

Abstract
Nature has long demonstrated the importance of chemical sequence to induce structure and tune physical interactions. Investigating macromolecular structure and dynamics is paramount to understand macromolecular binding and target recognition. To that end, we have synthesized and characterized flexible sulfonated oligothioetheramides (oligo-TEAs) by variable temperature pulse field gradient (PFG) NMR, double electron–electron resonance (DEER), and molecular dynamics (MD) simulations to capture their room temperature structure and dynamics in water. We have examined the contributions of synthetic length (2–12mer), pendant group charge, and backbone hydrophobicity. We observe significant entropic collapse, driven in part by backbone hydrophobicity. Analysis of individual monomer contributions revealed larger changes due to the backbone compared to pendant groups. We also observe screening of intramolecular electrostatic repulsions. Finally, we comment on the combination of DEER and PFG NMR measurements via Stokes–Einstein–Sutherland diffusion theory. Overall, this sensitive characterization holds promise to enable de novo development of macromolecular structure and sequence–structure–function relationships with flexible, but biologically functional macromolecules
Graphical abstract
INTRODUCTION
Biological macromolecules perform advanced functions by controlling the relationship between their chemical functionalities and structure. Higher-order structures including α-helices, β-sheets, stem-loops, and many others represent the potential of optimized sequence–structure–function relationships to generate sensitive stimuli-response, allostery, information management, and expression.1-5 Precise sequence control, as seen in biological scaffolds, can be used to control local enthalpic and global entropic contributions, important factors that dictate folding, self-assembly, and biological interactions.6 The ability to control structure and function by composition and sequence has prompted significant research in the development of sequence-defined polymers.7,8 Motivated by this call, we recently described sequence-defined oligothioetheramides (oligoTEAs) featuring a rapid and efficient synthesis, access to the backbone, and the use of diverse pendant groups.9,10 The synthetic strategy utilizes the reactive orthogonality of N-allyl acrylamide monomers, rather than protection/deprotection groups for iterative assembly.10 The accessibility of the oligoTEA backbone and pendant groups render them ideal for exploring macromolecular conformation and dynamics.
The solution-phase structure and dynamics of macromolecules are challenging to visualize because they are a complex product of intramolecular interactions, entropy, and solvent interactions derived from the chemical composition and sequence. One well-documented example of this challenge can be seen in the development of therapeutic glycosaminoglycans where sequence, sulfation pattern, and backbone structure are significant.11,12 Developing sequence-structure–function relationships of heparin pentasaccharide was challenging due to limited synthetic throughput to produce sequence-defined sulfated saccharine structures to elucidate the molecular basis of binding to antithrombin and develop similar therapeutics.13-15 Highly sulfated macromolecules have potential to show a diverse array of biological interactions and will continue to be explored.16 We hope ease of sulfated sequence control and macromolecular characterization of structure and dynamics can expedite future development of similarly complex macromolecules.
De novo development of design heuristics for functional oligoTEA structures requires an understanding of the contributions of monomer hydrogen bonding, hydrophobicity, and electrostatic interaction within the scaffold. Previous sequence-structure relationships can best be seen in the fields of foldamers17-19 and single-chain folding,20-23 which have pioneered relationships between chemical functionality, thermodynamics, self-assembly, and molecular ordering. Larger hydrophobic groups restrict conformational freedom; however, this strategy can limit water solubility.24-26 Hydrogen bonding is well-known to induce modes of structural formation as an attractive force (e.g., α-helices and β-sheets within peptides) 23,27 Last, macrocyclization restricts molecular conformational and lowers the entropic cost of interaction or binding.28,29 Overall, these examples demonstrate how a balance of intramolecular forces can regulate entropic chain collapse. However, it is also important to note the reduction of macromolecular conformation is not an explicit requirement of biological function. Evidence exists for flexible macromolecules that utilize multiple conformations in their bound states to maintain interactions with their target. These and other examples demonstrate the need and utility of precise macromolecular flexibility, also demonstrated by the functional modes of disordered protein domains.30-33
Solution-phase characterization of macromolecular structure and dynamics can be challenging due to the small length- and time-scales, which can limit common experimental structural elucidation techniques. Current techniques characterize structure at various resolution and throughput, depending on the size and flexibility of the macromolecule. NMR can serve as a high-resolution method for protein structure elucidation34,35 and primary ligand screening,36 but robust methods to study synthetic nonpeptide structure are lacking. Small-angle X-ray scattering (SAXS) has proven to be a robust low-resolution tool providing solution-phase ensembles, but can be limited by structure flexibility because of time- and space-averaged scattering.37-39 Comprehensive techniques have improved upon individual weaknesses by combining information to generally refine solution-phase techniques.40-44 Inspired by these methodologies, we sought to explore additional means of solution-phase characterization that was not limited by macromolecular flexibility.
Herein, we present the synthesis and solution-phase characterization of sulfonated oligoTEAs as a function of length (2–12mer). We also explored the effect of pendant group charge and backbone composition on dynamics of the oligoTEA chain. Structural investigation was performed by variable temperature pulsed field gradient (PFG) NMR, pulsed electron paramagnetic resonance (EPR), and molecular dynamics (MD) simulations. Variable temperature PFG NMR most directly measures the macromolecular hydrodynamic radius.45 EPR and MD can both observe solution-phase dynamics. Double electron–electron resonance (DEER) EPR can quantify the distance distribution between paramagnetic spin-labels46,47 and has been done for a few oligomer case studies.48-51 MD simulation visualizes oligoTEA time evolution and molecular configuration space. All characterization was carried out to capture room temperature structure and dynamics in water. We also comment on the combination of these data within the Stokes–Einstein–Sutherland equation with size and shape factors to quantify size and aspect ratio within simple geometries.52
RESULTS AND DISCUSSION
OligoTEA assembly as shown in Figure 1 begins with a fluorous-tagged soluble support functionalized with an allyl group. The first monomer is attached by a UV-initiated thiolene utilizing dithiol in the presence of a photoinitiator. Fluorous solid-phase extraction isolates the resulting fluorous thiol. The next monomer is attached via a thiol–Michael addition of an N-allyl acrylamide monomer in the presence of phosphine catalyst. The orthogonal reactivity ensures that the acrylamide reacts with the fluorous thiol with minimal cross-reactivity of the allyl group. To incorporate sulfonated pendant groups, a protected sulfonate N-allyl acrylamide monomer (PSM) was prepared utilizing an α-trifluoromethyltolyl (TFMT) group (Figure S1).53 With the attachment of an N-allyl acrylamide by the thiol–Michael addition, the allyl group is reestablished to begin the cycle again.
Figure 1.

Oligothioetheramide assembly. (ia). A fluorous olefin is reacted with a dithiol in a UV-initiated thiolene and the product is purified (ib) by fluorous solid-phase extraction (FSPE). (iia) A fluorous thiol is reacted with an N-allyl acrylamide monomer in a phosphine initiated thiol–Michael addition and purified by FSPE (iib). These two reactions are cycled until the desired oligomer length is reached. (iii) The fluorous support is cleaved with trifluoroacetic acid and HPLC purified to give the final desired oligomer. TMFT = α-trifluoromethyltolyl protecting group.
The first set of oligomers was synthesized by iterating dithiothreitol (DTT) dithiol and the protected sulfonate N-allyl acrylamide monomer and tracked by 1H NMR, 19F NMR, and a dithiodipyridine (DTDP) assay (Figure 2) As detailed in earlier reports, the synthetic progression of oligoTEAs can be tracked by 1H NMR by focusing on the disappearance of the allyl protons to confirm thiolene conversion.9 To track the completion of the thiol–Michael addition, 1H NMR has previously focused on the consumption of the thiol peak and appearance of allyl peaks. However, the monomer thiol peak of dithiolthreitol appears within the t-Boc of the fluorous support (Figure 2, methyl protons designated as “E”). Additional evidence of the Michael addition conversion can be observed with the growth of protons “C’ and ‘D” located on the aromatic tolyl group of the protected sulfonate N-allyl acrylamide. Also, a DTDP assay can determine the thiol concentration and subsequent consumption.54,55 Finally, sensitive 19F NMR can also be used to verify the completion of the Michael addition by observing the trifluoro group of the protected sulfonate N-allyl acrylamide (Figure 2, trifluoro group designated B). All monomer peak intensities increase and broaden with respect to the fluorous support as the oligomer is elongated as expected. Oligomers after each cycle were sequestered, cleaved, purified by HPLC, and confirmed by 1H NMR and LCMS (Figure S18–S58) providing synthetic lengths of 2–12 “mers.” To explore the effect of each chemical group, additional oligomers were synthesized at the 10mer length with (1) a positively charged guanidine (G) group, (2) a noncharged methyl sulfone (MeS) pendant group, and (3) a methylene backbone using butanedithiol (BDT) (Figure 1, Figure S59–64).
Figure 2.

Example of the synthetic progression of oligoTEA assembly of the hydroxylated sulfonated oligoTEA. Full molecular structures are shown with green trifluoro and orange proton groups on the fluorous support (parts A and E, respectively). and the PSM (part B and parts C, D, respectively). DTDP assay determination of thiol concentration, indicating thiol–Michael addition completion and qualitative yield. 19F NMR spectra are shown with fluorous trifluoro group A normalized. PSM trifluoro group “B” intensity increases and becomes broader as a function of oligomer length. Similarly, 1H NMR spectra normalized by Boc protons “E” shows intensity increases and broadening of PSM tolyl protons “C” and “D”.
Characterization of oligoTEAs by variable temperature PFG NMR visualizes macromolecular diffusion as a linear function of viscosity normalized temperature (Figure 3). This linearity rules out any intramolecular transitions as a function of temperature and constrains the prospective size within the Stokes–Einstein–Sutherland equation (eqs S2–S4). It additionally suggests minimal intermolecular interactions (e.g., aggregation, repulsion). Accurate measurements were obtained using 3 mm tubes, high gas flow rate, an optimized eddy current delay, convection compensation, and gradient pulse control experiments (Figure S2–S4).45 The translational diffusion of the backbone was compared to the diffusion of the oligomer end to discern any heterogeneity, revealing the uniform diffusion of each oligomer (Figure S5).
Figure 3.

(A) Translational diffusion coefficient versus viscosity normalized temperature of oligoTEAs over a range of 10–40 °C of 1–3 mM (DTT-Sulf)1–6 in D2O. (B) Diffusion coefficients of 1–3 mM oligomers in D2O with different pendant and backbone groups as a function of temperature, shown with the same scaling. Solid-line linear fits are shown with shaded regions representing the 90% CI.
Diffusion is affected by the hydrodynamic size, shape, and hydration state of a given macromolecule. The data in Figure 2 follow expected trends in that these macromolecules diffuse quicker at higher temperatures or shorter synthetic length. The oligomers with hydroxylated (DTT) backbone diffused slightly faster than the aliphatic backbone (BDT). This result can be rationalized by a number of factors including differences in shape, hydration effect caused by the more hydrophobic BDT backbone, or stronger hydrogen bonding in the DTT hydroxylated backbone resulting in a more compact faster diffusing oligomer. Additionally, the slope of the diffusion versus normalized temperature establishes a relationship between the hydrodynamic radius and aspect ratio depending on the assumed geometric model (eqs S5–S10).
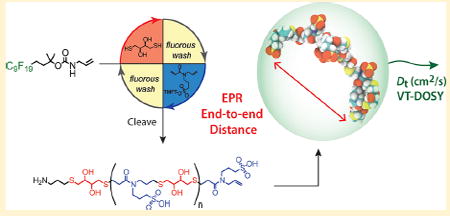
To observe the dynamics of these oligoTEAs in an aqueous environment double electron electron resonance (DEER) EPR distance measurements and molecular dynamics (MD) simulations were completed. DEER measurements were performed on dispin-labeled oligomers prepared by reaction of an oligoTEA diamine with a PROXYL NHS ester. The oligoTEA diamines were prepared by via an additional thiolene reaction of 2-(Boc-amino)ethanethiol on the terminal allyl group. TFA cleavage then liberates an amine at each end. Dispin-labeled oligomers were purified by HPLC, verified by LCMS (Figures S6 and S64–S94), measured by 4-pulse double electron–electron resonance (DEER), and reconstructed by Tikhonov regularization using the L-curve method. Molecular dynamics simulations of single oligomer chains were preformed for 50 ns in a water-filled box with periodic boundary conditions in an NVT ensemble to represent a dilute oligomer solution. Specifically, the DTT-Sulf, DTT-MeS, and BDT-Sulf oligoTEAs were simulated at all lengths (2–12mer) to aid in the visualization of trends as a function of synthetic length. To validate the simulations, the translational diffusion constant was calculated and compared to PFG NMR measurements, revealing good agreement (Figure S7). With respect to the (DTT-Sulf)1–6 series (Figure 4A), the probability distribution from EPR data shows an increase in end-to-end distance with increasing oligomer length, as well as a concomitant increase in conformational freedom as measured by the full width of half max (fwhm). The mean end-to-end distance of the oligomers calculated by MD is also in reasonable agreement with the EPR data, showing a modest increase with oligomer size. MD simulations observe a slightly greater end-to-end distance with the (DTT-Sulf)5,6, which falls within the experimentally observed conformational ensemble (Figure 4B). Overall, this data highlights oligomer flexibility, especially when considering they show an average ~50% collapse from their fully extended theoretical length (Figures 4B and S9).
Figure 4.

A. Distance reconstructions of the end-to-end distance distribution function generated from DEER EPR of 100 μM (DTT-Sulf)1–6 in 20% ethylene glycol in water vitrified (70K) from room temperature. B. Mean end-to-end distance versus synthetic length from DEER distance reconstruction and single-chain MD simulation (300 K, explicit water solvent). Data points represent the mean end-toend distance. Transparent bands represent the full-width at halfmaximum (FWHM) from EPR or standard deviation from the MD simulation time after equilibration. Supporting Information details the sample spin-labeling (Figure S6), DEER measurement, and MD simulation detail.
End-to-end distance measurements from DEER and MD were used to visualize the individual contributions of backbone hydroxylation and pendant groups, revealing a larger effect from the backbone. 10mers with a methylene backbone, i.e., (BDT-Sulf)5, positive charge (DTT-G)5, and neutral charge (DTT-MeS)5 were characterized (Figure 5). The EPR results show that the pendant groups have minimal influence on the average end-to-end distance and the conformational flexibility of the 10mer oligoTEAs. The length scale of these results are sensible given literature.56 The MD data on the other hand shows that the cationic (DTT-G)5 results in a relatively smaller end-to-end distance than predicted for the (DTT-Sulf)5 and (DTT-MeS)5. The methylene backbone of (BDT-Sulf)5 results in a smaller end-to-end distance in both DEER and MD experiments. This effect could be rationalized by greater hydrophobic collapse without backbone hydroxylation. However, PFG NMR suggests that the (BDT-Sulf)5 diffuses slower than all other 10-mers with pendant group modifications (Figure 3). Together these results can be rationalized by two possible explanations. The PROXYL spin-probes could be participating in hydrophobic collapse and thus locate closer to one another in BDT-Sulf. Though possible, the MD simulations predict similar collapse and were performed without spin probes on the molecular structure (Figure 5B), casting doubt on the ability of the spin probes to direct the collapse of the BDT-Sulf oligomer. Thus, the hydroxylated backbone likely participates in intramolecular interactions, aiding collapse by attractive forces (i.e., hydrogen bonding) to result in a smaller hydrodynamic size than the BDT-Sulf.
Figure 5.
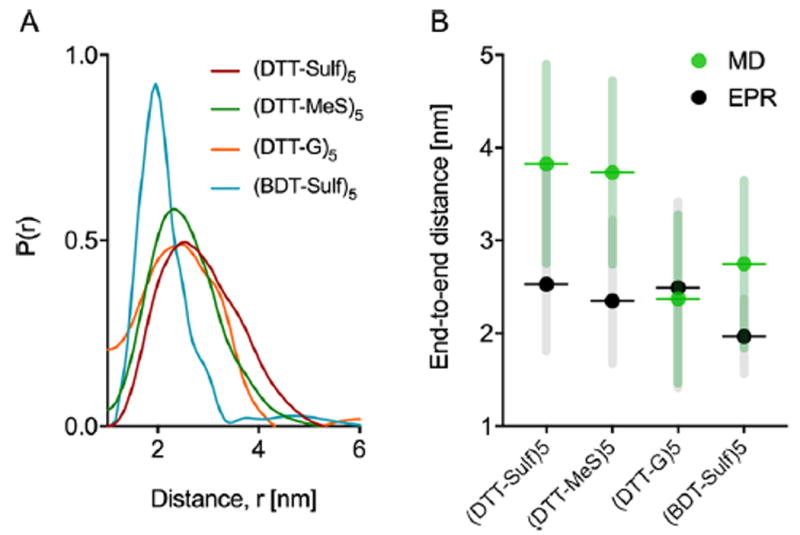
(A). Distance reconstructions of the end-to-end distance distribution function generated from DEER EPR of all 10mers at 100 μM in 20% ethylene glycol in water vitrified (70K) from room temperature. (B) Mean end-to-end distance from DEER distance reconstruction and single-chain molecular dynamics simulation (300 K, explicit water solvent). Data points represent the mean end-to-end distance. Error bars represent the full-width at half-maximum (EPR) or standard deviation (MD). Supporting Information details sample spinlabeling (Figure S12), DEER measurement, and MD simulation detail.
The effect of intramolecular electrostatic repulsion on chain dynamics can be visualized with DEER and MD by screening these interactions with ammonium cations (Figure 6). Aqueous ammonia was added to the oligoTEA sample preparation to give an estimated [NH4 +] concentration of 8.5 mM (Kb of 1.8 × 10−5), with oligoTEA concentration at 100 μM. The neutral oligoTEA (DTT-MeS)5 does not experience a change conformational flexibility or end-to-end distance with the addition of ammonium ions as would be expected (Figure 6E). However, the addition of ammonium ions in the (DTT-Sulf)5 oligoTEA results a slightly shorter end-to-end distance presumably due to screening of intramolecular electrostatic interactions (Figure 6A). Conversely, without salt present, intramolecular electrostatic repulsion is stronger, marginally broadening the conformational ensemble. This effect can is seen more prominently in the (DTT-Sulf)6 oligoTEA (Figure 6B). Addition of ammonium ions to the positively charged (DTT-G)5 oligoTEA leads to a slight narrowing of the distribution and little to no change in the average end-to-end distance (Figure 6C). The distribution and average end-to-end distance of the backbone modified (BDT-Sulf)5 oligoTEA showed similar results with slight narrowing of the distribution and little to no change in the average end-to-end distance (Figure 6D).
Figure 6.
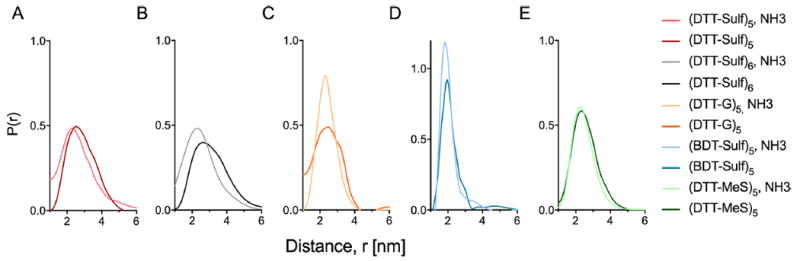
End-to-end distance distributions from DEER measurement of 100 μM oligoTEAs in 20% ethylene glycol in water vitrified (70 K) from room temperature revealing that all charged oligoTEAs experience electrostatic screening by the addition of ammonia during sample preparation. Lighter color lines indicate distributions measured with ammonia sample preparation.
DEER and PFG NMR measurements both provide an indication of macromolecular size; DEER gives the end-to-end distance while PFG NMR measures the translational diffusion coefficient. The Stokes–Einstein-Sutherland (SES) relation can be used to relate diffusivity to the macromolecular size and shape, given simple geometries (eqs S1–S5).52,57 Shape models have been developed by F. Perrin (oblate, prolate ellipsoids) and more recently by A. Ortega and J. Garcia de la Torre (rod) (eqs S6–S8).58-61 While an ellipsoid is geometrically preferred, the rod model is mathematically continuous over its range of aspect ratio, making it more robust. To solve for size and shape simultaneously, a constraint is needed outside of the data gathered from the PFG NMR (eq S4). DEER and MD end-to-end distances can be assumed to describe the length of the rod, allowing discrete solutions to be obtained for the hydrodynamic radius and aspect ratio (eqs S9–S10).
Similar to the end-to-end distance data (Figures 4, 5 and 6), this additional analysis confirms increases in size, as indicated by the hydrodynamic radii from the SES rod model, at longer oligomer lengths (Figure S12). However, the aspect ratio data derived from the MD end-to-end distances appear lower than what is observed experimentally (Figure 7). One possible explanation of this observation is that the addition of spin-labels for DEER measurement adds significant size or length to the smaller oligomers. This concern is compounded because the aspect ratio is exponentially sensitive to the difference between the end-to-end constraint and twice the hydrodynamics radius (Figure S13). To investigate this theory, the first three dispin labeled oligomers ((DTT-Sulf)1–3) were oxidized, measured by PFG NMR (Figure S14) and reanalyzed with the SES rod model. The new aspect ratio which takes into account the spin labels on (DTT-Sulf)1–3 confirms that the original aspect ratio of the smaller oligomers was inflated by the length added by the spin labels (Figure 7) Although the paramagnetic nitroxides can affect the NMR quality (i.e., oxidation is necessary), the shape and size analyses are best performed on the same macromolecule when shorter oligomer lengths are involved. Beyond a synthetic length of six monomers (three dithiol and three N-allylacrylamide monomers), the spin-probes have minimal (<10%) impact on diffusion (Figure S15). The overall SES shape analysis with the rod model utilizing PFG NMR and DEER data, and the MD data, indicate that all oligoTEAs prepared in this work have a low aspect ratio (1–2) as expected for relatively short and flexible macromolecules.
Figure 7.
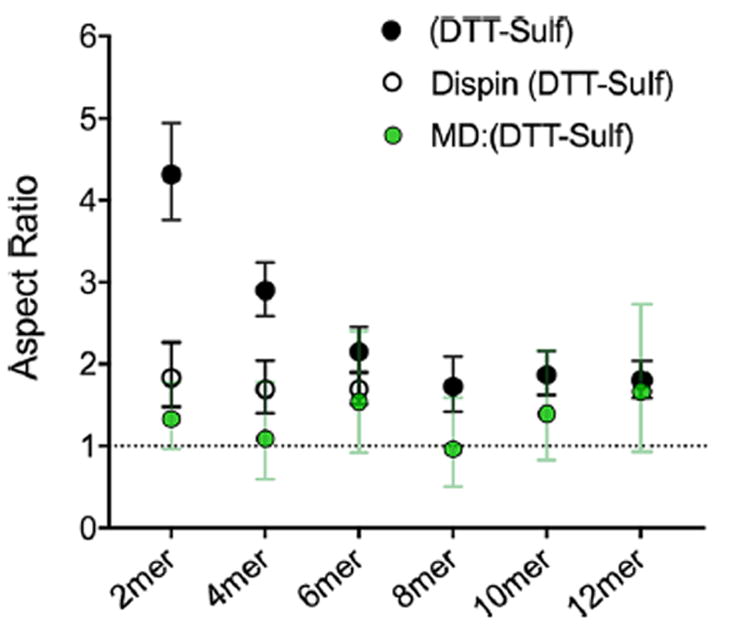
Aspect ratio of the (DTT-Sulf)1–6 series calculated from the solution to the SES rod model assuming the end-to-end distance data describes the long dimension (length) of the rod. Data in the black filled circles were computed using end-to-end distance measurements from EPR and the diffusion coefficient of oligomers without the spin probes present. The filled green circles represent solutions to the SES rod model using the end-to-end distance data obtained from MD simulations. The open circles represent the aspect ratio of the (DTT-Sulf)1–6 oligomers computed using the diffusion coefficient of the oligomers bearing the two spin probes. The error bars on the experimental (DTT-Sulf)1–6 data series represent errors from the PFG NMR data and a 95% confidence level in the DEER data, all propagated through the SES model.
CONCLUSION
In this work, we successfully synthesized sulfonated oligoTEAs and characterized their size, chain dynamics, and conformational ensemble by PFG NMR, DEER EPR, and MD simulations as a function of synthetic length (2–12mer) and individual monomer functional groups. We confirm the strength of entropy and hydrophobicity to create oligomer “collapse” of flexible structures. Our results suggest that this collapse can overwhelm intramolecular electrostatic repulsion. Analysis of individual monomer contributions revealed larger changes due to modulation of the backbone as opposed to pendant groups. Charged oligoTEAs were observed be affected by the addition of ammonium salt, resulting in fewer conformations of similar end-to-end distances, likely by ionic screening. While low resolution, these characterization techniques have provided insight into the ensemble of highly flexible oligoTEAs. We anticipate that monomers and/or sequence motifs that impart rigidity or directionality to the oligomer will also be resolved by this technique. The characterization of other short flexible oligomers will enable understanding of sequence-structure relationships toward the design of novel foldamers and macromolecular architectures. This methodology can also be applied to the structural characterization and development of flexible and biologically functional materials.
Supplementary Material
Acknowledgments
The authors sincerely thank ACERT personnel for training in using the EPR spectrometer, as well as invaluable guidance and support with the acquisition and processing of EPR data. Computational resources were provided by the Cornell Institute for Computational Science and Engineering (ICSE). Acknowledgment is made to the donors of the Petroleum Research Fund, administered by the American Chemical Society, for partial support of this research. This project received funding from the Army Research Office (W911NF-15-1-0179), Cornell University Startup funds and the Nancy and Peter Meinig Investigator Fellowship. J.S.B. acknowledges financial support from the National Science Foundation Graduate Research Fellowship Program (DGE-1144153). Equipment used to perform this research was funded in part by the Cornell Center for Materials Research (DMR-1120296), ACERT Grant NIH/NIGMS P41GM103521, and NSF-MRI (CHE-1531632).
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol. 7b01915.
Materials and methods, detailed monomer and oligomer assembly protocols, detailed protocols, acquisition parameters and processing details for PFG NMR and EPR, methods for molecular dynamics simulations, additional processed data, molecular diffusion theory, and 1H and 19F NMR and LCMS characterization data (PDF)
References
- 1.Lane DA, Philippou H, Huntington JA. Directing Thrombin. Blood. 2005;106(8):2605–2612. doi: 10.1182/blood-2005-04-1710. [DOI] [PubMed] [Google Scholar]
- 2.Siegert R, Leroux MR, Scheufler C, Hartl FU, Moarefi I. Structure of the Molecular Chaperone Prefoldin. Cell. 2000;103(4):621–632. doi: 10.1016/s0092-8674(00)00165-3. [DOI] [PubMed] [Google Scholar]
- 3.Harris LJ, Larson SB, Hasel KW, McPherson A. Refined Structure of an Intact IgG2a Monoclonal Antibody. Biochemistry. 1997;36(7):1581–1597. doi: 10.1021/bi962514+. [DOI] [PubMed] [Google Scholar]
- 4.Hingerty B, Brown RS, Jack A. Further Refinement of the Structure of Yeast tRNAPhe. J Mol Biol. 1978;124(3):523–534. doi: 10.1016/0022-2836(78)90185-7. [DOI] [PubMed] [Google Scholar]
- 5.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal Structure of the Nucleosome Core Particle at 2.8 A Resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 6.Rühmann E, Betz M, Heine A, Klebe G. Fragment Binding Can Be Either More Enthalpy-Driven or Entropy-Driven: Crystal Structures and Residual Hydration Patterns Suggest Why. J Med Chem. 2015;58(17):6960–6971. doi: 10.1021/acs.jmedchem.5b00812. [DOI] [PubMed] [Google Scholar]
- 7.Lutz J-F, Ouchi M, Liu DR, Sawamoto M. Sequence-Controlled Polymers. Science (Washington, DC, U S) 2013;341(6146):1238149–1238149. doi: 10.1126/science.1238149. [DOI] [PubMed] [Google Scholar]
- 8.Lutz J-F, Lehn J-M, Meijer EW, Matyjaszewski K. From Precision Polymers to Complex Materials and Systems. Nat Rev Mater. 2016;1:16024. [Google Scholar]
- 9.Porel M, Alabi CA. Sequence-Defined Polymers via Orthogonal Allyl Acrylamide Building Blocks. J Am Chem Soc. 2014;136(38):13162–13165. doi: 10.1021/ja507262t. [DOI] [PubMed] [Google Scholar]
- 10.Porel M, Brown JS, Alabi Ca. Sequence-Defined Oligothioetheramides. Synlett. 2015;26:565–571. [Google Scholar]
- 11.Bishop JR, Schuksz M, Esko JD. Heparan Sulphate Proteoglycans Fine-Tune Mammalian Physiology. Nature. 2007;446(7139):1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 12.Gama CI, Tully SE, Sotogaku N, Clark PM, Rawat M, Vaidehi N, Goddard WA, Nishi A, Hsieh-Wilson LC. Sulfation Patterns of Glycosaminoglycans Encode Molecular Recognition and Activity. Nat Chem Biol. 2006;2(9):467–473. doi: 10.1038/nchembio810. [DOI] [PubMed] [Google Scholar]
- 13.Avci FY, Karst NA, Linhardt RJ. Synthetic Oligosaccharides as Heparin-Mimetics Displaying Anticoagulant Properties. Curr Pharm Des. 2003;9:2323–2335. doi: 10.2174/1381612033453929. [DOI] [PubMed] [Google Scholar]
- 14.Petitou M, Van Boeckel CA. A A Synthetic Antithrombin III Binding Pentasaccharide Is Now a Drug! What Comes Next? Angew Chem Int Ed. 2004;43(24):3118–3133. doi: 10.1002/anie.200300640. [DOI] [PubMed] [Google Scholar]
- 15.Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The Anticoagulant Activation of Antithrombin by Heparin. Proc Natl Acad Sci U S A. 1997;94(26):14683–14688. doi: 10.1073/pnas.94.26.14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lever R, Page CP. Novel Drug Development Opportunities for Heparin. Nat Rev Drug Discovery. 2002;1(2):140–148. doi: 10.1038/nrd724. [DOI] [PubMed] [Google Scholar]
- 17.Hecht S, Huc I, editors. Foldamers. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2007. [Google Scholar]
- 18.Hill DJ, Mio MJ, Prince RB, Hughes TS, Moore JS. A Field Guide to Foldamers. Chem Rev. 2001;101(12):3893–4011. doi: 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- 19.Goodman CM, Choi S, Shandler S, DeGrado WF. Foldamers as Versatile Frameworks for the Design and Evolution of Function. Nat Chem Biol. 2007;3(5):252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ouchi M, Badi N, Lutz JF, Sawamoto M. Single-Chain Technology Using Discrete Synthetic Macromolecules. Nat Chem. 2011;3(12):917–924. doi: 10.1038/nchem.1175. [DOI] [PubMed] [Google Scholar]
- 21.Terashima T, Sugita T, Fukae K, Sawamoto M. Synthesis and Single-Chain Folding of Amphiphilic Random Copolymers in Water. Macromolecules. 2014;47(2):589–600. [Google Scholar]
- 22.Perez-Baena I, Barroso-Bujans F, Gasser U, Arbe A, Moreno AJ, Colmenero J, Pomposo J. A. Endowing Single-Chain Polymer Nanoparticles with Enzyme-Mimetic Activity. ACS Macro Lett. 2013;2(9):775–779. doi: 10.1021/mz4003744. [DOI] [PubMed] [Google Scholar]
- 23.Kulkarni C, Meijer EW, Palmans AR. A. Cooperativity Scale: A Structure-Mechanism Correlation in the Self-Assembly of Benzene-1,3,5-Tricarboxamides. Acc Chem Res. 2017;50(8):1928–1936. doi: 10.1021/acs.accounts.7b00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laursen JS, Harris P, Fristrup P, Olsen Ca. Triangular Prism-Shaped β-Peptoid Helices as Unique Biomimetic Scaffolds. Nat Commun. 2015 May;6:7013. doi: 10.1038/ncomms8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mannige RV, Haxton TK, Proulx C, Robertson EJ, Battigelli A, Butterfoss GL, Zuckermann RN, Whitelam S. Peptoid Nanosheets Exhibit a New Secondary-Structure Motif. Nature. 2015;526(7573):415–420. doi: 10.1038/nature15363. [DOI] [PubMed] [Google Scholar]
- 26.Sun J, Zuckermann RN. Peptoid Polymers: A Highly Designable Bioinspired Material. ACS Nano. 2013;7(6):4715–4732. doi: 10.1021/nn4015714. [DOI] [PubMed] [Google Scholar]
- 27.Pace CN, Scholtz JM. A Helix Propensity Scale Based on Experimental Studies of Peptides and Proteins. Biophys J. 1998;75(1):422–427. doi: 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Driggers EM, Hale SP, Lee J, Terrett NK. The Exploration of Macrocycles for Drug Discovery–an Underexploited Structural Class. Nat Rev Drug Discovery. 2008;7(7):608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 29.Porel M, Thornlow DN, Phan NN, Alabi CA. Sequence-Defined Bioactive Macrocycles via an Acid-Catalysed Cascade Reaction. Nat Chem. 2016;8(6):590–596. doi: 10.1038/nchem.2508. [DOI] [PubMed] [Google Scholar]
- 30.You W, Huang YM, Kizhake S, Natarajan A, Chang CenA. Characterization of Promiscuous Binding of Phosphor Ligands to Breast-Cancer-Gene 1 (BRCA1). C-Terminal (BRCT): Molecular Dynamics, Free Energy, Entropy and Inhibitor Design. PLoS Comput Biol. 2016;12(8):e1005057. doi: 10.1371/journal.pcbi.1005057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cozzini P, Kellogg GE, Spyrakis F, Abraham DJ, Costantino G, Emerson A, Fanelli F, Gohlke H, Kuhn La, Morris GM, et al. Target Flexibility: An Emerging Considertaion in Drug Discovery. J Med Chem. 2008;51(20):6237–6255. doi: 10.1021/jm800562d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, et al. Classification of Intrinsically Disordered Regions and Proteins. Chem Rev. 2014;114(13):6589–6631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright PE, Dyson HJ. Intrinsically Disordered Proteins in Cellular Signalling and Regulation. Nat Rev Mol Cell Biol. 2014;16(1):18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu G, Shen Y, Atreya HS, Parish D, Shao Y, Sukumaran DK, Xiao R, Yee A, Lemak A, Bhattacharya A, et al. NMR Data Collection and Analysis Protocol for High-Throughput Protein Structure Determination. Proc Natl Acad Sci U S A. 2005;102(30):10487–10492. doi: 10.1073/pnas.0504338102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laskowski RA. Structural Quality Assurance. Structural Bioinformatics. 2005;44:273. [PubMed] [Google Scholar]
- 36.Pellecchia M, Bertini I, Cowburn D, Dalvit C, Giralt E, Jahnke W, James TL, Homans SW, Kessler H, Luchinat C, et al. Perspectives on NMR in Drug Discovery: A Technique Comes of Age. Nat Rev Drug Discovery. 2008;7(9):738–745. doi: 10.1038/nrd2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mertens HDT, Svergun DI. Structural Characterization of Proteins and Complexes Using Small-Angle X-Ray Solution Scattering. J Struct Biol. 2010;172(1):128–141. doi: 10.1016/j.jsb.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 38.Volkov VV, Svergun DI. Uniqueness of Ab Initio Shape Determination in Small-Angle Scattering. J Appl Crystallogr. 2003;36(3I):860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Svergun DI, Koch MHJ. Small-Angle Scattering Studies of Biological Macromolecules in Solution. Rep Prog Phys. 2003;66(10):1735–1782. [Google Scholar]
- 40.Tsutakawa SE, Hura GL, Frankel KA, Cooper PK, Tainer JA. Structural Analysis of Flexible Proteins in Solution by Small Angle X-Ray Scattering Combined with Crystallography. J Struct Biol. 2007;158(2):214–223. doi: 10.1016/j.jsb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 41.Snyder DA, Chen Y, Denissova NG, Acton T, Aramini JM, Ciano M, Karlin R, Liu J, Manor P, Rajan PA, et al. Comparisons of NMR Spectral Quality and Success in Crystallization Demonstrate That NMR and X-Ray Crystallography Are Complementary Methods for Small Protein Structure Determination. J Am Chem Soc. 2005;127(47):16505–16511. doi: 10.1021/ja053564h. [DOI] [PubMed] [Google Scholar]
- 42.Kojima M, Timchenko AA, Higo J, Ito K, Kihara H, Takahashi K. Structural Refinement by Restrained Molecular-Dynamics Algorithm with Small-Angle X-Ray Scattering Constraints for a Biomolecule. J Appl Crystallogr. 2004;37(1):103–109. [Google Scholar]
- 43.Putnam CD, Hammel M, Hura GL, Tainer JA. X-Ray Solution Scattering (SAXS). Combined with Crystallography and Computation: Defining Accurate Macromolecular Structures, Conformations and Assemblies in Solution. Q Rev Biophys. 2007;40(03):191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 44.Curtis JE, Raghunandan S, Nanda H, Krueger S. SASSIE: A Program to Study Intrinsically Disordered Biological Molecules and Macromolecular Ensembles Using Experimental Scattering Restraints. Comput Phys Commun. 2012;183(2):382–389. [Google Scholar]
- 45.Antalek B. Using Pulsed Gradient Spin Echo NMR for Chemical Mixture Analysis: How to Obtain Optimum Results. Concepts Magn Reson. 2002;14(4):225–258. [Google Scholar]
- 46.Borbat PP, Freed JH. Pulse Dipolar Electron Spin Resonance: Distance Measurements. Struct Bonding. 2013;152:1–82. [Google Scholar]
- 47.Sahu ID, McCarrick RM, Lorigan GA. Use of Electron Paramagnetic Resonance to Solve Biochemical Problems. Biochemistry. 2013;52(35):5967–5984. doi: 10.1021/bi400834a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fafarman AT, Borbat PP, Freed JH, Kirshenbaum K. Characterizing the Structure and Dynamics of Folded Oligomers: Pulsed ESR Studies of Peptoid Helices. Chem Commun. 2007;4:377–379. doi: 10.1039/b612198e. [DOI] [PubMed] [Google Scholar]
- 49.Godt A, Schulte M, Zimmermann H, Jeschke G. How Flexible Are Poly(para-Phenyleneethynylene)s? Angew Chem. 2006;118(45):7722–7726. doi: 10.1002/anie.200602807. [DOI] [PubMed] [Google Scholar]
- 50.Pfannebecker V, Klos H, Hubrich M, Volkmer T, Heuer A, Wiesner U, Spiess HW. Determination of End-to-End Distances in Oligomers by Pulsed EPR. J Phys Chem. 1996;100(32):13428–13432. [Google Scholar]
- 51.Banham JE, Baker CM, Ceola S, Day IJ, Grant GH, Groenen EJJ, Rodgers CT, Jeschke G, Timmel CR. Distance Measurements in the Borderline Region of Applicability of CW EPR and DEER: A Model Study on a Homologous Series of Spin-Labelled Peptides. J Magn Reson. 2008;191(2):202–218. doi: 10.1016/j.jmr.2007.11.023. [DOI] [PubMed] [Google Scholar]
- 52.Macchioni A, Ciancaleoni G, Zuccaccia C, Zuccaccia D. Determining Accurate Molecular Sizes in Solution through NMR Diffusion Spectroscopy. Chem Soc Rev. 2008;37(3):479–489. doi: 10.1039/b615067p. [DOI] [PubMed] [Google Scholar]
- 53.Pauff SM, Miller SC. A Trifluoroacetic Acid-Labile Sulfonate Protecting Group and Its Use in the Synthesis of a near-IR Fluorophore. J Org Chem. 2013;78(2):711–716. doi: 10.1021/jo302065u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Egwim IO, Gruber HJ. Spectrophotometric Measurement of Mercaptans with 4,4′-dithiodipyridine. Anal Biochem. 2001;288(2):188–194. doi: 10.1006/abio.2000.4891. [DOI] [PubMed] [Google Scholar]
- 55.Grassetti DR, Murray JF. Determination of Sulfhydryl Groups with 2,2′- or 4,4′-dithiodipyridine. Arch Biochem Biophys. 1967;119(1):41–49. doi: 10.1016/0003-9861(67)90426-2. [DOI] [PubMed] [Google Scholar]
- 56.Murnen HK, Rosales AM, Dobrynin AV, Zuckermann RN, Segalman RA. Persistence Length of Polyelectrolytes with Precisely Located Charges. Soft Matter. 2013;9(1):90–98. [Google Scholar]
- 57.Schulze BM, Watkins DL, Zhang J, Ghiviriga I, Castellano RK. Estimating the Shape and Size of Supramolecular Assemblies by Variable Temperature Diffusion Ordered Spectroscopy. Org Biomol Chem. 2014;12(40):7932–7936. doi: 10.1039/c4ob01373e. [DOI] [PubMed] [Google Scholar]
- 58.Perrin F. Mouvement Brownien D’un Ellipsoide - I. Dispersion Diélectrique Pour Des Molécules Ellipsoidales. J Phys Radium. 1934;5(10):497–511. [Google Scholar]
- 59.Perrin F. Mouvement Brownien D’un Ellipsoide (II). Rotation Libre et Dépolarisation Des Fluorescences. Translation et Diffusion de Molécules Ellipsoidales. J Phys Radium. 1936;7(1):1–11. [Google Scholar]
- 60.Koenig SH. Brownian Motion of an Ellipsoid: A Correction to Perrin’s Results. Biopolymers. 1975;14:2421–2423. [Google Scholar]
- 61.Ortega A, García de la Torre J. Hydrodynamic Properties of Rodlike and Disklike Particles in Dilute Solution. J Chem Phys. 2003;119(18):9914. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.