

Summary
The size of all cells, from bacteria to vertebrates, is proportional to the growth rate set by nutrient availability, but the underlying mechanisms are unknown. Here, we show that nutrients modulate cell size and growth rate via the TORC2 signaling network in budding yeast. An important function of the TORC2 network is to modulate synthesis of ceramide lipids, which play roles in signaling. TORC2-dependent control of ceramide signaling strongly influences both cell size and growth rate. Thus, cells that cannot make ceramides fail to modulate their growth rate or size in response to changes in nutrients. PP2A associated with the Rts1 regulatory subunit (PP2ARts1) is embedded in a feedback loop that controls TORC2 signaling and helps set the level of TORC2 signaling to match nutrient availability. Together, the data suggest a model in which growth rate and cell size are mechanistically linked by ceramide-dependent signals arising from the TORC2 network.
Keywords: Cell Size, Cell Growth, Growth Rate, TORC2, ceramide, Rts1
Graphical Abstract
Introduction
All cells control their size. It is likely that cell size is controlled by highly conserved mechanisms, since size control would have been essential for the survival of the earliest cells. However, it has not yet been possible to define conserved mechanisms that broadly explain cell size control in diverse cell types. Control of cell size is relevant to cancer because severe defects in size are a nearly universal feature of cancer cells, yet nothing is known about the underlying causes.
The size of a cell ultimately reflects the outcome of mechanisms that control cell growth. One might therefore imagine that control of cell growth and size evolved together and are mechanistically linked. Indeed, there is evidence for a close relationship between cell growth and size. For example, in budding yeast, cell size at all cell cycle transitions is proportional to the growth rate during the previous growth interval [1, 2]. Moreover, the size of cells, from bacteria to vertebrates, is proportional to the growth rate set by nutrients [3–5]. Thus, cells growing in poor nutrients can be nearly half the size of cells growing in rich nutrients. The close relationship between cell size and growth rate could be explained by a model in which the same signals that control growth rate also control the amount of growth that occurs during the cell cycle. Alternatively, cells could measure the rate of growth and set the amount of growth required for cell division accordingly.
Distinguishing models for control of cell growth and size will require a better understanding of cell growth. Control of cell growth represents an extraordinary challenge. The central processes of growth - ribosome biogenesis and membrane expansion - must be coordinated with each other, and the rates of each process must be matched to the availability of building blocks and energy derived from nutrients. In budding yeast, the transcription of over a thousand genes is proportional to growth rate, which suggests that global top-down signals ensure that the rate of growth is matched to nutrient availability [6].
A conserved signaling network controls growth in all eukaryotic cells. At the heart of the network are the Tor kinases, which assemble distinct multiprotein kinase complexes called TORC1 and TORC2 [7–9]. The Tor kinase complexes control members of the AGC kinase family, which play essential roles in growth control [10–12]. The AGC kinases are also controlled by phosphoinositide-dependent kinase 1 (PDK1) [13, 14]. TORC1/2 and PDK1 phosphorylate distinct sites on AGC kinases that promote activation.
Little is known about how the diverse events of cell growth are coordinated with each other, or how growth rate and cell size are controlled by nutrients. We previously discovered that a form of PP2A associated with the Rts1 regulatory subunit (PP2ARts1), is required for nutrient modulation of cell size in budding yeast [15]. Here, we investigate the mechanisms by which PP2ARts1 influences cell growth and size.
Results
PP2ARts1 restrains the activity of Mss4, a conserved PI(4)P kinase
To investigate how PP2ARts1 influences cell growth and size, we first searched for proteins controlled by PP2ARts1. We previously used proteome-wide mass spectrometry to identify proteins that are hyperphosphorylated in rts1Δ cells, which would suggest that they are direct or indirect targets of PP2ARts1 [16]. Mss4, a phosphatidylinositol-4-phosphate 5-kinase, emerged from this analysis as a strong candidate. The mass spectrometry identified 6 high-confidence sites on Mss4 that undergo hyperphosphorylation in rts1Δ cells.
Mss4 promotes TORC2-dependent activation of Ypk1 and Ypk2, a pair of redundant AGC kinase paralogs related to vertebrate SGK kinases. To do this, Mss4 produces PI(4,5)P2, which recruits both TORC2 and proteins called Slm1 and Slm2 to the plasma membrane [17–19]. The Slm1/2 proteins bind both TORC2 and Ypk1/2, serving as a scaffold to drive phosphorylation of Ypk1/2 by TORC2. Full activation of Ypk1/2 also requires phosphorylation by Pkh1 and Pkh2, another pair of redundant kinase paralogs that are homologs of vertebrate PDK1 (Figure 1A) [14].
Figure 1. PP2ARts1 controls the TORC2 signaling network via the PI(4)P kinase Mss4.

(A) A summary of signaling events in the TORC2 network. (B) Wildtype and rts1Δ cells were grown to early log phase at 22°C in YPD medium. Mss4-3XHA was detected by western blotting. (C) rts1Δ and wildtype control cells containing Mss4-GFP were grown at 22°C in the same culture in complete synthetic medium containing dextrose. Wildtype controls cells were marked with Spc42-mRuby2 so they could be distinguished from rts1Δ cells. Cells were imaged by fluorescence microscopy in a field of view that includes both strains. Signal intensity was quantified along a line that bisected the plasma membrane. (D) The Mss4-GFP signal at the plasma membrane was quantified in wild type (21 cells) and rts1Δ (22 cells). *** denotes p< 0.0001 in Student’s t-test. (E) Bar plots showing levels of PI(4,5)P2 in wildtype and rts1Δ cells grown to early log phase at 22°C in YPD medium. Measurements were expressed as a mole percentage of total anionic phospholipids normalized to WT levels. Error bars represent the standard deviation of the mean of two biological replicates. * denotes p= 0.025 in Student’s t-test. (F) Wildtype and rts1Δ cells were grown to early log phase at 22°C in YPD medium. Western blotting with phosphospecific antibodies was used to detect a TORC2-dependent phosphorylation site (Ypk-pT662) and a Pkh1/2-dependent site (Ypk-pT504) on Ypk1/2. Total Ypk1 was detected using an anti-Ypk1 antibody. (G) Cells of the indicated genotypes were grown overnight to early log phase at 22°C in YPD medium. The cultures were then shifted to 30°C for 2 hours. Western blotting with phosphospecific antibodies was used to detect a TORC2-dependent phosphorylation site on Ypk1/2 (Ypk-pT662) and a Pkh1/2-dependent site (Ypk-pT504). Total Ypk1 was detected using an anti-Ypk1 antibody. The asterisk indicates a non-specific band. See also Figure S1 and Table S1.
Mss4 undergoes phosphorylation that causes an electrophoretic mobility shift that can be detected by western blot. Mss4 was hyperphosphorylated in rts1Δ cells, consistent with the proteome-wide mass spectrometry data (Figure 1B). Levels of the Mss4 protein were unchanged in rts1Δ cells (Figure S1A). Mss4 phosphorylation is thought to drive its association with the plasma membrane, although this has never been directly tested in vivo [20]. We found rts1Δ caused increased association of Mss4 with the plasma membrane, consistent with the idea that phosphorylation of Mss4 drives membrane association (Figures 1C and 1D). Increased association of Mss4 with the plasma membrane should drive increased production of PI(4,5)P2. We quantified PI(4,5)P2 in wildtype and rts1Δ cells, which confirmed that rts1Δ causes increased levels of PI(4,5)P2 (Figure 1E). Thus, the data suggest that Mss4 is hyperactive in rts1Δ cells.
PP2ARts1 influences signaling in the TORC2 network
The discovery that Mss4 is hyperactive in rts1Δ cells suggested that it could drive increased TORC2-dependent phosphorylation of Ypk1/2. To test this, we utilized a phosphospecific antibody that recognizes a TORC2-dependent site found on both Ypk1 and Ypk2 (Figures S1B and S1C). An antibody that recognizes Ypk1 was used to control for changes in Ypk1 levels. Both Ypk1 and Ypk2 underwent increased TORC2-dependent phosphorylation in rts1Δ cells (Figures 1F and S1D). To test whether hyperphosphorylation of Ypk1/2 in rts1Δ cells is dependent upon Mss4, we generated a new temperature-sensitive allele of MSS4 with a low restrictive temperature (30°C), which allowed inactivation of Mss4 without the complication of heat shock effects (Figures S1E, F). Inactivation of Mss4 caused loss of TORC2-dependent phosphorylation of Ypk1/2 and eliminated increased TORC2-dependent phosphorylation of Ypk1/2 in rts1Δ cells (Figures 1G and S1G).
A phosphospecific antibody that recognizes the site on Ypk1/2 that is phosphorylated by Pkh1/2 revealed that rts1Δ also caused an Mss4-dependent increase in phosphorylation of Ypk1/2 by Pkh1/2 (Figures 1F and 1G). Previous studies suggested that phosphorylation of Ypk1/2 by TORC2 promotes further activating phosphorylation of Ypk1/2 by Pkh1/2 [17, 21].
To further test whether PP2ARts1-dependent control of Mss4 influences signaling to Ypk1/2, we generated a mutant version of MSS4 that lacks phosphorylation sites controlled by PP2ARts1. Our previous proteome-wide mass spectrometry analysis identified candidate sites that show evidence of hyperphosphorylation in rts1Δ cells [16]. A mutant that lacks these sites (mss4-ps1) showed reduced phosphorylation in a wild type background, but still underwent partial hyperphosphorylation in rts1Δ cells, which suggested that the mass spectrometry analysis missed sites (Figure S1H) (see Methods). Nevertheless, the mss4-ps1 mutant blocked hyperactivation of TORC2 in rts1Δ cells, consistent with a model in which PP2ARts1 influences TORC2 signaling via Mss4.
PP2ARts1 modulates the TORC2 network in response to nutrients
PP2ARts1 is required for nutrient modulation of cell size, which suggests that it relays nutrient-dependent signals [15]. We therefore considered the possibility that PP2ARts1 relays nutrient-dependent signals to the TORC2 network. To investigate further, we tested whether Mss4 undergoes a PP2ARts1-dependent response to changes in carbon source. Wildtype cells and rts1Δ cells were shifted from rich carbon (2% glucose) to poor carbon (2% glycerol/ethanol) and Mss4 phosphorylation was assayed by western blot. Mss4 underwent rapid dephosphorylation in response to poor carbon that was partially dependent upon PP2ARts1 (Figure 2A). Purified PP2ARts1 does not dephosphorylate Mss4 in vitro, which suggests that it acts indirectly.
Figure 2. The TORC2 network is modulated by nutrients.

(A) Wildtype and rts1Δ cells were grown to early log phase at 22°C in YPD medium. Cells were washed into YPG/E medium and the behavior of Mss4-3XHA was assayed by western blotting at the indicated times. (B) Wildtype and rts1Δ cells were shifted from rich to poor carbon as in (A). Ypk-pT662, Ypk-pT504 and Ypk1 were assayed by western blot. (C) Wildtype and rts1Δ cells were grown at 22°C to early log phase in YEP media containing glucose (Glu), galactose (Gal) or glycerol/ethanol (GE). Ypk-pT662, Ypk-pT504 and Ypk1 were assayed by Western blotting. An asterisk indicates a non-specific band. See also Figure S2 and Table S1.
We next tested whether TORC2 or Pkh1/2 are modulated in response to changes in carbon source. In wildtype cells, a shift to poor carbon caused rapid loss of TORC2-dependent phosphorylation of Ypk1/2 (Figure 2B). As cells adapted to the new carbon source, TORC2 activity recovered but remained below levels observed in rich carbon. Similar behavior was observed for Pkh1/2-dependent phosphorylation of Ypk1/2. The shift to poor carbon also caused increased electrophoretic mobility of Ypk1/2 (Figure 2B). Electrophoretic mobility shifts in Ypk1/2 are due to redundant protein kinase paralogs called Fpk1 and Fpk2, which play poorly understood roles in controlling Ypk1/2 [22]. Thus, the data suggest that Fpk1/2 activity is reduced in poor carbon.
In rts1Δ cells, the acute response to poor carbon occurred normally, but TORC2-dependent phosphorylation of Ypk1/2 recovered to abnormally high levels after cells adapted to the poor carbon. Pkh1/2-dependent phosphorylation of Ypk1/2 also recovered to abnormally high levels in rts1Δ cells (Figure 2B).
To investigate TORC2 signaling after long term adaptation to poor carbon, we grew wildtype and rts1Δ cells for 16 hours in carbon sources of varying quality. Glycerol/ethanol was again used as a poor carbon source, while galactose served as a carbon source of intermediate quality. Again, TORC2 and Pkh1/2 signaling were elevated in rts1Δ cells under all conditions, and poor nutrients caused large shifts in the electrophoretic mobility of Ypk1/2 (Figures 2C and S2). Thus, the TORC2 network is modulated by carbon source, and normal modulation of the network requires PP2ARts1.
The level of Ypk1 signaling strongly influences cell size
We next tested whether cell size defects caused by loss of PP2ARts1 could be caused by misregulation of Ypk1/2. Since Ypk1/2 are essential for viability, we analyzed effects of partial loss of function caused by ypk1Δ or ypk2Δ. ypk1Δ caused a large decrease in cell size (Figure 3A). Moreover, ypk1Δ was epistatic to rts1Δ, since rts1Δ failed to increase the size of ypk1Δ cells. Loss of Ypk2 had no effect on cell size (Figure S3). An analog-sensitive allele of YPK1 in a ypk2Δ background (ypk1-as ypk2Δ) caused reduced cell size in the absence of inhibitor, consistent with previous work showing that analog-sensitive alleles often cause reduced kinase activity (Figure 3B). Addition of a low dose of inhibitor further reduced cell size, which suggested that cell size is proportional to Ypk1/2 activity.
Figure 3. Ypk1/2 signaling strongly influences cell size.

(A–D) Cells of the indicated genotypes were grown to log phase at 22°C and cell size distributions were determined using a Coulter counter. Each plot is the average of 3 biological replicates. For each biological replicate, 3 technical replicates were analyzed and averaged. (A) Cells were grown in YPD medium. (B) Cells were grown in YPD medium for 16 hours in the presence of 0.1 μM 3-MOB-PP1 or DMSO control. (C) Cells were grown in YPD medium. (D) Cells were grown in YPGal medium. See also Figure S3 and Table S1.
We also tested whether Pkh1/2, critical upstream activators of Ypk1/2, influence cell size. To achieve partial loss of function we utilized cells dependent upon a temperature sensitive allele of pkh1 (pkh1-ts pkh2Δ) and grew the cells at a semi-restrictive temperature. Partial loss of Pkh1/2 caused reduced cell size and also strongly reduced the size of rts1Δ cells (Figure 3C). Overexpression of Pkh1/2 caused increased cell size (Figure 3D). Previous studies in Drosophila and mice found that reduced PDK1 activity causes reduced cell size, while overexpression causes increased cell size, which suggests that the relationship between PDK1 signaling and cell size is conserved [23, 24]. The Pkh1/2 kinases also target the AGC kinases Sch9 and Pkc1, so it is possible that they influence cell size by multiple mechanisms.
Ceramides are required for normal control of cell size
Ypk1/2 control synthesis of sphingolipids, which play roles in signaling and are used to synthesize minor structural components of membranes. The first step of sphingolipid synthesis is catalyzed by serine palmitoyltransferase, which joins a long fatty acid tail to serine to create lipids called long chain bases (Figure 4A) [25, 26]. Further processing steps use long chain bases to generate ceramides, which include two fatty acid tails linked to a polar head group. The polar head group of ceramides can be further modified to generate complex sphingolipids. In budding yeast, these include inositol-phosphorylceramide (IPC), mannosyl-inositol-phosphorylceramide (MIPC), and mannosyl-diinositol-phosphorylceramide (M(IP)2C). Together, complex sphingolipids constitute approximately 10% of total lipids. In contrast, ceramides constitute 0.1% of total lipids [27, 28].
Figure 4. Ceramide is required for normal control of cell size.

(A) A summary of sphingolipid synthesis pathways. Small-molecule inhibitors are indicated in red. DHS: dihydrosphingosine, PHS: phytosphingosine, DHS-P/PHS-P: dihydrosphingosine/phytosphingosine-1-phosphate. (B,C) Wildtype or rts1Δ cells were grown in YPD media at 22°C for 16 hours to early log phase in the presence of varying concentrations of myriocin. Cell size distributions were determined using a Coulter counter. (D–G) Cells were grown to early log phase in YPG/E medium and small unbudded cells were isolated by centrifugal elutriation. Cells were released into YPD medium in the presence of varying concentrations of myriocin at 25°C and samples were taken at 10 minute intervals. (D) Mean cell volume was measured with a Coulter counter and plotted as a function of time. (E) The percentage of budded cells was plotted as a function of time. (F) A plot of growth rate versus myriocin concentration. (G) A plot of cell size at cell cycle entry versus myriocin concentration. See also Figure S4 and Table S1.
Ypk1/2 promote sphingolipid synthesis by inhibiting Orm1 and Orm2, a pair of redundant paralogs that inhibit serine palmitoyltransferase [29, 30]. Ypk1/2 also directly phosphorylate and stimulate ceramide synthase, which uses long chain bases to synthesize ceramides [31, 32]. Ceramide synthase is encoded by redundant paralogs called LAC1 and LAG1. Inhibition of sphingolipid synthesis causes increased TORC2-dependent phosphorylation of Ypk1/2, as well as increased phosphorylation of the Orm1/2 proteins [29, 33]. Thus, it is thought that sphingolipid synthesis is controlled by negative feedback in the TORC2 network.
Since Ypk1/2 control sphingolipid synthesis, we tested whether modulation of sphingolipid synthesis contributes to cell size defects caused by decreased Ypk1/2 signaling. To modulate sphingolipid synthesis, we first utilized myriocin, an inhibitor of serine palmitoyltransferase [34]. Cells were grown for 16 hours in sub-lethal concentrations of myriocin ranging from 0.2 to 1.0 μg/ml. At these concentrations, myriocin causes a dose-dependent decrease in levels of long chain bases [35]. We found that myriocin caused a dose-dependent decrease in cell size (Figure 4B). The decrease in size caused by myriocin was largely eliminated in rts1Δ cells, consistent with the fact that rts1Δ cells have increased Ypk1/2 activity and are resistant to myriocin (Figure 4C) [30]. The decrease in size was also largely eliminated by overexpression of Pkh2, which should promote increased activity of Ypk1/2 (Figure S4A). Myriocin also decreased the size of fission yeast, which suggests that it influences a conserved mechanism (Figure S4B).
To investigate further, we tested the effects of myriocin on cell growth and size at a specific stage of the cell cycle. We focused on G1 phase because previous studies suggested that cell size at completion of G1 phase is tightly controlled [4, 36]. Centrifugal elutriation was used to isolate small unbudded cells in G1 phase. Before elutriation, cells were grown in poor carbon to obtain very small unbudded cells. After elutriation, cells were released into medium containing rich carbon, which resulted in a prolonged interval of growth to achieve the increased size of cells in rich carbon. Myriocin was added at the same range of doses used in Figure 4B. Average cell size and the percentage of cells with buds were measured at 10 minute intervals during growth in G1 phase (Figures 4D and 4E). Growth rate was measured as the increase in volume before 25% bud emergence, divided by the time (Figure 4F). The time of cell cycle entry was defined as the time at which 25% of the cells had buds, which was used to determine cell volume at cell cycle entry (Figure 4G).
Control cells grew at a rate of 0.15 fL/min during G1 and initiated bud emergence at a size of 37 fL. Myriocin caused a dose-dependent decrease in growth rate, yet had no effect on the timing of bud emergence. It also caused a dose-dependent decrease in the size at which cells entered the cell cycle. The maximal dose of myriocin decreased growth rate to 0.074 fL/min and reduced cell size at cell cycle entry to 23.7 fL.
To test how sphingolipid production influences cell cycle entry, we analyzed how myriocin affects the behavior of Cln3 and Whi5, which are required for normal control of cell size in G1 phase. Current models suggest that Cln3 is an early G1 cyclin that activates Cdk1, which then phosphorylates and inactivates Whi5, a transcriptional repressor that blocks transcription of genes needed for cell cycle entry [37, 38]. It has been proposed that growth-dependent dilution of Whi5 plays a critical role in triggering cell cycle entry [39]. Thus, myriocin could induce early cell cycle entry by reducing levels of Whi5, or by increasing levels of Cln3. However, the highest dose of myriocin did not cause reduced Whi5 levels or increased levels of Cln3 (Figure S4C).
We next determined which products of sphingolipid synthesis influence cell size. Deletion of the genes that encode ceramide synthase (lac1Δ lag1Δ) caused a large reduction in cell size (Figure 5A). An inhibitor of ceramide synthase (fumonisin B1) also caused reduced cell size (Figure S5A). As with myriocin, rts1Δ reduced the effects of fumonisin B1 on cell size. We were unable to recover viable lac1Δ lag1Δ rts1Δ cells to test the effects of rts1Δ on lac1Δ lag1Δ. Blocking production of MIPC or M(IP)2C had no effect on cell size (Figure S5B). To test for a role of IPC, we used aureobasidin A, a small molecule inhibitor that blocks synthesis of IPC. However, the results were difficult to interpret. Below 5 ng/ml, aureobasidin A had no effect on proliferation or size, whereas above 15 ng/ml it completely blocked proliferation. At intermediate concentrations, there appeared to be a sharp cutoff between lethality versus no effect. More importantly, intermediate doses of aureobasidin A (12.5 ng/ml) caused approximately 50% lethality (Figure S5C). Thus, in contrast to myriocin, aureobasidin A could not be used to observe the effects of modulating levels of IPC. A further concern is that aureobasidin is poorly characterized and could have off-target effects.
Figure 5. Ceramides are required for nutrient modulation of growth rate and cell size.

(A) Wildtype and lac1Δ lag1Δ cells were grown to log phase in YPD medium at 22°C. Cell size distributions were determined using a Coulter counter. (B) lac1Δ lag1Δ cells were grown to log phase in YPD (Rich) or YPG/E (poor) medium for 16 hours at 22°C. Cell size distributions were determined using a Coulter counter. (C–H) Wildtype or lac1Δ lag1Δ cells were grown to log phase in YPG/E medium. Small unbudded cells in G1 were isolated by centrifugal elutriation and released into either YPD media (Rich) or YPG/E medium (Poor) at 25°C. (C,E) Mean cell volume was analyzed using a Coulter counter and plotted as a function of time. (D,F) The percentage of budded cells was plotted as a function of time. (G) A plot of growth rate versus carbon source. (H) A plot of cell volume at cell cycle entry versus carbon source. See also Figure S5 and Table S1.
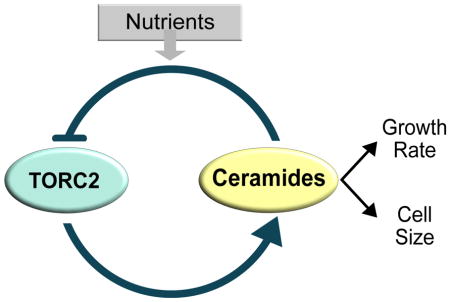
Ceramides are required for nutrient modulation of growth rate and cell size
PP2ARts1 is required for nutrient modulation of cell size and for normal modulation of signals that control ceramide synthesis. We therefore hypothesized that nutrient modulation of cell size is dependent upon modulation of ceramide synthesis. Cells that lack ceramide synthase failed to increase their size in response to rich nutrients, indicating a failure in nutrient modulation of cell size (Figure 5B). To investigate further, we compared growth of wildtype and lac1Δ lag1Δ cells during G1 phase in rich or poor carbon. Small unbudded wildtype cells growing in poor carbon were isolated by centrifugal elutriation. After elutriation, half of the culture was shifted to rich carbon. The cells that remained in poor carbon grew at a rate of 0.085 fL/min and initiated bud emergence at 26 fL (Figures 5C, 5D and 5G). The cells shifted to rich carbon increased their growth rate to 0.127 fL/minute and initiated bud emergence at 36 fL. Bud emergence in rich carbon was delayed 40 minutes, presumably to allow cells to reach the new threshold amount of growth required for cell cycle entry.
In contrast, lac1Δ lag1Δ cells failed to increase their growth rate in rich carbon (Figures 5E and 5G). Moreover, the delay in bud emergence was reduced nearly two-fold and the cells initiated cell cycle entry at nearly identical small sizes in rich and poor carbon (Figures 5F and 5H). Thus, loss of ceramide synthase caused a nearly complete failure in modulation of growth rate and cell size in response to carbon source.
Ceramides are required for negative feedback signaling in the TORC2 network
Previous work found that TORC2 signaling is influenced by negative feedback [29, 33, 35]. Thus, inhibition of sphingolipid synthesis triggers increased TORC2 activity. However, the mechanisms by which feedback signals are relayed are unknown. Since PP2ARts1 and Mss4 influence signaling in the TORC2 network, we hypothesized that they play roles in feedback signaling. To investigate, we tested whether Mss4 responds to sphingolipid-dependent signals. To do this, we took advantage of the fact that exogenously added phytosphingosine is taken up by cells and utilized in sphingolipid synthesis pathways. Addition of phytosphingosine at concentrations above 5 μM caused dephosphorylation of Mss4 (Figure 6A). In a time course, addition of 20 μM phytosphingosine triggered dephosphorylation of Mss4 within 10 minutes (Figure 6B). The strongest effects of phytosphingosine were transient, which suggests that the active lipids generated from phytosphingosine are short-lived, or that the signaling network adapts to elevated levels of phytosphingosine. Dephosphorylation of Mss4 in response to phytosphingosine was strongly attenuated in rts1Δ cells (Figure 6B).
Figure 6. Ceramides are required for negative feedback in the TORC2 network.

(A) Cells were grown to early log phase in YPD medium. Varying concentrations of phytosphingosine (PHS) were added and cells were incubated at 25°C. Samples were taken 15 minutes after addition of phytosphingosine and Mss4-3XHA was detected by western blot. (B) Wildtype and rts1Δ cells were grown to early log phase in YPD. 20 μM phytosphingosine (PHS) was added to each culture followed by incubation at 25°C. Samples were taken at the indicated times and Mss4-3XHA was detected by western blot. (C) Wildtype and lac1Δ lag1Δ cells were grown to early log phase in YPD at 22°C. Mss4-3XHA was detected by western blot. (D) lac1Δ lag1Δ cells were grown to early log phase in YPD at 25°C. Samples were taken at the indicated times after addition of 20 μM phytosphingosine (PHS) and Mss4-3XHA was detected by western blot. (E) Wildtype and lac1Δ lag1Δ cells were grown to early log phase in YPD at 22°C. Ypk-pT662, Ypk-pT504 and Ypk1 were assayed by western blot. (F) Wildtype and lac1Δ lag1Δ cells were grown to early log phase in YPD at 22°C. Samples were taken at the indicated times after addition of 20 μM phytosphingosine (PHS) and Ypk-pT662, Ypk-pT504 and Ypk1 were assayed by western blot. For panels E and F, an asterisk indicates a non-specific band. See also Figure S6 and Table S1.
We next tested whether phytosphingosine must be further modified to influence Mss4 phosphorylation. Phytosphingosine can be phosphorylated to produce sphingosine-1-phosphate, or it can be used to synthesize ceramides. Blocking phosphorylation of phytosphingosine had no effect on Mss4 phosphorylation (Figure S6A). In contrast, Mss4 was hyperphosphorylated in lac1Δ lag1Δ cells, and dephosphorylation of Mss4 in response to phytosphingosine failed to occur (Figure 6C, D).
Hyperphosphorylation of Mss4 in lac1Δ lag1Δ cells suggested that the TORC2 network should be hyperactive. Indeed, phosphorylation of Ypk1/2 by both TORC2 and Pkh1/2 was increased in lac1Δ lag1Δ cells (Figure 6E and Figures S6B). Inhibition of Lac1/Lag1 with fumonisin B1 also caused increased phosphorylation of Ypk1/2 by TORC2 and Pkh1/2 (Figure S6C). We further discovered that negative feedback signaling could be triggered by addition of exogenous phytosphingosine. Thus, addition of phytosphingosine to wildtype cells triggered rapid loss of TORC2 and Pkh1/2-dependent phosphorylation of Ypk1/2 (Figure 6F). In contrast, addition of phytosphingosine to lac1Δ lag1Δ cells failed to trigger feedback (Figure 6F). Addition of phytosphingosine triggered negative feedback in rts1Δ cells, which again suggests the existence of both PP2ARts1-dependent and independent mechanisms for modulation of TORC2 (Figure S6D).
We used aureobasidin A to test whether ceramides must be converted to complex sphingolipids to influence TORC2 signaling. To do this, we pre-incubated cells with aureobasidin A for 30 minutes to block production of IPC. This triggered a slight increase in phosphorylation of Ypk1/2 by both TORC2 and Pkh1/2, as previously reported [33]. However, aureobasidin A at 0.5 μg/ml failed to block negative feedback signaling in response to added phytosphingosine (Figure S6E). Aureobasidin A also failed to block dephosphorylation of Mss4 in response to exogenous phytosphingosine (Figure S6F).
Together, these observations suggest that ceramides relay negative feedback signals, and that negative feedback works, in part, by ceramide-dependent control of PP2ARts1. The data further suggest that ceramide-dependent signals stimulate PP2ARts1 to dephosphorylate Mss4.
Ceramides are required for nutrient modulation of the TORC2 signaling network
Since ceramides play a role in modulating the TORC2 network, we hypothesized that they are required for nutrient modulation of TORC2 signaling. Dephosphorylation of Mss4 in response to poor carbon was strongly reduced in lac1Δ lag1Δ cells (Figure 7A). In addition, the dramatic decrease in TORC2 and Pkh1/2 signaling to Ypk1/2 failed to occur (Figure 7B). Thus, ceramide-dependent signals play an essential role in modulation of the TORC2 network in response to carbon source.
Figure 7. Ceramides are required for nutrient modulation of the TORC2 signaling network.

(A–B) Wildtype and lac1Δ lag1Δ cells were grown to early log phase in YPD at 22°C. After washing into YPG/E, cells were incubated at 25°C and samples were taken at the indicated times. Mss4-3XHA was detected by western blot. Ypk-pT662, Ypk-pT504 and Ypk1 were assayed by western blot. An asterisk indicates a non-specific band. (C) Quantification of ceramides and complex sphingolipids in wild type and rts1Δ cells growing in rich or poor carbon. Data represent the average of three biological replicates. A Welch Two Sample t-test is used to estimate the p-values: * p < 0.05, ** p < 0.01. (D) A model for how nutrient-dependent modulation of the TORC2 signaling network could influence cell growth and size. See also Figure S7, Table S1 and Data S1.
Levels of ceramides and complex sphingolipids are modulated by carbon source and PP2ARts1
Together, the data suggest that ceramide-dependent signals are reduced in poor carbon and elevated in cells that lack PP2ARts1. A previous study found that ceramides and IPC are reduced in poor carbon, consistent with the model [27]. To investigate further, we used mass spectrometry to quantify levels of ceramides and complex sphingolipids in wild type and rts1Δ cells growing in rich or poor carbon. Ceramides are difficult to quantify because they are present at very low quantities. In contrast, IPC, which is built directly from ceramides, is present at 10-fold higher levels and yields more confident quantitative data, so it could provide a more reliable, although indirect, readout of the rate of ceramide synthesis.
We used mass spectrometry to quantify ceramides, IPC, MIPC and M(IP)2C in wild type and rts1Δ cells growing in rich or poor carbon (Figure 7C and Data S1). Each of these classes of lipids is comprised of several subspecies that differ in length and hydroxylation of their fatty acid chains. Here, we show the sum of all subspecies for each class. Data for the subspecies within each class are shown in Figure S7A and Data S1. It is unknown whether there are functional differences between subspecies. Data for all lipid classes are shown in Figure S7B and Data S1.
The behavior of IPC closely parallels the observed effects of carbon source and rts1Δ on cell size and ceramide-dependent signaling. Thus, levels of IPC were significantly reduced in poor carbon and elevated in rts1Δ cells. Ceramides were also reduced in poor carbon and elevated in rts1Δ cells in rich carbon. However, there was not a significant difference in ceramide levels between wild type and rts1Δ cells in poor carbon, which could be due to the lower coverage and lower certainty of quantification of low abundant ceramide subspecies.
Together, data from diverse experimental approaches point to ceramides as the most likely mediator of ceramide-dependent signals that control cell growth and size, but the data cannot rule out a role for IPC.
Discussion
PP2ARts1 modulates TORC2 signaling in response to carbon source
It is well known that TORC1 is modulated by nutrients in both yeast and vertebrates. However, only a few studies in yeast have found evidence for modulation of TORC2 by nutrients [40–42]. Here, we discovered that a shift from rich to poor carbon triggers an immediate decrease in TORC2 signaling to Ypk1/2, as well as a decrease in Pkh1/2 signaling. As cells adjust to the new carbon source, TORC2 signaling recovers, but is strongly reduced relative to TORC2 signaling in rich carbon.
Diverse observations demonstrate that PP2ARts1 is required for normal signaling in the TORC2 network, as well as normal modulation of TORC2 signaling in response to carbon source. The data suggest a model in which PP2ARts1 relays information regarding carbon source to Mss4, which could modulate the level of TORC2 signaling to match the quality of the carbon source. However, carbon source also modulates TORC2 signaling by PP2ARts1-independent mechanisms. For example, the immediate decrease in TORC2 signaling that occurs in response to a shift to poor carbon is independent of PP2ARts1. This acute PP2ARts1-independent response to poor carbon could function to shut down growth as cells retool for utilization of a poor carbon source, while the long term PP2ARts1-dependent response could reflect a regulated decrease in TORC2 signaling to ensure that rates of growth processes controlled by TORC2 are matched to the carbon source.
PP2ARts1 relays ceramide-dependent signals in the TORC2 network
Previous studies found that the TORC2 network is influenced by negative feedback. Thus, the TORC2 network promotes production of sphingolipids, while sphingolipids inhibit the TORC2 network [29, 33]. However, it was unknown how feedback signals are relayed. Here, ceramides built from sphingolipids emerge as a likely mediator of feedback. Inactivation of ceramide synthase causes hyperactivity of the TORC2 network. Furthermore, exogenous phytosphingosine, a precursor for synthesis of ceramides, fails to trigger negative feedback in cells that lack ceramide synthase, which suggests that it must be converted to ceramide to influence TORC2 signaling. A number of observations suggest that ceramide-dependent feedback signals are relayed by PP2ARts1. However, it is clear that there are also PP2ARts1-independent mechanisms of feedback signaling. For example, the observation that lac1Δ lag1Δ causes stronger effects on TORC2 signaling than rts1Δ suggests that ceramides do not signal solely via PP2ARts1. Previous studies have suggested that ceramides could directly modulate the activity of PP2A [43].
The data suggest that PP2ARts1 relays both nutrient-dependent and ceramide-dependent signals. A model that could explain both roles is that PP2ARts1 is embedded in the TORC2 network feedback loop where it can relay nutrient-dependent signals that set the overall level of TORC2 network signaling.
Ceramides are required for nutrient modulation of TORC2 signaling
In addition to functioning in feedback signaling, ceramide-dependent signals are strictly required for nutrient modulation of the TORC2 network. This discovery has important implications. Previous studies suggested that negative feedback in the TORC2 network could be a homeostatic mechanism that helps maintain constant levels of sphingolipids [35]. However, sphingolipid homeostasis was only detected in cells treated with very low doses of myriocin. At higher doses, myriocin caused a dose-dependent decrease in sphingolipids [35]. Moreover, another study found that the TORC2 network responds to delivery of sphingolipids to the plasma membrane, rather than to synthesis of sphingolipids at the endoplasmic reticulum [44]. Therefore, an alternative model is that feedback ensures that the level of TORC2 signaling at the plasma membrane, which is strongly influenced by sphingolipid synthesis at the endoplasmic reticulum, is maintained at a constant level that is appropriate for the carbon source. In this model, ceramide-dependent feedback ensures that TORC2 signaling is maintained at a constant level, whereas nutrient-dependent signals relayed by PP2ARts1 help set the overall level of TORC2 signaling, and thus the level of ceramide-dependent signaling, to a level that is appropriate for the carbon source.
Normal control of cell size is dependent upon ceramides
We previously found that PP2ARts1 is required for nutrient modulation of cell size [15]. Here, we found that PP2ARts1 controls signaling in the TORC2 network, and that signals from the TORC2 network are required for normal control of cell size. Thus, decreased signaling in the TORC2 network causes reduced cell size. Conversely, increased signaling, as seen in rts1Δ cells, causes increased cell size. We further discovered that TORC2-dependent control of ceramide synthesis is required for normal control of cell size.
Together, the data demonstrate that an output of the TORC2 network influences cell size. Several observations point to ceramide-dependent signals as the output that most directly influences cell size. For example, inactivation of ceramide synthase causes decreased cell size. Similarly, decreased activity of the TORC2 network, which should cause decreased production of ceramides, also causes decreased cell size. Conversely, increased activity of the network caused by rts1Δ or GAL1-PKH1/2 causes increased cell size. Most importantly, TORC2, Pkh1/2 and Ypk1/2 are all hyperactive in both lac1Δ lag1Δ cells and in rts1Δ cells. However, lac1Δ lag1Δ cells are unusually small, whereas rts1Δ cells are unusually large. A key difference is that lac1Δ lag1Δ cells fail to produce ceramides, whereas rts1Δ cells appear to produce unusually high levels of ceramides and IPC due to hyperactivity of Ypk1/2. The implication is that levels of ceramides or IPC, rather than the activity of signaling proteins in the TORC2 network, are most closely correlated with cell size. However, it is not yet possible to rule out the possibility that changes in levels of ceramides or IPC control TORC2 network kinases by changing their localization, rather than their kinase activities.
Previous work found that loss of SCH9, an AGC kinase controlled by TORC1, causes a reduction in cell size [45]. Sch9 controls ribosome biogenesis, which suggested that cell size could be linked to ribosome biogenesis. A more recent study discovered that sch9Δ causes reduced ceramide synthesis, which suggests that the reduced size of sch9Δ cells could be a consequence of reduced ceramide synthesis [46].
Nutrient modulation of growth rate and cell size in G1 phase is dependent upon ceramides
To better understand how carbon source and ceramide-dependent signals influence cell growth and size, we analyzed cells growing in G1 phase. Consistent with previous reports, we found that cells shifted from poor to rich carbon in early G1 phase increase their growth rate and undergo a prolonged delay in bud emergence compared to their counterparts in poor carbon. They also complete G1 phase at a much larger size. Thus, a shift from poor to rich carbon appears to trigger an immediate resetting of the threshold amount of growth required for cell cycle entry [47].
Ceramide-dependent signals strongly influence cell growth and size in G1 phase. An inhibitor of sphingolipid synthesis caused dose-dependent decreases in growth rate and cell size at cell cycle entry, yet had no effect on the timing of cell cycle entry. Moreover, cells that lack ceramide synthase failed to increase their growth rate when shifted from poor to rich carbon. They also failed to increase the threshold amount of growth for cell cycle entry. Thus, ceramides are required for nutrient modulation of growth rate and cell size in G1 phase.
Why does loss of ceramides cause a decrease in growth rate and cell size? One might imagine that decreased production of ceramides causes decreased growth rate simply because ceramides are precursors for complex sphingolipids that are building blocks for membranes. Decreased growth rate, in turn, could lead to decreased cell size. However, we found no evidence that complex sphingolipids play roles in ceramide-dependent signals that modulate cell size or growth rate. In addition, complex sphingolipids constitute only 10% of total lipids in cells growing on rich carbon, and inhibiting production of MIPC and M(IP)2C, which constitute 50% of complex sphingolipids [27, 28], had no effect on growth rate or size. Thus, the data suggest that ceramide-dependent signals could play a direct role in modulating growth rate and cell size. The idea that ceramides are the key signaling molecules in this context is appealing because ceramides are conserved in eukaryotic cells, whereas complex sphingolipids show significant differences between yeast and vertebrates [26].
Previous work suggested a model in which cell size at cell cycle entry is measured via the concentration of the Whi5 protein [39]. In this model, dilution of Whi5 by growth triggers cell cycle entry. Here, we found that inhibition of sphingolipid synthesis causes a large reduction in cell size at cell cycle entry without effects on Whi5 protein levels. Thus, cells treated with myriocin enter the cell cycle with higher concentrations of Whi5 relative to control cells. At the least, this observation suggests that there are mechanisms that control cell size in G1 phase that work independently of changes in Whi5 protein concentration. It remains to be seen whether these mechanisms play a general role in cell size control that is more important than Whi5 dilution, or whether they operate only under conditions of changing nutrients.
Are growth rate and cell size coordinately controlled by common signals?
What kind of model could explain the remarkable effects of ceramide-dependent signaling on cell growth and size? Also, why is cell size proportional to growth rate? One possibility is that a nutrient modulated timer determines the duration of growth in G1 phase. In this model, nutrient-dependent signals would set a timer that determines the duration of growth, while ceramide-dependent signals would set the growth rate. However, this model is difficult to reconcile with several observations. For example, when cells are shifted from poor to rich carbon in early G1 phase, the duration of G1 phase is increased compared to cells that remain in poor carbon. In contrast, cells that have been growing continuously in rich carbon have a shorter G1 phase than cells growing in poor carbon, which is thought to be a consequence of cells in poor carbon being born at a smaller size and therefore having to undergo more growth to reach a threshold size [2, 48, 49]. Thus, the duration of growth in G1 phase is most closely correlated with the initial size of the cell and its rate of growth, rather than the carbon source, which argues against a timer model.
An alternative model is that ceramide-dependent signals set growth rate, as well as the threshold amount of growth required for cell cycle entry. A model for how PP2ARts1 and TORC2 could influence cell growth and size is shown in Figure 7D. The model refers to signaling in cells that have adapted to growth in different carbon sources and is not relevant to the acute PP2ARts1- independent response to a shift to poor carbon. In the model, inhibition of ceramide synthesis should cause decreased growth rate, as well as a lowering of the threshold amount of growth required for cell cycle entry. As a result, cells to undergo cell cycle entry with the same timing as normal cells, but at a reduced size. The observation that lac1Δ lag1Δ cells undergo a partial G1 delay when shifted from poor to rich carbon indicates that there are also ceramide-independent signals that control G1 duration. Growth in G1 phase can be divided into two intervals referred to as T1 and T2 [50]. T1 is strongly influenced by cell size and growth rate, whereas T2 is not. Thus, one possibility is that growth during T1 is modulated by nutrients in a ceramide-dependent manner, whereas T2 is modulated in a ceramide-independent manner.
The idea that signals originating in the TORC2 network coordinately control growth rate and size would explain why loss of PP2ARts1 disrupts normal TORC2 signaling, as well as the normal relationship between growth rate and cell size [15]. Distinguishing alternative models will require filling in a number of gaps in our understanding of the TORC2 network. For example, the data thus far suggest that ceramide-dependent signals are the output of the TORC2 network that most directly influences growth rate and size, yet little is known about the targets of ceramide-dependent signaling. Candidate targets of ceramides have been identified [51], but thus far there are no biochemically defined ceramide-binding proteins in yeast. Thus, an important next step will be to define the targets of ceramide-dependent signaling in the TORC2 network. Another important goal will be to connect ceramide-dependent signals to the downstream effectors of cell size control. Finally, discovery of the signals that control PP2ARts1 could provide insight into how nutrients and ceramide-dependent signals influence TORC2 signaling.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Douglas R Kellogg (dkellogg@ucsc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experiments were performed using Saccharomyces cerevisiae or Schizosaccharomyces pombe yeast strains listed in Table S1. All Saccharomyces cerevisiae strains are in the W303 background (leu2-3,112 ura3-1 can1-100 ade2-1 his3-11,15 trp1-1 GAL+ ssd1-d2).
METHOD DETAILS
Yeast strains and culture conditions
One-step PCR-based gene replacement was used for construction of deletions and epitope tags at the endogenous locus [52, 53]. Cells were grown in YP medium (1% yeast extract, 2% peptone, 40 mg/L adenine) supplemented with 2% dextrose (YPD), 2% galactose (YPGal), or 2% glycerol/ethanol (YPG/E). Schizosaccharomyces pombe strains were grown in standard YE media with supplements. Microscopy experiments were carried out using complete synthetic media with dextrose (CSM).
Myriocin was dissolved in 100% methanol to make a 500 μg/ml stock solution. Aureobasidin A was dissolved in 100% methanol to make a 5 mg/ml stock solution. Phytosphingosine was dissolved in 100% ethanol to make a 10 mM stock solution. Fumonisin B1 was made as a 14 mM stock solution dissolved in 100% methanol. All experiments using fumonisin B1 were carried out in strains carrying yor1Δ, which is required for sensitivity to fumonisin B1 [54].. In Figure 4, methanol was added to control samples at concentrations ranging from 0.04% to 0.2%. In Figure S4A, B and C, methanol was added to the control samples at 0.08%, 0.06% and 0.2%, respectively.
For experiments using analog-sensitive alleles, cells were grown in YPD medium without supplemental adenine. The adenine analog inhibitor 3-MOB-PP1 stock was prepared in 100% DMSO at 10 mM and added to cultures at a final concentration of 0.1 μM. In Figure 3B, 0.002% DMSO was added to the control experiment.
To create the mss4–8 allele, the His3MX6 marker was integrated downstream of the MSS4 open reading frame ]52]. A fragment that contains the MSS4 kinase domain, the HisMX6 marker, and a short region downstream of MSS4 was then amplified with Taq polymerase to introduce mutations and transformed into a wildtype strain (Primers: GATCAGAGTCTGCAACGGCAG and GTTCACCATCGGCCTCGAGC). Transformants were selected on -HIS media and further screened for temperature sensitivity at 30°C and 37°C. To verify that the temperature-sensitive phenotype was due to mutations in MSS4, candidates were tested for rescue by a plasmid containing the wildtype MSS4 gene (pMH1). We selected a mutant (mss4–8) that was unable to grow at 30°C. DNA sequencing identified 3 mutations within the kinase domain (L506P, S539L, I672V).
To create the mss4-ps1 allele, we used mass spectrometry data to identify sites that show evidence of hyperphosphorylation in rts1Δ cells, including sites found in only one biological replicate [16], which identified 10 sites on MSS4. A fragment of MSS4 that includes 10 phosphorylation site mutations (T125A, S140A, T222A, S225A, S262A, S269A, T271A, S272A, S341A, T343A) was synthesized by Atum (www.atum.bio) and used to replace the original MSS4 sequence in pMH2 by gap repair. The new plasmid pMA19 was verified by DNA sequencing and was used to precisely replace the endogenous MSS4 gene to generate the mss4-ps1 mutant.
Western blotting
To ensure that protein loading was normalized in time course experiments, we determined optical densities of cultures from each strain that yield equal amounts of extracted protein. This was necessary because large cell mutants (i.e. rts1Δ) scatter light differently. We found that optical densities of 0.7 for rts1Δ and 0.5 for the rest of the strains yielded normalized protein loadings.
To analyze proteins from cells growing in early log phase, cultures were grown overnight at 25°C to an OD600 of less than 0.8. After adjusting optical densities to normalize protein loading, 1.6-ml samples were collected and centrifuged at 13,000 rpm for 30 s. The supernatant was removed and 250 μl of glass beads were added before freezing in liquid nitrogen.
To analyze cells shifted from rich to poor nutrients, cultures were grown in YPD medium overnight at 25°C to an OD600 of less than 0.8. After adjusting optical densities to normalize protein loading, cells were washed three times with a large volume of YPG/E medium and then incubated at 30°C in YPG/E for the time course. 1.6-ml samples were collected at each time point.
Cells were lysed into 140 μl of sample buffer (65 mM Tris-HCl, pH 6.8, 3% SDS, 10% glycerol, 50 mM NaF, 100 mM glycerophosphate, 5% 2-mercaptoethanol, and bromophenol blue). PMSF was added to the sample buffer to 2 mM immediately before use. Cells were lysed in a Mini-bead-beater 16 (BioSpec) at top speed for 2 min. The samples were removed and centrifuged for 15 s at 13,000 rpm in a microfuge and placed in boiling water for 5 min. After boiling, the samples were centrifuged for 5 min at 13,000 rpm and loaded on an SDS polyacrylamide gel.
Samples were analyzed by western blotting as previously described [55]. SDS-PAGE gels were run at a constant current of 20 mA and electrophoresis was performed on gels containing 10% polyacrylamide and 0.13% bis-acrylamide. Proteins were transferred to nitrocellulose using the Trans-Blot Turbo system. Blots were probed with primary antibody overnight at 4°C. Proteins tagged with the HA epitope were detected with the 12CA5 anti-HA monoclonal antibody. Rabbit anti-phospho-T662 antibody was used to detect TORC2-dependent phosphorylation of YPK1/2 at a dilution of 1:20,000 in TBST (10 mM Tris-Cl, pH 7.5, 100 mM NaCl, and 0.1% Tween 20) containing 3% dry milk. Pkh1/2-dependent phosphorylation of Ypk1/2 was detected using an anti-SGK phospho-specific antibody at a dilution of 1/1000. Total Ypk1 was detected using anti-Ypk1 antibody at a dilution of 1:2000.
All blots were probed with an HRP-conjugated donkey anti-rabbit secondary antibody or HRP-conjugated donkey anti–mouse antibody or HRP-conjugated donkey anti-goat for 45–90 min at room temperature. Secondary antibodies were detected via chemiluminescence with Advansta ECL reagents using a Chemi-Doc imaging system.
Densitometric quantification of western blot signals was performed using ImageJ [56]. Quantification of TORC2- and Pkh1/2-dependent phosphorylation of Ypk1/2 was calculated as the ratio of the phospho-specific signal over the total Ypk1 protein signal, with wild type signal normalized to a value 1. At least three biological replicates were analyzed and averaged to obtain quantitative information.
Synchronization by centrifugal elutriation
Cells were grown overnight at 25°C in YPG/E medium to increase the fraction of very small unbudded cells. Centrifugal elutriation was performed as previously described [57, 58]. In brief, cells were elutriated at 4°C at a speed of 2,800 rpm in a Beckman Coulter J6-MI centrifuge with a JE-5.0 rotor. Small unbudded cells were released into fresh YPD or YPG/E media at 25°C and samples were taken at 10-min intervals.
Analysis of cell size distributions and bud emergence
Cell cultures were grown overnight to early log phase at 25°C. A 900 μl sample of each culture was fixed with 100 μl of 37% formaldehyde for 1 h and then washed twice with PBS + 0.04% sodium azide + 0.02% Tween-20. Cell size was measured using a Coulter counter (Channelizer Z2; Beckman Coulter) as previously described [59]. In brief, 30 μl of fixed culture was diluted in 10 ml diluent (Isoton II; Beckman Coulter) and sonicated for 3 s before cell sizing. Each plot is the average of three independent biological replicates in which three independent technical replicates were analyzed. The size of elutriated cells was measured in the same manner except that the cells were not sonicated. The percentage of budded cells was measured by counting the number of small unbudded cells over a total of ≥200 cells using a Zeiss Axioskop 2 (Carl Zeiss) and an AxioCam HRm camera with a 63×1.4 numerical aperture objective.
Microscopy
To visualize Mss4-GFP in wild type and rts1Δ cells under identical conditions, we marked wild type cells with Spc42-mRuby2 so they could be cultured and visualized together with rts1Δ cells. Wildtype and rts1Δ cells containing MSS4-GFP were grown together to early log phase. Fields of view containing both genotypes were imaged using a Solamere spinning disk confocal system equipped with a Yokogawa CSUX-1 scan head, Nikon TE2000-E inverted stand, Hamamatsu ImageEM ×2 camera, and Plan Apo 60×/1.4-n.a. oil objective and controlled by Micro-Manager software. Cells were imaged in 12 z-series with a step size of 0.5 μm and analyzed using ImageJ. Signal at the plasma membrane was quantified using ImageJ.
Cell viability was measured by trypan blue dye exclusion. 100 μl of log phase cells were added to a 100 μl trypan blue solution (0.4% in PBS). At least 300 cells were counted for each condition.
To measure cell size in Schizosaccharomyces pombe, cells were fixed with calcofluor-white (Sigma) and length at septation was calculated using ImageJ.
Anionic phospholipid analysis
The analysis of anionic phospholipid content of cells was carried out as described [60, 61]. Samples were prepared by adding 0.8 mL of ice-cold CH3OH/12M HCl (96:4) supplemented with 2 mM AlCl3, to 107 cells. Lipids were then extracted by the addition of 0.6 mL ice-cold CHCl3 and vortexing. Separation of aqueous and organic phases was performed by addition of 0.3 mL ice-cold H2O, followed by centrifugation for 2 min at 9,300 g at 4°C. The lower organic phase was collected and transferred to new tubes containing 1 mL ice-cold CH3OH:2 mM C2H2O4 in H2O (1:0.9). An additional separation step was performed by vortexing the sample, followed by centrifugation for 2 min at 9,300 g at 4°C. The resulting lower organic phase was transferred to a new tube and dried. Lipids were then deacylated by resuspension with 0.5 mL of methylamine reagent [CH3OH/40% methylamine in H2O/1-butanol/H2O (47:36:9:8)] and incubation at 50°C for 45 min. The resulting solution was dried, then resuspended in 0.5 mL of 1-butanol/petroleum ether/ethyl formate (20:40:1), and extracted twice with an equal volume of water. Aqueous extracts were dried, resuspended in water, and subjected to anion-exchange HPLC on an Ionpac AS11-HC column (Dionex, Sunnyvale, CA). Negatively charged glycerol head groups were eluted with a 1.5–86 mM KOH gradient and detected online by suppressed conductivity 75 in a Dionex ion chromatography system equipped with an ASRS-ultra II self-regenerating suppressor. Individual peaks were identified and peak areas were calculated using the Chromeleon software version 6.60 (Dionex). Using deacylated anionic phospholipids as standards, lipid masses were calculated, and were expressed as molar fractions of total anionic phospholipids present in the sample.
Lipid extraction for mass spectrometry lipidomics
To quantify lipids by mass spectrometry, wild type and rts1Δ cells were grown for 16 hours in YPD or YPG/E at 22°C. 50 ml of cells at an OD600nm of 0.8 were harvested by centrifugation at 5000×g and washed twice with ice-cold water. The pellet was resuspended in 1 ml of ice-cold water and transferred to a fresh 2 ml Eppendorf tube. Cells were lysed with 200 μl of glass beads using a Mini-bead-beater 16 (BioSpec) at top speed for 5 min at 4°C. 500 μl of yeast lysate were transferred to a fresh tube and frozen on liquid nitrogen. Sample identities were blinded for analysis so there could be no possibility of bias in the analysis. Three biological replicates were analyzed for each genotype and condition.
Mass spectrometry-based lipid analysis was performed by Lipotype GmbH (Dresden, Germany) as described [27, 28]. Lipids were extracted using a two-step chloroform/methanol procedure [28]. Samples were spiked with internal lipid standard mixture containing: CDP-DAG 17:0/18:1, ceramide 18:1;2/17:0 (Cer), diacylglycerol 17:0/17:0 (DAG), lyso-phosphatidate 17:0 (LPA), lyso-phosphatidylcholine 12:0 (LPC), lyso-phosphatidylethanolamine 17:1 (LPE), lyso-phosphatidylinositol 17:1 (LPI), lyso-phosphatidylserine 17:1 (LPS), phosphatidate 17:0/14:1 (PA), phosphatidylcholine 17:0/14:1 (PC), phosphatidylethanolamine 17:0/14:1 (PE), phosphatidylglycerol 17:0/14:1 (PG), phosphatidylinositol 17:0/14:1 (PI), phosphatidylserine 17:0/14:1 (PS), ergosterol ester 13:0 (EE), triacylglycerol 17:0/17:0/17:0 (TAG), stigmastatrienol, inositolphosphorylceramide 44:0;2 (IPC), mannosyl-inositolphosphorylceramide 44:0;2 (MIPC) and mannosyl-di-(inositolphosphoryl)ceramide 44:0;2 (M(IP)2C). After extraction, the organic phase was transferred to an infusion plate and dried in a speed vacuum concentrator. 1st step dry extract was re-suspended in 7.5 mM ammonium acetate in chloroform/methanol/propanol (1:2:4, V:V:V) and 2nd step dry extract in 33% ethanol solution of methylamine in chloroform/methanol (0.003:5:1; V:V:V). All liquid handling steps were performed using Hamilton Robotics STARlet robotic platform with the Anti Droplet Control feature for organic solvents pipetting.
MS data acquisition
Samples were analyzed by direct infusion on a QExactive mass spectrometer (Thermo Scientific) equipped with a TriVersa NanoMate ion source (Advion Biosciences). Samples were analyzed in both positive and negative ion modes with a resolution of Rm/z=200=280000 for MS and Rm/z=200=17500 for MSMS experiments, in a single acquisition. MSMS was triggered by an inclusion list encompassing corresponding MS mass ranges scanned in 1 Da increments [62]. Both MS and MSMS data were combined to monitor EE, DAG and TAG ions as ammonium adducts; PC as an acetate adduct; and CL, PA, PE, PG, PI and PS as deprotonated anions. MS only was used to monitor LPA, LPE, LPI, LPS, IPC, MIPC, M(IP)2C as deprotonated anions; Cer and LPC as acetate adducts.
QUANTITATION AND STATISTICAL ANALYSIS
A minimum of three independent biological replicates were performed for each experimental condition. For the statistical analyses, one-tailed unpaired t-test was performed using Prism 5 (Grahpad). P values are described in each figure legend.
Mass spectrometry lipidomics data were analyzed with in-house developed lipid identification software based on LipidXplorer [63, 64]. Data post-processing and normalization were performed using an in-house developed data management system. Only lipid identifications with a signal-to-noise ratio >5, and a signal intensity 5-fold higher than in corresponding blank samples were considered for further data analysis.
Supplementary Material
Acknowledgments
We thank members of the laboratory for advice and support. We also thank Ted Powers (UC Davis) for the Ypk-pT662 phosphospecific antibody and John Tamkun (UC Santa Cruz) for 12CA5 antibody. This work was supported by National Institutes of Health grant GM053959.
Footnotes
Declaration of Interests
C.K. and M.A.S. have paid employment at Lipotype GmbH. This does not alter the authors’ adherence to all policies on sharing data and materials.
Author Contributions
Conceptualization, R.L., M.A-G., D.K.; Methodology, R.L., M.A-G, K.S., M.H., M.D., C.M., C.K. and M.A.S; Writing- Original draft, R.L., M.A-G. and D.K; Writing- Review & Editing, R.L., M.A-G, C.K., M.A.S and D.K.; Funding Acquisition, D.K; Supervision, D.K.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ferrezuelo F, Colomina N, Palmisano A, Garí E, Gallego C, Csikász-Nagy A, Aldea M. The critical size is set at a single-cell level by growth rate to attain homeostasis and adaptation. Nat Commun. 2012;3:1012. doi: 10.1038/ncomms2015. [DOI] [PubMed] [Google Scholar]
- 2.Leitao RM, Kellogg DR. The duration of mitosis and daughter cell size are modulated by nutrients in budding yeast. J Cell Biol. 2017;216:3463–3470. doi: 10.1083/jcb.201609114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaechter M, Maaloe O, Kjeldgaard NO. Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J Gen Microbiol. 1958;19:592–606. doi: 10.1099/00221287-19-3-592. [DOI] [PubMed] [Google Scholar]
- 4.Johnston GC, Pringle JR, Hartwell LH. Coordination of growth with cell division in the yeast Saccharomyces cerevisiae. Exp Cell Res. 1977;105:79–98. doi: 10.1016/0014-4827(77)90154-9. [DOI] [PubMed] [Google Scholar]
- 5.Hirsch J, Han PW. Cellularity of rat adipose tissue: effects of growth, starvation, and obesity. J Lipid Res. 1969;10:77–82. [PubMed] [Google Scholar]
- 6.Airoldi EM, Huttenhower C, Gresham D, Lu C, Caudy AA, Dunham MJ, Broach JR, Botstein D, Troyanskaya OG. Predicting cellular growth from gene expression signatures. PLoS Comput Biol. 2009;5:e1000257. doi: 10.1371/journal.pcbi.1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbet NC, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN. TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell. 1996;7:25–42. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 9.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 10.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 11.Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, Broach JR, De Virgilio C, Hall MN, Loewith R. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell. 2007;26:663–674. doi: 10.1016/j.molcel.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 12.Kamada Y, Fujioka Y, Suzuki NN, Inagaki F, Wullschleger S, Loewith R, Hall MN, Ohsumi Y. Tor2 directly phosphorylates the AGC kinase Ypk2 to regulate actin polarization. Mol Cell Biol. 2005;25:7239–7248. doi: 10.1128/MCB.25.16.7239-7248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science. 1998;279:707–710. doi: 10.1126/science.279.5351.707. [DOI] [PubMed] [Google Scholar]
- 14.Casamayor A, Torrance PD, Kobayashi T, Thorner J, Alessi DR. Functional counterparts of mammalian protein kinases PDK1 and SGK in budding yeast. Curr Biol. 1999;9:186–197. doi: 10.1016/s0960-9822(99)80088-8. [DOI] [PubMed] [Google Scholar]
- 15.Artiles K, Anastasia S, McCusker D, Kellogg DR. The Rts1 regulatory subunit of protein phosphatase 2A is required for control of G1 cyclin transcription and nutrient modulation of cell size. PLoS Genet. 2009;5:e1000727. doi: 10.1371/journal.pgen.1000727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zapata J, Dephoure N, Macdonough T, Yu Y, Parnell EJ, Mooring M, Gygi SP, Stillman DJ, Kellogg DR. PP2ARts1 is a master regulator of pathways that control cell size. J Cell Biol. 2014;204:359–376. doi: 10.1083/jcb.201309119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niles BJ, Mogri H, Hill A, Vlahakis A, Powers T. Plasma membrane recruitment and activation of the AGC kinase Ypk1 is mediated by target of rapamycin complex 2 (TORC2) and its effector proteins Slm1 and Slm2. Proc Natl Acad Sci USA. 2012;109:1536–1541. doi: 10.1073/pnas.1117563109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Audhya A, Loewith R, Parsons AB, Gao L, Tabuchi M, Zhou H, Boone C, Hall MN, Emr SD. Genome-wide lethality screen identifies new PI4,5P2 effectors that regulate the actin cytoskeleton. EMBO J. 2004;23:3747–3757. doi: 10.1038/sj.emboj.7600384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berchtold D, Walther TC. TORC2 plasma membrane localization is essential for cell viability and restricted to a distinct domain. Mol Biol Cell. 2009;20:1565–1575. doi: 10.1091/mbc.E08-10-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Audhya A, Emr SD. Regulation of PI4, 5P2 synthesis by nuclear–cytoplasmic shuttling of the Mss4 lipid kinase. EMBO J. 2003;22:4223–4236. doi: 10.1093/emboj/cdg397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 2001;20:4380–4390. doi: 10.1093/emboj/20.16.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roelants FM, Baltz AG, Trott AE, Fereres S, Thorner J. A protein kinase network regulates the function of aminophospholipid flippases. Proc Natl Acad Sci USA. 2010;107:34–39. doi: 10.1073/pnas.0912497106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR. Essential role of PDK1 in regulating cell size and development in mice. EMBO J. 2002;21:3728–3738. doi: 10.1093/emboj/cdf387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rintelen F, Stocker H, Thomas G, Hafen E. PDK1 regulates growth through Akt and S6K in Drosophila. Proc Natl Acad Sci USA. 2001;98:15020–15025. doi: 10.1073/pnas.011318098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dickson RC. Thematic review series: sphingolipids. New insights into sphingolipid metabolism and function in budding yeast. J Lipid Res. 2008;49:909–921. doi: 10.1194/jlr.R800003-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Breslow DK, Weissman JS. Membranes in balance: mechanisms of sphingolipid homeostasis. Mol Cell. 2010;40:267–279. doi: 10.1016/j.molcel.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klose C, Surma MA, Gerl MJ, Meyenhofer F, Shevchenko A, Simons K. Flexibility of a eukaryotic lipidome--insights from yeast lipidomics. PLoS ONE. 2012;7:e35063. doi: 10.1371/journal.pone.0035063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ejsing CS, Sampaio JL, Surendranath V, Duchoslav E, Ekroos K, Klemm RW, Simons K, Shevchenko A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc Natl Acad Sci USA. 2009;106:2136–2141. doi: 10.1073/pnas.0811700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roelants FM, Breslow DK, Muir A, Weissman JS, Thorner J. Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2011;108:19222–19227. doi: 10.1073/pnas.1116948108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Y, Miao Y, Yamane Y, Zhang C, Shokat KM, Takematsu H, Kozutsumi Y, Drubin DG. Orm protein phosphoregulation mediates transient sphingolipid biosynthesis response to heat stress via the Pkh-Ypk and Cdc55-PP2A pathways. Mol Biol Cell. 2012;23:2388–2398. doi: 10.1091/mbc.E12-03-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muir A, Ramachandran S, Roelants FM, Timmons G, Thorner J. TORC2-dependent protein kinase Ypk1 phosphorylates ceramide synthase to stimulate synthesis of complex sphingolipids. eLife. 2014;3:1–34. doi: 10.7554/eLife.03779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aronova S, Wedaman K, Aronov PA, Fontes K, Ramos K, Hammock BD, Powers T. Regulation of ceramide biosynthesis by TOR complex 2. Cell Metab. 2008;7:148–158. doi: 10.1016/j.cmet.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berchtold D, Piccolis M, Chiaruttini N, Riezman I, Riezman H, Roux A, Walther TC, Loewith R. Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat Cell Biol. 2012;14:542–547. doi: 10.1038/ncb2480. [DOI] [PubMed] [Google Scholar]
- 34.Miyake Y, Kozutsumi Y, Nakamura S, Fujita T, Kawasaki T. Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem Biophys Res Commun. 1995;211:396–403. doi: 10.1006/bbrc.1995.1827. [DOI] [PubMed] [Google Scholar]
- 35.Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, Ejsing CS, Weissman JS. Orm family proteins mediate sphingolipid homeostasis. Nature. 2010;463:1048–1053. doi: 10.1038/nature08787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartwell LH, Unger MW. Unequal division in Saccharomyces cerevisiae and its implications for the control of cell division. J Cell Biol. 1977;75:422–435. doi: 10.1083/jcb.75.2.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costanzo M, Nishikawa JL, Tang X, Millman JS, Schub O, Breitkreuz K, Dewar D, Rupes I, Andrews B, Tyers M. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell. 2004;117:899–913. doi: 10.1016/j.cell.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 38.de Bruin RAM, McDonald WH, Kalashnikova TI, Yates J, Wittenberg C. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell. 2004;117:887–898. doi: 10.1016/j.cell.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 39.Schmoller KM, Turner JJ, Kõivomägi M, Skotheim JM. Dilution of the cell cycle inhibitor Whi5 controls budding-yeast cell size. Nature. 2015 doi: 10.1038/nature14908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hatano T, Morigasaki S, Tatebe H, Ikeda K, Shiozaki K. Fission yeast Ryh1 GTPase activates TOR Complex 2 in response to glucose. Cell Cycle. 2015;14:848–856. doi: 10.1080/15384101.2014.1000215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yerlikaya S, Meusburger M, Kumari R, Huber A, Anrather D, Costanzo M, Boone C, Ammerer G, Baranov PV, Loewith R. TORC1 and TORC2 work together to regulate ribosomal protein S6 phosphorylation in Saccharomyces cerevisiae. Mol Biol Cell. 2016;27:397–409. doi: 10.1091/mbc.E15-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen A, Kupiec M, Weisman R. Glucose activates TORC2-Gad8 protein via positive regulation of the cAMP/cAMP-dependent protein kinase A (PKA) pathway and negative regulation of the Pmk1 protein-mitogen-activated protein kinase pathway. J Biol Chem. 2014;289:21727–21737. doi: 10.1074/jbc.M114.573824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nickels JT, Broach JR. A ceramide-activated protein phosphatase mediates ceramide-induced G1 arrest of Saccharomyces cerevisiae. Genes Dev. 1996;10:382–394. doi: 10.1101/gad.10.4.382. [DOI] [PubMed] [Google Scholar]
- 44.Clarke J, Dephoure N, Horecka I, Gygi S, Kellogg D. A conserved signaling network monitors delivery of sphingolipids to the plasma membrane in budding yeast. Mol Biol Cell. 2017;28:2589–2599. doi: 10.1091/mbc.E17-01-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jorgensen P, Tyers M. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 2004;18:2491–2505. doi: 10.1101/gad.1228804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swinnen E, Wilms T, Idkowiak-Baldys J, Smets B, De Snijder P, Accardo S, Ghillebert R, Thevissen K, Cammue B, De Vos D, Bielawski J, Hannun YA, Winderickx J. The protein kinase Sch9 is a key regulator of sphingolipid metabolism in Saccharomyces cerevisiae. Mol Biol Cell. 2014;25:196–211. doi: 10.1091/mbc.E13-06-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnston GC, Ehrhardt CW, Lorincz A, Carter BL. Regulation of cell size in the yeast Saccharomyces cerevisiae. J Bacteriol. 1979;137:1–5. doi: 10.1128/jb.137.1.1-5.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lord PG, Wheals AE. Variability in individual cell cycles of Saccharomyces cerevisiae. J Cell Sci. 1981;50:361–376. doi: 10.1242/jcs.50.1.361. [DOI] [PubMed] [Google Scholar]
- 49.Di Talia S, Wang H, Skotheim JM, Rosebrock AP, Futcher B, Cross FR. Daughter-Specific Transcription Factors Regulate Cell Size Control in Budding Yeast. PLoS Biol. 2009;7:e1000221. doi: 10.1371/journal.pbio.1000221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Talia S, Skotheim JM, Bean JM, Siggia ED, Cross FR. The effects of molecular noise and size control on variability in the budding yeast cell cycle. Nature. 2007;448:947–951. doi: 10.1038/nature06072. [DOI] [PubMed] [Google Scholar]
- 51.Gallego O, Betts MJ, Gvozdenovic-Jeremic J, Maeda K, Matetzki C, Aguilar-Gurrieri C, Beltran-Alvarez P, Bonn S, Fernández-Tornero C, Jensen LJ, Kuhn M, Trott J, Rybin V, Müller CW, Bork P, Kaksonen M, Russell RB, Gavin AC. A systematic screen for protein-lipid interactions in Saccharomyces cerevisiae. Mol Syst Biol. 2010;6:430. doi: 10.1038/msb.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Longtine MS, McKenzie A, DeMarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 53.Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, Knop M. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- 54.Martínez-Montañés F, Schneiter R. Following the flux of long-chain bases through the sphingolipid pathway in vivo using mass spectrometry. J Lipid Res. 2016;57:906–915. doi: 10.1194/jlr.D066472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harvey SL, Enciso G, Dephoure N, Gygi SP, Gunawardena J, Kellogg DR. A phosphatase threshold sets the level of Cdk1 activity in early mitosis in budding yeast. Mol Biol Cell. 2011;22:3595–3608. doi: 10.1091/mbc.E11-04-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Futcher B. Cell cycle synchronization. Methods Cell Sci. 1999;21:79–86. doi: 10.1023/a:1009872403440. [DOI] [PubMed] [Google Scholar]
- 58.McCusker D, Royou A, Velours C, Kellogg D. Cdk1-dependent control of membrane-trafficking dynamics. Mol Biol Cell. 2012;23:3336–3347. doi: 10.1091/mbc.E11-10-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jorgensen P, Nishikawa JL, Breitkreutz BJ, Tyers M. Systematic identification of pathways that couple cell growth and division in yeast. Science. 2002;297:395–400. doi: 10.1126/science.1070850. [DOI] [PubMed] [Google Scholar]
- 60.Nasuhoglu C, Feng S, Mao J, Yamamoto M, Yin HL, Earnest S, Barylko B, Albanesi JP, Hilgemann DW. Nonradioactive analysis of phosphatidylinositides and other anionic phospholipids by anion-exchange high-performance liquid chromatography with suppressed conductivity detection. Anal Biochem. 2002;301:243–254. doi: 10.1006/abio.2001.5489. [DOI] [PubMed] [Google Scholar]
- 61.Marquer C, Tian H, Yi J, Bastien J, Armi CDA, Yang-Klingler Y, Zhou B, Chan RB, Di Paolo G. Arf6 controls retromer traffic and intracellular cholesterol distribution via a phosphoinositide-based mechanism. Nat Commun. 2016;7:1–14. doi: 10.1038/ncomms11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Surma MA, Herzog R, Vasilj A, Klose C, Christinat N, Morin-Rivron D, Simons K, Masoodi M, Sampaio JL. An automated shotgun lipidomics platform for high throughput, comprehensive, and quantitative analysis of blood plasma intact lipids. Eur J Lipid Sci Technol. 2015;117:1540–1549. doi: 10.1002/ejlt.201500145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Herzog R, Schwudke D, Schuhmann K, Sampaio JL, Bornstein SR, Schroeder M, Shevchenko A. A novel informatics concept for high-throughput shotgun lipidomics based on the molecular fragmentation query language. Genome Biol. 2011;12:R8. doi: 10.1186/gb-2011-12-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Herzog R, Schuhmann K, Schwudke D, Sampaio JL, Bornstein SR, Schroeder M, Shevchenko A. LipidXplorer: a software for consensual cross-platform lipidomics. PLoS ONE. 2012;7:e29851. doi: 10.1371/journal.pone.0029851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.