801 was used to investigate its expression in SNpc and examine the role of RTP801 in the effects of CRS treatment on the PD process. RTP801+ cells were significantly up‐regulated in SNpc after CRS treatment, especially in the A53T group, indicating that αSyn intensified the up‐regulation of RTP801 with CRS treatment (Figure 3A). Western blots of RTP801 in the mesencephalon confirmed these results (Figure 3B). Rat pheochromocytoma (PC12) cells and PC12 cells stably overexpressing αSyn A53T (PC12‐A53T) were treated with corticosterone, the major stress hormone in rodents, to mimic the effects of stress on dopaminergic cells. The levels of RTP801 in the PC12 cells demonstrated an effect of CRS treatment, similar to that in vivo, as up‐regulation of RTP801 negatively correlated with cell viability (Figure 3C). We constructed the pcDNA3.1‐RTP801 vector and transfected it into PC12 cells to confirm the effect of RTP801 on cell viability. The expression of RTP801 after transfection was confirmed by Western blot. Cell viability was reduced in a dose‐dependent manner, suggesting that increased levels of RTP801 induced degeneration of the PC12 cells (Figure 3D).
Figure 3.
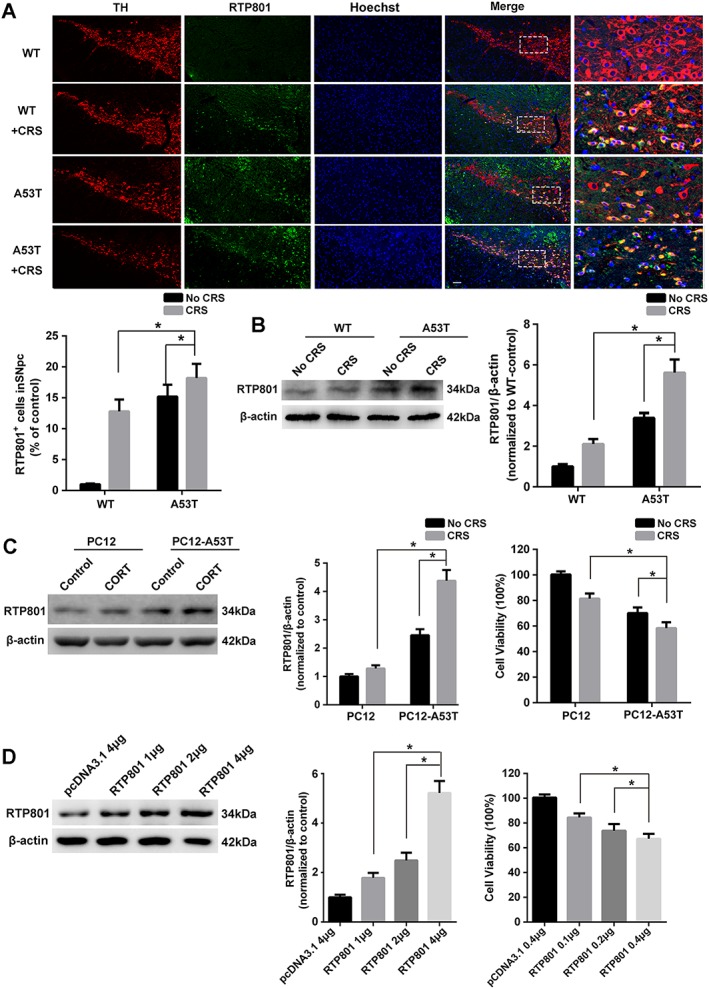
Up‐regulated RTP801 with stress treatment reduces cell viability. (A) Immunofluorescence staining of TH (red) and RTP801 (green) in SNpc. Bar = 20 μm. RTP801+ cell in SNpc was calculated (mean ± SEM, n = 5). (B) The expression of RTP801 in mesencephalon (mean ± SEM, n = 5). (C) Cells were treated with or without corticosterone (CORT; 10 μM) for 48 h, and RTP801 was measured. Cell viability was evaluated using MTT in 96‐well plates (mean ± SEM, n = 5). (D) Different amounts of pcDNA‐RTP801 were transfected into cells in 6‐well plates. RTP801 expression and cell viability were evaluated 48 h after transfection. Data shown are means ± SEM, n = 5. *P<0.05, significantly different as indicated.
Post‐transcriptional down‐regulation of RTP801 by miR‐7 is suppressed by CRS
Post‐transcriptional regulation by microRNAs (miRNAs) is critical in PD pathogenesis (Zhou et al., 2016). Post‐transcription regulation of RTP801 by miRNAs has not been well studied. In our experiments, TargetScan predicted that the 3′‐untranslated region (3′‐UTR) of DDIT4 could be targeted by miR‐7 (Figure 4A), which is associated with the pathophysiology of PD (Horsham et al., 2015). The expression of miR‐7 in mesencephalon was down‐regulated after the CRS treatment (Figure 4B). We then transfected PC12 cells with miR‐7 and found a notable down‐regulation of RTP801 (Figure 4C). DDIT4 3′‐UTR and miR‐7 binding site mutated DDIT4–3′‐UTR‐MUT were constructed to investigate the binding site (Figure 4D). Dual luciferase reporter assays suggested the post‐transcriptional regulation of RTP801 through the 3′‐UTR (Figure 4E). Treatment of the cells with corticosterone enhanced the luciferase activity, which was abolished with miR‐7, compared to the base level. However, DDIT4–3′‐UTR‐MUT groups exhibited no significant differences (ns) from one another, which confirmed the binding and effective site of miR‐7 (Figure 4F). The effects of anti‐miR‐7were similar to those of corticosterone (Figure 4G) and demonstrated that the down‐regulation induced by miR‐7 could be triggered by exposure to stress.
Figure 4.
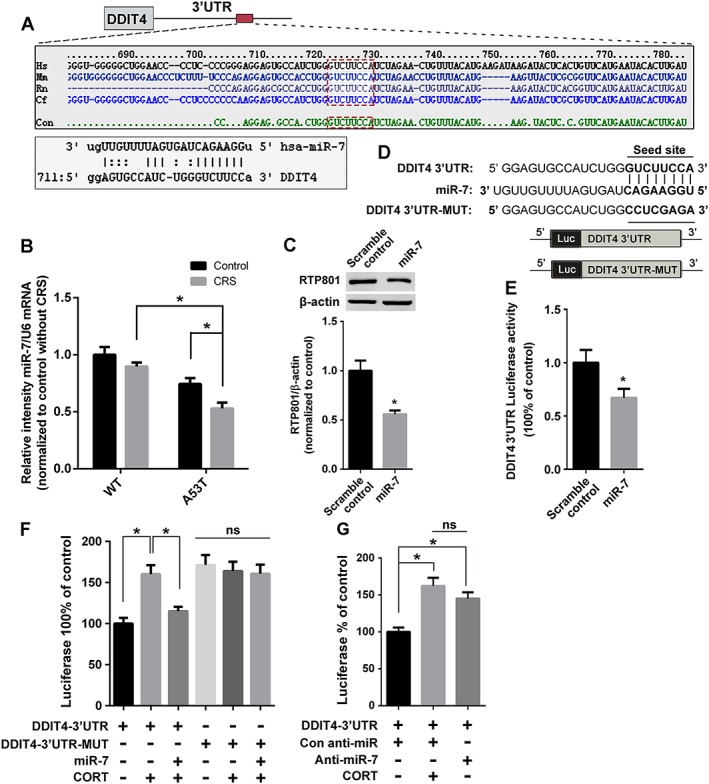
CRS up‐regulates RTP801 via miR‐7 suppression. (A) Prediction of miR‐7 binding site in DDIT4 3′‐UTR by TargetScan. The consensus seed sequence in species is emphasized by the red box. (B) Expression of miR‐7 in mesencephalon was assessed using Q‐PCR. (C) MiR‐7 or scramble control transfected cells were lysed to examine the expression of RTP801. (D) The schematic diagram of DDIT4 3′‐UTR and DDIT4‐3′‐UTR‐MUT, which were constructed into a pmirGLO vector. (E) Cells transfected with DDIT4 3′‐UTR were treated with scramble control or miR‐7 and lysed for dual‐luciferase activity test. (F) DDIT4 3′‐UTR or DDIT4‐3′‐UTR‐MUT transfected cells were treated with or without miR‐7 and corticosterone (CORT) to examine dual‐luciferase activity. (G) Cells were treated with an anti‐miR‐7 and corticosterone to examine the effect on DDIT4 3′‐UTR. Data shown are means ± SEM, n = 5. *P<0.05, significantly different as indicated.
Proteasome degradation of RTP801 is retarded by A53T αSyn
The biological half‐life of RTP801 protein is short. Therefore, we also investigated changes in the cellular levels of RTP801 after stress. PC12 or PC12‐A53T cells were transfected with RTP801 and treated with CHX for up to 120 min. The half‐life of RTP801 in PC12 cells was less than 90 min, which was clearly extended beyond 120 min in PC12‐A53T stable cells, suggesting that overexpression of A53T αSyn may protect RTP801 from degradation (Figure 5A). We therefore treated PC12 or PC12‐A53T cells overexpressing RTP801 with a proteasome inhibitor, MG‐132. Figure 5B shows that MG‐132 increased RTP801 protein levels in PC12 cells, compatible with proteasomal degradation of RTP801. In contrast, no significant increase in RTP801 protein was induced by MG‐132 in PC12‐A53T stable cells, indicating that αSyn A53T overexpression blocked the proteasomal degradation of RTP801 and thus increased the total amount of RTP801 protein present in these cells.
Figure 5.
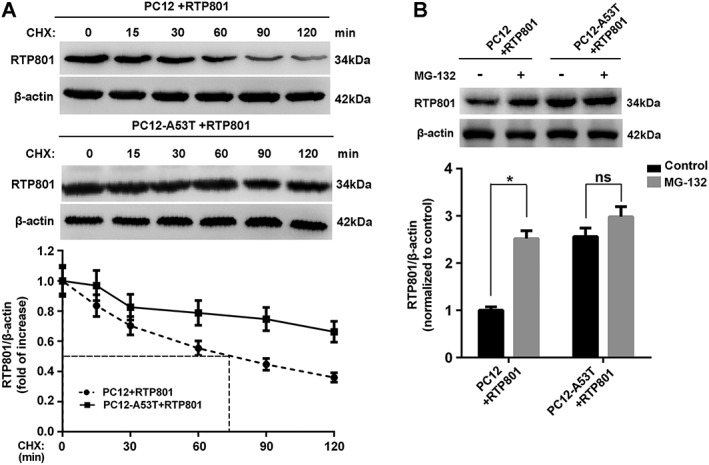
Inhibition of proteasomal degradation by αSyn A53T prolongs the biological half‐life of RTP801. (A) Cells transfected with pcDNA‐RTP801 were treated with CHX (10 μg·mL−1) for the indicated periods and lysed to examine the content of RTP801. (B) Cells transfected with pcDNA‐RTP801 were treated with or without MG‐132 (10 mM) for 24 h and subjected to Western blotting for RTP801. Data shown are means ± SEM, n = 5. *P<0.05, significantly different as indicated.
Block of autophagy induced by RTP801 contributes to the accumulation of αSyn in A53T mice
Abnormality of RTP801 disrupts energy homeostasis via inhibition of mTOR complex 1 signalling during stress (Ellisen, 2005; Katiyar et al., 2009). We first determined the autophagic flux in the mesencephalon of mice by measuring the levels of LC3‐II and p62, which are reliable markers of autophagic activity (Lamark et al., 2009; Klionsky et al., 2016). Significant elevations of LC3‐II and p62 levels were observed in A53T mice after CRS treatment, which suggests disrupted autophagy (Figure 6A). Elevated LC3‐II indicated an increased initiation of autophagy or a hindered autophagic flux, and the accumulated p62 showed disorder of autophagic flux. PC12‐A53T cells were transiently transfected with RTP801 siRNA or control siRNA to examine the role of RTP801 in this perturbation of autophagy. Both RTP801 and LC3‐II were elevated after corticosterone treatment but LC3‐II was down‐regulated when RTP801 was inhibited. Notably, the accumulation of p62 was almost abolished after RTP801 inhibition, which indicated that RTP801 may affect the autophagic flux. The accumulation of αSyn 15 and 55 kDa was reduced, especially the 55 kDa protein (Figure 6B). Therefore, we hypothesized that RTP801 contributed to αSyn accumulation by blocking autophagy. 3‐MA is an autophagy inhibitor and treatment of the cells with 3‐MA almost eliminated the difference between groups with or without RTP801 overexpression, confirming that it was a block of autophagy caused by RTP801 that primarily caused the accumulation of αSyn (Figure 6C).
Figure 6.
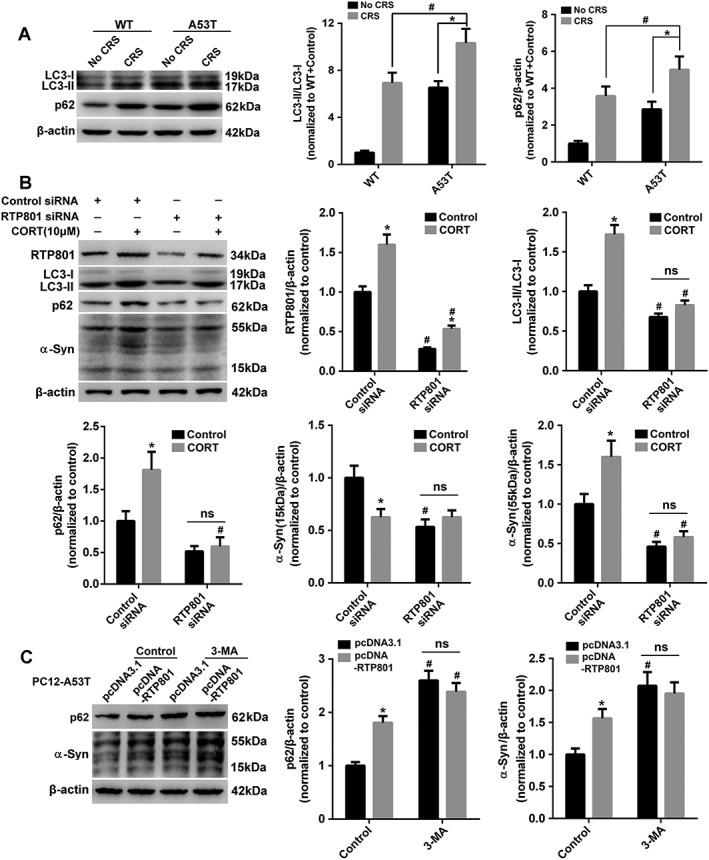
RTP801‐triggered block of autophagy causes aggregation of αSyn. (A) Mesencephalic LC3‐II and p62 were examined. (B) Cells transfected with or without RTP801 siRNA were lysed to examine RTP801, LC3‐II, p62 and αSyn after corticosterone (CORT) treatment. Data shown are means ± SEM, n = 5. *P<0.05, significant effect of corticosterone, # P<0.05, significant effect of RTP801 siRNA. (C) Cells transfected with or without pcDNA‐RTP801 were subjected to p62 and αSyn testing after 24 h of 3‐MA (10 mM) treatment. Data shown are means ± SEM, n = 5. *P<0.05, significant effect of pcDNA‐RTP801, # P<0.05, significant effect of 3‐MA.
ER stress and up‐regulated RTP801 intensified each other after CRS treatment
Endoplasmic reticulum (ER) stress was observed in human tissue derived from PD patients post mortem (Slodzinski et al., 2009; Mercado et al., 2016). ER stress in CRS‐treated A53T mice was also investigated. Grp78, ATF4 and CHOP were clearly up‐regulated after CRS treatment in A53T mice (Figure 7A). PC12 and PC12‐A53T cells provided results similar to those obtained in vivo (Figure 7B). Inhibition of ER stress by salubrinal, which attenuates A53T αSyn‐induced toxicity in PC12 cells (Smith et al., 2005), significantly attenuated the up‐regulation of RTP801 (Figure 7C), which suggests that the activated ER stress contributes to RTP801 expression. Notably, RTP801 inhibition inversely weakened the ER stress response in CORT‐treated A53T‐PC12 cells (Figure 7D), which suggests a positive feedback between the ER stress and RTP801 expression to intensify the response after stress treatment.
Figure 7.
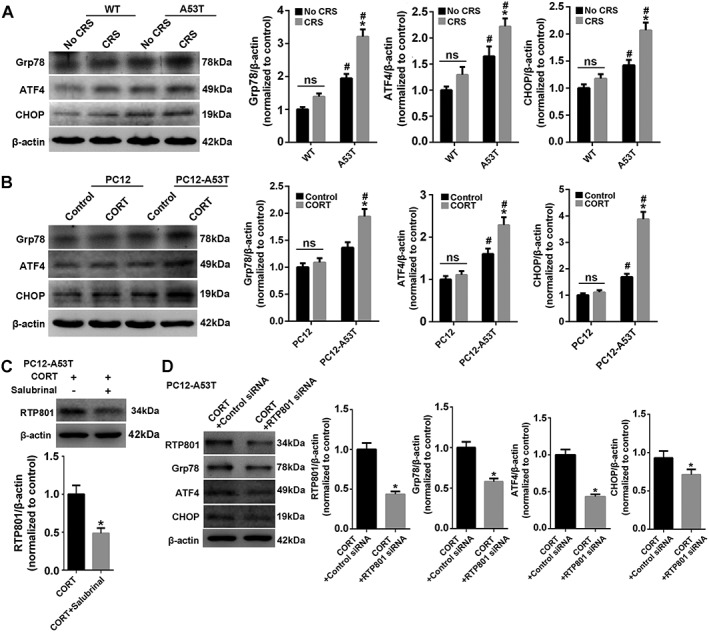
ER stress and up‐regulated RTP801 reinforce each other in CRS‐treated A53T mice. (A) Mesencephalic Grp78, ATF4 and CHOP were tested. Data shown are means ± SEM, n = 5. *P<0.05, significant effect of CRS, #*P<0.05, significantly different from WT . (B) Cells were treated with or without corticosterone (CORT) and lysed to examine Grp78, ATF4 and CHOP. *P<0.05, significant effect of corticosterone; # P<0.05, significantly different from PC12. (C) RTP801 was investigated after corticosterone (CORT; 10 μM) or corticosterone plus salubrinal (5 μM) treated PC12‐A53T. (D) PC12‐A53T cells transfected with or without RTP801 siRNA were lysed to examine RTP801, Grp78, ATF4 and CHOP after corticosterone treatment. Data shown are means ± SEM, n = 5. *P<0.05, significantly different from control siRNA.
RTP801 down‐regulation alleviates PD‐like neurodegeneration in CRS‐treated A53T mice
We injected lentivirus‐mediated RTP801‐shRNA into the SNpc of A53T mice to validate the role of RTP801 in the neurodegeneration of CRS‐treated A53T mice (Figure 8A). RTP801 inhibition significantly improved the behavioural defects, compared to the effects of scramble shRNA, following CRS treatment (Figure 8B). The content of dopamine in striatum was also higher after RTP801 inhibition (Figure 8C). A significant decrease of te toxic oligomeric form of αSyn was also found in the RTP801 shRNA‐injected group (Figure 8D). Immunofluorescence of RTP801 and TH in SNpc revealed that the inhibition of the up‐regulated RTP801 caused by CRS treatment clearly alleviated the damage to neurites and the loss of TH+ neural cells, which indicates improvement in the neurodegeneration of dopaminergic cells (Figure 8E).
Discussion
In recent years, the incidence of PD has exhibited a trend towards earlier onset i.e., towards a younger age. These patients will suffer more pain and cause more financial burden to their family and society than older patients. Middle‐aged PD patients often suffer greater life pressures and live with a higher self‐requirement. Mounting evidence has confirmed that increased stress was a risk factor, and this constantly increasing stress in modern society may have a causal relationship with the earlier onset of PD. However, there is no strong evidence to support this correlation.
Stress and hormones affect function of the motor system, because motor control‐related brain regions express glucocorticoid receptors (Ahima and Harlan, 1990; Ahima et al., 1992). Izzo et al. reported that the dopaminergic system was especially susceptible to the effects of stress (Izzo et al., 2005). Cortisol levels were higher in Parkinsonism and positively associated with gait deficits (Charlett et al., 1998). These results suggest stress as a critical factor that affects the neurodegenerative processes and symptoms of PD. The follow‐up study in veterans who experienced long‐term stress during war demonstrated that chronic stress increased the incidence of PD (Gibberd and Simmonds, 1980), but it was not a sufficient condition to trigger the occurrence of PD. αSyn is a soluble, natively unfolded protein in the CNS. Aggregates of αSyn is the major component of Lewy bodies and the hallmark of synucleinopathy. The mutated form of αSyn (A53T) forms oligomers faster than WT αSyn and contributes to the early onset of familial PD (Conway et al., 1998). Therefore, we chose αSyn A53T mice as a model for our research into PD. CRS caused weight loss without inducing PD‐like behavioural disorders in WT mice in our study. However, CRS treatment in A53T mice markedly induced PD‐like changes, including motor and non‐motor dysfunction, which suggests that CRS aggravated neurodegeneration in PD‐sensitive mice. We also used corticosterone, the major glucocorticoid in rodents, to mimic the effects of stress, in PC12 cells.
RTP801 is a stress‐responsive protein that was highly elevated in PD patients, with specific expression in neuromelanin‐containing neurons of the SN, but not cerebellar neurons (Malagelada et al., 2006), which indicated its specific expression in pathological locations of PD. RTP801 was essential for stress‐induced synaptic loss and depressive behaviour (Ota et al., 2014), which is consistent with our immunofluorescent results (Figure 8C). Notably, RTP801 exerts both pro‐ and anti‐apoptotic effects, depending on the cell context (Shoshani et al., 2002; Malagelada et al., 2006; Canal et al., 2014). The present study used differentiated PC12‐A53T stable cells to investigate the crosstalk between RTP801 and αSyn A53T in PD progression. The content of RTP801 increased markedly after CRS treatment in WT and A53T mice, but especially in the A53T mice.
The production and degradation of RTP801 are strictly regulated to produce its short half‐life in the stress response. MicroRNAs are important post‐transcriptional genetic regulators. At least three miRNAs are regulators of RTP801/REDD1 expression in the context of tumourigenesis: miR‐495 (Hwang‐Verslues et al., 2011), miR‐221 (Pineau et al., 2010) and micro‐RNA30c (Li et al., 2012). No miRNAs that modulate RTP801 expression in the context of neurodegeneration have been reported (Canal et al., 2014). Here, we have found that miR‐7 targeted the 3′‐UTR of RTP801, which was inhibited in CRS‐treated A53T mice (Figure 4). Notably, miR‐7 targets αSyn and NLRP3 in dopaminergic neurons, which is associated with the pathophysiology of PD. Our findings suggest that miR‐7 plays an additional role beyond αSyn regulation. There may also be a combined effect of αSyn and RTP801 regulation by miR‐7 in the PD process, but this hypothesis requires further testing. The degradation of RTP801 is also important for maintaining its content. RTP801 may be targeted for the ubiquitin proteasome system. Our results distinctly demonstrated that the biological half‐life of RTP801 was notably longer in PC12‐A53T‐treated cells than controls. We confirmed that degradation of RTP801 via the ubiquitin proteasome system was inhibited by A53T αSyn overexpression (Figure 5).
PD is characterized by aggregates of αSyn protein, which are degraded by the autophagy lysosome pathway (Dinter et al., 2016). Numerous studies suggested that mTOR inhibition caused autophagy. The effect of the mTOR inhibitor RTP801 on autophagy was also investigated in our research. The accumulation of p62 and αSyn was significantly decreased after RTP801 inhibition, which suggests a role for RTP801 in inducing autophagic dysfunction. 3‐MA was used to confirm this conclusion, but further studies are needed to clarify the underlying mechanism.
ER stress triggers an evolutionarily conserved series of signal transduction events to ameliorate the accumulation of unfolded protein in the ER. However, ER stress causes cell damage and death during overreactions (Kim et al., 2008). Signs of ER stress were observed in post‐mortem tissue from sporadic human PD cases and most animal models of the disease, which indicates that ER stress is important for the manifestations of α‐synucleinopathy in vivo (Colla et al., 2012). Smith et al. reported that ER stress‐mediated A53T mutant αSyn induced toxicity (Smith et al., 2005). Our results demonstrated that ER stress was aggravated after CRS exposure in vivo and in vitro. RTP801 served as a mediator of the outer and inner stress of the cells but also exhibited crosstalk with the ER stress overreaction. The ER stress and up‐regulated RTP801 intensified each other to intensify the response after CRS treatment (Figure 7).
In conclusion, RTP801 plays an essential role in the progression of CRS‐induced PD‐like neurodegeneration (Figure 9). The complex connection between RTP801 and αSyn affects many pathways that are involved in the pathology of PD. Our research clarified the reactive factor in stress‐induced PD process, which indicated that control of RTP801 up‐regulation may be a promising target for reversing the tendency towards earlier onset of PD in a younger population with constant social stress.
Figure 9.
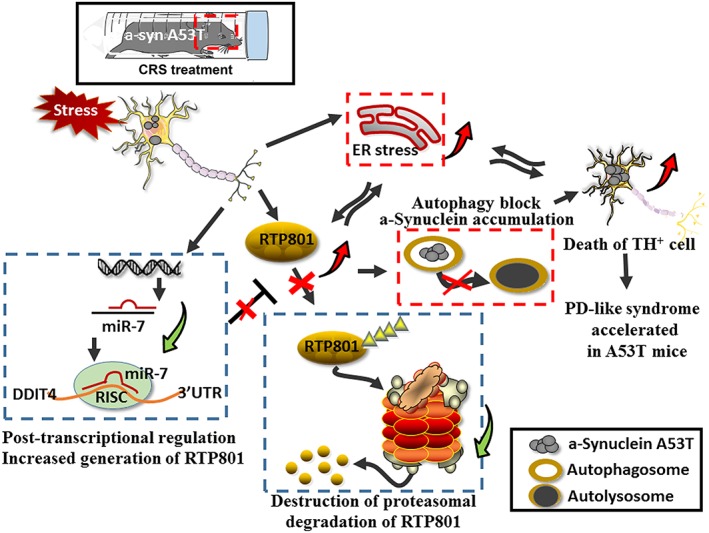
Schematic summary of the results. CRS treatment induced RTP801 up‐regulation and caused neurodegeneration, or even death, of TH+ cells in A53T mice. PD‐like pathogenesis was mediated by the robust enhancement of RTP801. Increased content of RTP801 occurred via inhibition of miR‐7, which post‐transcriptionally inhibited RTP801 expression and retarded proteasomal degradation. Up‐regulation of RTP801 intensified the toxicity of αSyn A53T for dopaminergic neurons. The crosstalk between ER stress and up‐regulated RTP801 reinforced the extra stress exposure. The block of autophagy that increased accumulation of oligomeric forms of αSyn aggravated the pathogenesis.
Author contributions
Z.Z. and S.‐F.C. designed and performed all the experiments, analysed data and wrote the manuscript; S.‐S.W. and Y.‐N.J. performed animal experiments; Y.G., P.‐F.Y. and Q.‐D.A. provided intellectual contributions; N.‐H.C. supervised all experiments and helped prepare the manuscript. All co‐authors have read and edited the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81603316, 81603315, 81274122, 81373997, 81273629, 81473376, U1402221, 81573640, 81573636 and 81560663), CAMS Innovation Fund for Medical Sciences (CIFMS) (2016‐I2M‐1‐004), Beijing Natural Science Foundation (7131013 and 7161011), Beijing Key Laboratory of New Drug Mechanisms and Pharmacological Evaluation Study (BZ0150), Key Research and Development Project of Hunan Province (2015SK2029‐1), the Scientific Research Foundation of the Higher Education Institutions of Hunan Province (15K091), PUMC Youth Fund (3332016058), the Fundamental Research Funds for the Central Universities (2014RC03 and 2016RC350002).
Zhang, Z. , Chu, S.‐F. , Wang, S.‐S. , Jiang, Y.‐N. , Gao, Y. , Yang, P.‐F. , Ai, Q.‐D. , and Chen, N.‐H. (2018) RTP801 is a critical factor in the neurodegeneration process of A53T α‐synuclein in a mouse model of Parkinson's disease under chronic restraint stress. British Journal of Pharmacology, 175: 590–605. doi: 10.1111/bph.14091.
References
- Ahima RS, Harlan RE (1990). Charting of type II glucocorticoid receptor‐like immunoreactivity in the rat central nervous system. Neuroscience 39: 579–604. [DOI] [PubMed] [Google Scholar]
- Ahima RS, Tagoe CN, Harlan RE (1992). Type II corticosteroid receptor‐like immunoreactivity in the rat cerebellar cortex: differential regulation by corticosterone. Neuroendocrinology 55: 683–694. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2015a). The Concise Guide To PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2015b). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba Y, Kuroiwa A, Uitti RJ, Wszolek ZK, Yamada T (2005). Alterations of T‐lymphocyte populations in Parkinson disease. Parkinsonism Relat Disord 11: 493–498. [DOI] [PubMed] [Google Scholar]
- Baida G, Bhalla P, Kirsanov K, Lesovaya E, Yakubovskaya M, Yuen K et al (2014). REDD1 functions at the crossroads between the therapeutic and adverse effects of topical glucocorticoids. EMBO Mol Med 7: 42–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal M, Romani‐Aumedes J, Martin‐Flores N, Perez‐Fernandez V, Malagelada C (2014). RTP801/REDD1: a stress coping regulator that turns into a troublemaker in neurodegenerative disorders. Front Cell Neurosci 8: 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlett A, Dobbs RJ, Purkiss AG, Wright DJ, Peterson DW, Weller C et al (1998). Cortisol is higher in parkinsonism and associated with gait deficit. Acta Neurol Scand 97: 77–85. [DOI] [PubMed] [Google Scholar]
- Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T et al (2012). Endoplasmic reticulum stress is important for the manifestations of alpha‐synucleinopathy in vivo . J Neurosci 32: 3306–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT (1998). Accelerated in vitro fibril formation by a mutant alpha‐synuclein linked to early‐onset Parkinson disease. Nat Med 4: 1318–1320. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SS, Bodine C, Matafonova E, Pantazides BG, Bernoud‐Hubac N, Harrison FE et al (2011). Treatment with a gamma‐ketoaldehyde scavenger prevents working memory deficits in hApoE4 mice. J Alzheimers Dis 27: 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinter E, Saridaki T, Nippold M, Plum S, Diederichs L, Komnig D et al (2016). Rab7 induces clearance of alpha‐synuclein aggregates. J Neurochem 138: 758–774. [DOI] [PubMed] [Google Scholar]
- Djamshidian A, O'Sullivan SS, Papadopoulos A, Bassett P, Shaw K, Averbeck BB et al (2011). Salivary cortisol levels in Parkinson's disease and its correlation to risk behaviour. J Neurol Neurosurg Psychiatry 82: 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellisen LW (2005). Growth control under stress: mTOR regulation through the REDD1‐TSC pathway. Cell Cycle 4: 1500–1502. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM (2002). Neuronal alpha‐synucleinopathy with severe movement disorder in mice expressing A53T human alpha‐synuclein. Neuron 34: 521–533. [DOI] [PubMed] [Google Scholar]
- Gibberd FB, Simmonds JP (1980). Neurological disease in ex‐Far‐East prisoners of war. Lancet 2: 135–137. [DOI] [PubMed] [Google Scholar]
- Goldstein DS (2011). Stress, allostatic load, catecholamines, and other neurotransmitters in neurodegenerative diseases. Endocr Regul 45: 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Tischler AS (1976). Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A 73: 2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad JJ, Saade NE, Safieh‐Garabedian B (2002). Cytokines and neuro‐immune‐endocrine interactions: a role for the hypothalamic‐pituitary‐adrenal revolving axis. J Neuroimmunol 133: 1–19. [DOI] [PubMed] [Google Scholar]
- Heng Y, Zhang QS, Mu Z, Hu JF, Yuan YH, Chen NH (2016). Ginsenoside Rg1 attenuates motor impairment and neuroinflammation in the MPTP‐probenecid‐induced parkinsonism mouse model by targeting alpha‐synuclein abnormalities in the substantia nigra. Toxicol Lett 243: 7–21. [DOI] [PubMed] [Google Scholar]
- Horsham JL, Ganda C, Kalinowski FC, Brown RA, Epis MR, Leedman PJ (2015). MicroRNA‐7: a miRNA with expanding roles in development and disease. Int J Biochem Cell Biol 69: 215–224. [DOI] [PubMed] [Google Scholar]
- Huang RR, Hu W, Yin YY, Wang YC, Li WP, Li WZ (2015). Chronic restraint stress promotes learning and memory impairment due to enhanced neuronal endoplasmic reticulum stress in the frontal cortex and hippocampus in male mice. Int J Mol Med 35: 553–559. [DOI] [PubMed] [Google Scholar]
- Hwang‐Verslues WW, Chang PH, Wei PC, Yang CY, Huang CK, Kuo WH et al (2011). miR‐495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E‐cadherin and REDD1. Oncogene 30: 2463–2474. [DOI] [PubMed] [Google Scholar]
- Izzo E, Sanna PP, Koob GF (2005). Impairment of dopaminergic system function after chronic treatment with corticotropin‐releasing factor. Pharmacol Biochem Behav 81: 701–708. [DOI] [PubMed] [Google Scholar]
- Katiyar S, Liu E, Knutzen CA, Lang ES, Lombardo CR, Sankar S et al (2009). REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A‐DDB1 ubiquitin ligase. EMBO Rep 10: 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC (2008). Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 7: 1013–1030. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A et al (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12: 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T, Kirkin V, Dikic I, Johansen T (2009). NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8: 1986–1990. [DOI] [PubMed] [Google Scholar]
- Li XH, Ha CT, Fu D, Xiao M (2012). Micro‐RNA30c negatively regulates REDD1 expression in human hematopoietic and osteoblast cells after gamma‐irradiation. PLoS One 7: e48700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhuang X, Gou L, Ling X, Tian X, Liu L et al (2013). Protective effects of nizofenone administration on the cognitive impairments induced by chronic restraint stress in mice. Pharmacol Biochem Behav 103: 474–480. [DOI] [PubMed] [Google Scholar]
- Lohr KM, Bernstein AI, Stout KA, Dunn AR, Lazo CR, Alter SP et al (2014). Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease‐related neurodegeneration in vivo . Proc Natl Acad Sci U S A 111: 9977–9982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malagelada C, Ryu EJ, Biswas SC, Jackson‐Lewis V, Greene LA (2006). RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson's disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci 26: 9996–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado G, Castillo V, Soto P, Sidhu A (2016). ER stress and Parkinson's disease: pathological inputs that converge into the secretory pathway. Brain Res 1648: 626–632. [DOI] [PubMed] [Google Scholar]
- Ota KT, Liu RJ, Voleti B, Maldonado‐Aviles JG, Duric V, Iwata M et al (2014). REDD1 is essential for stress‐induced synaptic loss and depressive behavior. Nat Med 20: 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Q, Zetterstrom T, Fillenz M (1990). Tail pinch‐induced changes in the turnover and release of dopamine and 5‐hydroxytryptamine in different brain regions of the rat. Neuroscience 35: 133–138. [DOI] [PubMed] [Google Scholar]
- Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B et al (2010). miR‐221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A 107: 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE (2010). Regulatory T cells attenuate Th17 cell‐mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson's disease. J Immunol 184: 2261–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Nuber S, Overk CR, Ubhi K, Mante M, Patrick C et al (2014). Accumulation of oligomer‐prone alpha‐synuclein exacerbates synaptic and neuronal degeneration in vivo . Brain 137: 1496–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, Takeda S et al (2011). Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab 13: 170–182. [DOI] [PubMed] [Google Scholar]
- Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S et al (2002). Identification of a novel hypoxia‐inducible factor 1‐responsive gene, RTP801, involved in apoptosis. Mol Cell Biol 22: 2283–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slodzinski H, Moran LB, Michael GJ, Wang B, Novoselov S, Cheetham ME et al (2009). Homocysteine‐induced endoplasmic reticulum protein (herp) is up‐regulated in parkinsonian substantia nigra and present in the core of Lewy bodies. Clin Neuropathol 28: 333–343. [PubMed] [Google Scholar]
- Smith LK, Jadavji NM, Colwell KL, Katrina Perehudoff S, Metz GA (2008). Stress accelerates neural degeneration and exaggerates motor symptoms in a rat model of Parkinson's disease. Eur J Neurosci 27: 2133–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A et al (2005). Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha‐synuclein‐induced toxicity. Hum Mol Genet 14: 3801–3811. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kubica N, Ellisen LW, Jefferson LS, Kimball SR (2006). Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J Biol Chem 281: 39128–39134. [DOI] [PubMed] [Google Scholar]
- Zhang S, Shao SY, Song XY, Xia CY, Yang YN, Zhang PC et al (2016a). Protective effects of Forsythia suspense extract with antioxidant and anti‐inflammatory properties in a model of rotenone induced neurotoxicity. Neurotoxicology 52: 72–83. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chu SF, Mou Z, Gao Y, Wang ZZ, Wei GN et al (2016b). Ganglioside GQ1b induces dopamine release through the activation of Pyk2. Mol Cell Neurosci 71: 102–113. [DOI] [PubMed] [Google Scholar]
- Zhou JY, Zhong HJ, Yang C, Yan J, Wang HY, Jiang JX (2010). Corticosterone exerts immunostimulatory effects on macrophages via endoplasmic reticulum stress. Br J Surg 97: 281–293. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Lu M, Du RH, Qiao C, Jiang CY, Zhang KZ et al (2016). MicroRNA‐7 targets Nod‐like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson's disease. Mol Neurodegener 11: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou K, Guo W, Tang G, Zheng B, Zheng Z (2013). A case of early onset Parkinson's disease after major stress. Neuropsychiatr Dis Treat 9: 1067–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]