

Abstract
Strategies for musculoskeletal tissue regeneration apply adult mesenchymal stem/stromal cells (MSCs) that can be sourced from bone marrow‐ and lipo‐aspirates. Adipose tissue‐derived MSCs are more easily harvested in the large quantities required for skeletal tissue‐engineering approaches, but are generally considered to be less osteogenic than bone marrow MSCs. Therefore, we tested a new molecular strategy to improve their osteogenic lineage‐differentiation potential using the fungal metabolite cytochalasin D (CytoD). We show that CytoD, which may function by redistributing the intracellular location of β‐actin (ACTB), is a potent osteogenic stimulant as reflected by significant increases in alkaline phosphatase activity, extracellular matrix mineralization, and osteoblast‐related gene expression (e.g., RUNX2, ALPL, SPARC, and TGFB3). RNA sequencing analyses of MSCs revealed that acute CytoD treatment (24 hours) stimulates a broad program of osteogenic biomarkers and epigenetic regulators. CytoD decreases mRNA and protein levels of the Polycomb chromatin regulator Enhancer of Zeste Homolog 2 (EZH2), which controls heterochromatin formation by mediating trimethylation of histone 3 lysine 27 (H3K27me3). Reduced EZH2 expression decreases cellular H3K27me3 marks indicating a global reduction in heterochromatin. We conclude that CytoD is an effective osteogenic stimulant that mechanistically functions by blocking both cytoplasmic actin polymerization and gene‐suppressive epigenetic mechanisms required for the acquisition of the osteogenic phenotype in adipose tissue‐derived MSCs. This finding supports the use of CytoD in advancing the osteogenic potential of MSCs in skeletal regenerative strategies. Stem Cells Translational Medicine 2018;7:197–209
Keywords: Mesenchymal stem cells, Tissue regeneration, Bone, Epigenetics, Adipose stem cells
Significance Statement.
Mesenchymal stem cells (MSC)‐based therapy for aging‐related bone disorders still needs to be optimized for controlled and consistent outcomes. Adipose tissue‐derived MSCs are easily isolated and are available in large numbers, making them ideal candidates for large scale cell transplantation strategies. The present study shows that the bone‐forming capacity of adipose MSCs can be successfully improved using a mold‐derived compound which potently modulates the cells to attain an osteogenic phenotype. This approach also highlights epigenetically tuning stem cell fate for efficacious stem cell therapy in bone regenerative medicine.
Introduction
Stem cell therapies in regenerative medicine require an optimal source of stem cells which must be capable of self‐renewal and reparative properties to restore tissue integrity and function. The therapeutic importance of mesenchymal stem/stromal cells (MSCs) is evident from their use in over 300 ongoing clinical trials that use either allogeneic or autologous MSCs (see www.clinicaltrials.gov) for a broad range of clinical conditions, including osteoarthritis, myocardial infarction, diabetes mellitus, as well as pulmonary, neurodegenerative, and immunological disorders 1, 2, 3. Based on recent concerns regarding variability and inconsistency in tissue healing responses, there is considerable focus on improved characterization and optimization of these cells prior to their application for efficacious tissue regeneration 4, 5, 6, 7. Strategies using MSCs for treating bone defects (e.g., to support fracture repair in age‐related osteoporosis) require that their quality is validated and optimized. For example, development of methods to rejuvenate senescent stem cells from aging patients or to modulate the osteogenic capacity of MSC without altering their stem cell properties, remain important stem cell engineering objectives.
MSCs from various sources have been isolated and characterized, of which adipose tissue‐derived MSCs are particularly advantageous because of their ease of isolation and the high starting cell numbers acquired from primary tissue digests or after brief culture. The stromal fraction of adipose tissue contains between 1% and 10% MSCs which possess the ability for multi‐lineage differentiation and immunomodulation. Hence, these cells are useful for cell transplantations to treat skeletal degenerative disorders such as osteoarthritis 2 and for interactions with biopolymer scaffolds to support skeletal tissue engineering applications 8. Our group has previously validated in vitro differentiation as well as cell surface marker expression and the transcriptome of clinical grade adipose‐derived MSCs 4, 9, 10, 11. Osteogenic cells, growth factors, and biomaterials are important elements that form the foundation of bone tissue engineering and skeletal regenerative therapies. Unlike bone marrow derived MSCs, which generally differentiate more readily into skeletal tissues such as bone and cartilage 12, 13, the osteogenic capacity of lipo‐aspirate derived MSCs is less evident. This limitation provided the motivation for studies that seek to enhance the osteogenic capacity of adipose MSCs.
Strategies to modulate adipose‐derived MSC osteogenesis include the use of osteogenic factors (e.g., bone morphogenic protein, parathyroid hormone) 14, 15, differentiation agents (dexamethasone, l‐ascorbate, and glycerol‐2‐phosphate) 16, and other cocktails (melatonin, strontium, vitamins D and K) 17. More recently, epigenetic drugs and inhibitors that control the biological fate of human mesenchymal stromal cells have been studied. Preliminary work from our research group and others has established the bone‐anabolic properties of the epigenetic drugs GSK126 and sulforaphane both of which may have potential use as stem cell modulating agents in autologous therapy for aging patients 18, 19, 20. Inhibition of the Polycomb group protein Enhancer of Zeste Homolog 2 (EZH2), a histone methyltransferase which catalyzes tri‐methylation of H3K27, enhances osteogenesis in MSCs and its conditional genetic loss generates bone‐related phenotypes 21, 22, including accelerated cranial suture closure that results from accelerated membranous bone formation in mice. In addition, EZH2 decreases during terminal osteoblast differentiation and bone extra‐cellular matrix (ECM) production 19. In vitro inhibition of EZH2 using the EZH2‐selective inhibitor GSK126, also improves osteogenic differentiation in both precommitted osteoblasts (MC3T3s) and in human adipose MSCs 18. Thus, modulation of chromatin‐related epigenetic mechanisms represents a viable method for enhancing osteoblastic differentiation of mesenchymal progenitor cells.
Beyond chromatin‐related methods for altering stem cell phenotypes, studies on matrix environment and cytoskeletal responses to external force cues have highlighted the importance of cell stretching and stiffness in determining stem cell lineage specification 23. More recently, investigations on cytoskeletal reorganization and its associated actin structural dynamics using the fungal metabolite CytoD (an actin‐cytoskeleton modifying drug derived from molds) have revealed that intranuclear actin promotes phenotype conversion of MSCs into the osteogenic lineage 24. Nuclear import of actin is regulated by its co‐transporters importin‐9 (IPO9) and cofilin 25, and both are required for osteogenic effects of CytoD. One model by which actin polymerization exerts an osteogenic stimulus to differentiation of bone marrow derived MSCs is via YAP relocalization to the cytoplasm and the subsequent de‐repression of the osteogenic transcription factor RUNX2 24.
In this study, we tested this novel actin‐based molecular strategy to improve their osteogenic lineage‐differentiation potential of adipose MSCs to support orthopedic applications requiring musculoskeletal tissue regeneration. We show that osteogenic differentiation of adipose‐derived MSCs can be improved by conditioning cells with CytoD. Mechanistically, we show that CytoD alters chromatin organization by affecting the Polycomb‐related epigenetic regulator EZH2 and the architectural bone‐specific transcription factor RUNX2, that collectively play key roles osteogenic lineage commitment of MSCs toward osteogenesis. In addition, RNA‐sequencing revealed that CytoD stimulated adipose MSCs into the osteogenic lineage by activation and repression of novel genes. The findings of this work show promise in advancing stem cell therapeutics for musculoskeletal repair and may impact on bone tissue engineering applications to accelerate bone fracture repair in aging patients.
Materials and Methods
Isolation and Expansion of Adipose‐Derived Mesenchymal Stem Cells
Mesenchymal stromal cells were isolated from the adipose tissue obtained from healthy donors with their consent and with approval from the Mayo Clinic Institutional Review Board (IRB) as previously described 9, 26. Briefly, following the enzymatic digestion of fat tissue for 1.5 hours at 37°C with 0.075% collagenase Type I (Worthington Biochemicals, Lakewood, NJ) the adipocytes were removed from the stromal vascular fraction by low speed centrifugation (400g for 5 minutes). After aspirating the supernatant, the cell pellet was rinsed with phosphate‐buffered saline (PBS) and passed through 70 µm and 40 µm cell strainers (BD Biosciences, San Jose, CA). The cell suspension was then incubated in T‐175 flasks at 37°C in 5% CO2 at a cell density of 1.0–2.5 × 103 cells per cm2 in maintenance medium (Advanced MEM) with 5% platelet lysate, PLTMax (MillCreek LifeSciences, Rochester, MN), 2 U/ml heparin (Novaplus), 2 mM l‐glutamine (Invitrogen, Waltham, MA), and antibiotics (100 U/ml penicillin, 100 g/ml streptomycin, Sigma‐Aldrich, St. Louis, MO). Cells were harvested as described previously 9.
Osteogenic Differentiation
Adipose MSCs were seeded at 3,000 cells per cm2 in maintenance medium in 6‐well plates and incubated under standard culture conditions for 24 hours before being changed to osteogenic medium containing vehicle (DMSO) or 0.1 µg/ml cytochalasin D (Sigma‐Aldrich). Osteogenic medium maintenance media supplemented with 10 nM dexamethasone, 25 µg/ml ascorbic acid, and 10 mM β‐glycerophosphate. Cells were maintained for up to a maximum of 14 days, with media changed every 3 to 4 days. RNA and protein were collected at various time points (days 1, 2, 3, and 6) during the course of differentiation. CytoD or vehicle was present in osteogenic media throughout the course of the experiments.
Immunofluorescence Microscopy
Adipose MSCs were seeded at a density of 5,000 cells per cm2 on round glass cover slips placed in 12‐well plates. The following day, the cells were treated in osteogenic medium containing drug or vehicle as detailed above. At day 1 post‐treatment, the cover slips were fixed in 3.7% paraformaldehyde solution in PBS for 10 minutes at room temperature. Each coverslip was placed in a petri dish and extracted with 0.1% Triton X‐100 in PBS for 3–5 minutes. After two washes with PBS, each coverslip was stained with 200 µl of diluted Rhodamine Phalloidin (Molecular Probes, Waltham, MA; 5 µl methanolic stock solution in 500 µl PBS containing 1% BSA) and incubated for 20 minutes at room temperature. Bovine serum albumin was added to the staining solution to reduce background staining that might occur with fluorescent phallotoxins. The cover slips were washed twice with PBS and then mounted onto a glass slide using a permanent mountant containing DAPI. The cover slips were sealed with clear nail polish and digital images were generated using a Zeiss laser scanning microscope.
Alkaline Phosphatase Quantification
Alkaline phosphatase (ALP/ALPL) expression was monitored to assess osteogenic differentiation of adipose MSCs at day 6 post‐treatment both by histological staining and colorimetric analysis. In situ histological staining was performed with cell monolayers that were first washed with PBS and then fixed for 3 minutes in 100% methanol. After three washes with PBS to remove the fixative, ALPL staining was performed by addition of 5 ml of staining buffer (100 mM Tris‐HCl, pH 9.0, 150 mM NaCl, 1 mM MgCl2) containing chromogen substrate solution composed of 33 µl of 50 mg/ml nitro blue tetrazolium (NBT) and 16.5 µl of 50 mg/ml 5‐bromo‐4‐chloro‐3‐indolyl phosphate (BCIP). Cell layers were stained with BCIP/NBT substrate for 30 minutes. The substrate solution was removed, cells were washed twice with water and plates were dried by exposure to ambient air. Staining was visualized by scanning plates on an Epson scanner and bright‐field microscopy.
Additional quantitative analysis of ALPL staining was performed using a para‐nitrophenyl phosphate (pNPP) substrate‐based chromogenic assay of cultures in 6‐well plates. At day 6 post‐induction, media was removed and the cell layers were washed three times with PBS. For each well of a 6‐well plate, 300 µl of 0.1% TE buffer was added and the plates were subjected to a freeze‐thaw cycle to disrupt the cell membrane. To each well, 500 µl of 0.5 µg/ml Hoechst dye (Sigma) was added and the plates were incubated in the dark for 15 minutes at room temperature. DNA content of the wells was quantified by measuring the fluorescence at 340/460 nm on a TECAN reader. ALPL standards were prepared with dilutions ranging from 1 to 10−5 U. The substrate was prepared by dissolving 2.5 mg/ml pNPP in 0.1M diethanolamine buffer (pH 10.5) containing 150 mM NaCl and 2 mM MgCl2. To each well, 300 µl of pNPP substrate was added to the plate with gentle agitation. The plates were incubated at room temperature for 30 minutes for development of the yellow water‐soluble reaction product which absorbs light at 405 nm. The reaction was stopped by adding 200 μl of 0.1M NaOH. The absorbance of the samples and standards at 405 nm was measured using a SoftmaxPro plate reader to obtain the calculated activity of the ALPL enzyme (expressed as ALPL activity per unit DNA).
Alizarin Red and von Kossa Staining
After 10 or 14 days in osteogenic differentiation with drug or vehicle, cells were stained with Alizarin Red S to detect mineralization. Cell layers were washed with PBS after removal of media, fixed in 4% paraformaldehyde for 10 minutes, and incubated in 0.1% solution of Alizarin Red S for 30 minutes with gentle agitation. Excess staining solution was removed by rinses with PBS and distilled water. The presence of calcified deposits was quantified by densitometry as previously described 5, 27. For detecting calcium phosphate by von Kossa staining, cells were fixed as described above and incubated in 1% silver nitrate solution for 45 minutes under UV illumination. The plates were washed twice in distilled water, then treated with 5% sodium thiosulphate for 2 minutes and finally washed with ultrapure water. The black‐colored deposits of calcium phosphate were then identified and quantified by scanning on an Epson scanner at a resolution of 300 dpi. Images were exported to the Quantity One (Bio‐Rad) software tool and converted to gray scale. Seven regions per well were arbitrarily chosen and the same region of interest was applied across all the three wells per condition, thus totaling 21 regions of measurement per treatment group. Data are shown as mean intensity ± SEM. A darker stain implies a higher level of mineralization.
Real Time Polymerase Chain Reaction
To evaluate osteogenesis at the mRNA level, total RNA before treatment (day 0) and at days 1, 2, 3, and 6 post‐treatment was extracted using Trizol. RNA was then reverse transcribed into cDNA using the SuperScript III First‐Strand Synthesis System (Invitrogen). Gene expression was quantified using real‐time polymerase chain reaction PCR. Real‐time qPCR was performed with 10 ng cDNA per 10 µl reaction using the QuantiTect SYBR Green PCR Kit (Qiagen) and a CFX384 Real‐Time qPCR System (BioRad, Hercules, CA). Previously validated primer sequences were used to amplify cDNAs for genes of interest (Supporting Information Table 1). Data are represented as relative expression units (REU) of the gene of interest normalized to GAPDH. Gene expression levels were quantified using the 2−ΔΔCt method.
Western Blotting
Cells were seeded at a density of 5,000 per cm2 and allowed to adhere for 24 hours before treatment with vehicle or CytoD for three days in osteogenic media. Protein was collected by scraping the cells with 3% SDS in Tris/HCl buffer followed by heating for 5 minutes at 95°C and sonication. The lysates were quantified using the bicinchoninic acid assay (BCA assay) and equal quantities of protein (10 µg) were loaded onto Mini‐PROTEAN Precast Gels (Bio‐Rad) for electrophoretic separation. Proteins were blotted onto nitro‐cellulose membranes. The membranes were then blocked for 1hr in 5% milk in TBST and incubated overnight at 4°C using primary antibodies for EZH2, RUNX2, ACTB, Cyclin A, Cyclin B1, EZH1, Lamin B, GAPDH, H3K27me3, and total Histone H3. The following day, blots were washed with TBST and incubated with the corresponding secondary antibodies for 1hr at room temperature followed by development using ECL Prime Western Blotting Detection Reagent (GE Healthcare, Chicago, IL).
RNA‐Interference
Cells were transfected with siRNA (50 nM) in serum‐free Opti‐MEM overnight before replacing the medium and adding reagents for control or treatment groups. Importin‐9 siRNA smart pools were purchased from Dharmacon (GE Healthcare) and transfection was carried out as recommended by the manufacturer.
RNA‐Sequencing
High‐resolution RNA‐sequencing (RNA‐seq) was performed using three distinct adipose tissue‐derived MSCs from different human donors 9. MSC cultures were each treated in triplicate for 24 hours with or without Cytochalasin D. The resulting biological triplicates for each of the three donors were pooled prior to RNA‐seq to yield technically robust datasets. RNA‐seq analysis of MSCs was performed as described previously 9, 18 with oligo dT purified mRNA and indexed cDNAs (standard TruSeq Kits 12‐Set A and 12‐Set B) on the Illumina platform (Illumina's RTA version 1.17.21.3). Raw reads were mapped using a well‐established pipeline (Bioinformatics Core standard tool) that includes analysis using the MAPRSeq v.1.2.1 system, alignment with TopHat 2.0.6, gene counting with the HTSeq application, as well as normalization and expression analysis using edgeR. Gene expression is expressed in reads per kilobase pair per million mapped reads (RPKM). The sequencing data are available at Gene Expression Omnibus accession number GSE96788.
Statistical Analyses
Results are represented as mean ± SD, unless specified otherwise. Data were typically collected from experiments performed in triplicate and were analyzed by applying Student's t test or one‐way analysis of variance (ANOVA) where necessary using GraphPad Prism 7 software and a p‐value of .05 was considered significant.
Results
Adipose MSC Osteogenesis and Mineralization Is Improved with Cytochalasin D
CytoD has been previously shown to enhance osteogenic differentiation of bone marrow‐derived MSCs that possess the inherent property to differentiate into skeletal lineage cells 24. In this study, we investigated whether CytoD causes similar changes in adipose tissue‐derived MSCs that are adapted to adipokine microenvironments and, intrinsically, are less osteogenic than bone marrow MSCs. As evident from ALPL measurements, CytoD increases ALPL levels in adipose tissue‐derived MSCs (Fig. 1A, 1B). The scanned plates show marked differences in the gross morphology of cells treated with CytoD compared with control cells treated with vehicle. A closer look at the cell layers shows that CytoD treated adipose MSCs contain higher ALPL levels as indicated by increased purple staining. Cells treated with CytoD exhibit higher staining intensities with BCIP/NBT, whereas the control cells had minimally positive staining. To corroborate and quantify the CytoD enhanced increase in ALPL levels, we expressed ALPL activity per unit DNA and found that ALPL activity is significantly higher in CytoD‐treated cells at day 6 post‐treatment (Fig. 1B). Analysis of mineralization at days 10 and 14 by Alizarin Red staining and the von Kossa method revealed that CytoD stimulated robust mineralization of MSCs, as reflected by fourfold higher intensity of Alizarin Red staining and increased appearance of black calcium deposits as detected by von Kossa staining (Fig. 1B). CytoD‐treated adipose MSCs produced significantly higher levels of calcium phosphates in the extracellular matrix (Fig. 1A). Together, in situ histological analysis of cell cultures revealed that CytoD enhanced the osteogenic differentiation and mineralization of adipose MSCs.
Figure 1.
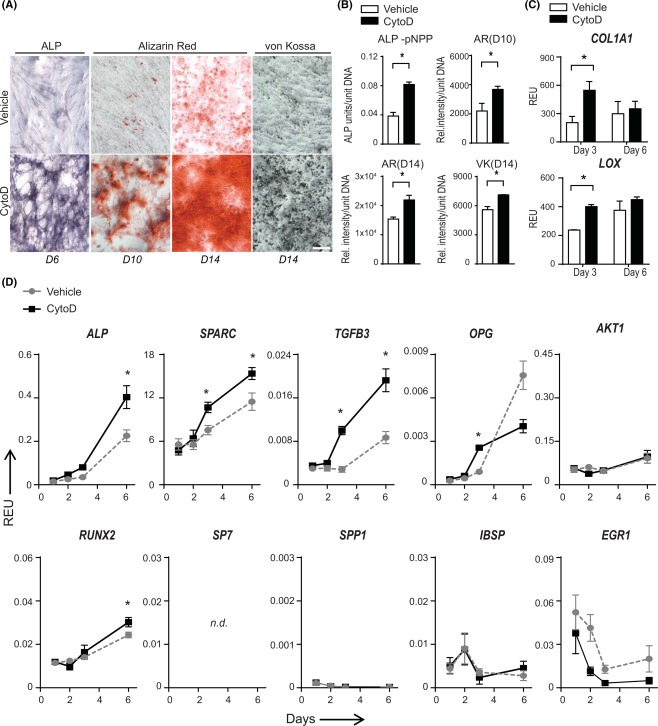
CytoD accelerates osteogenic differentiation and mineralization of adipose mesenchymal stem/stromal cells (MSCs). (A): Histological staining of cell layers for detection of ALPL content at day 6 (D6) post‐induction and mineralization at day 10 (D10) and day 14 (D14) with ARS and VK showed increased staining for ALPL and calcium deposition in the extracellular matrix; Scale bar: 100 µm. (B): Quantification and densitometric analysis of staining normalized to DNA content. *, p < .05. (C): Quantitative PCR analysis at day 3 and day 6 post‐treatment with CytoD showed significant upregulation of matrix markers, COL1A1 and LOX (collagen crosslinking enzyme); *, p < .05. (D): Quantitative PCR analysis of osteogenic markers ALP, SPARC (osteonectin), TGFB3, OPG, RUNX2, SP7, SPP1, IBSP, EGR1, and AKT1 normalized to GAPDH over a 6‐day period following induction with CytoD. SP7 was not detected (n.d) by qPCR in adipose MSCs. *, p < .05. Abbreviations: ALPL, alkaline phosphatase; AR, Alizarin Red; pNPP, para nitro phenyl phosphate; Rel.intensity, relative staining intensity; REU, relative expression units; VK, von Kossa.
CytoD Enhances Expression of Bone Extracellular Matrix Markers in Adipose MSCs
Considering the notable increase in mineralization with CytoD at the histological level, we next examined if changes were reflected in the mRNA levels of collagenous and noncollagenous bone ECM markers. CytoD significantly increased COL1A1 expression as early as day 3 post‐treatment. Similarly, the expression of lysyl oxidase, LOX, an enzyme involved in crosslinking of extracellular matrix proteins, was upregulated with CytoD (Fig. 1C). This correlates with the CytoD‐induced differences in matrix deposition and mineralization visualized by cell staining (Fig. 1A). The finding that CytoD affects matrix deposition was further confirmed by increases in the levels of other relevant bone markers such as ALPL, SPARC (osteonectin) that play important roles in osteogenic differentiation of MSCs, as well as TGFB3 (Fig. 1D). CytoD‐induced ALPL and RUNX2 transcript levels were higher than control at day 6 post‐treatment, but not at earlier time points. Interestingly, adipose MSCs become highly positive for ALPL but not osteocalcin or BGLAP (Supporting Information Fig. 1). Osteonectin or SPARC was upregulated as early as day 3 and continued to increase until day 6, whereas OPG expression was significantly higher at an earlier time point of day 3. Similarly, the levels of YAP, a potent transcription co‐activator showed an early increase with CytoD induction at day 3 and reached a plateau thereafter (data not shown). Notably, CytoD‐enhanced osteogenic differentiation proceeded independently of SP7 in adipose MSCs. Similarly, SPP1 and BGLAP expression were minimally detected in all donors (Fig. 1D; Supporting Information Fig. 1). No significant changes in expression were seen for mRNA levels of AKT1 or HPRT1, which are two reference genes that as expected do not change relative to GAPDH (Fig. 1D and data not shown). The observation that CytoD‐induced mineralization is accompanied by increased gene expression of key bone ECM markers collectively indicate that osteogenic differentiation of adipose MSCs is significantly enhanced with CytoD treatment.
Enhanced Osteogenic Differentiation of Adipose MSCs by CytoD Is Accompanied by Changes in Actin Dynamics
Because CytoD is known to disrupt filamentous actin and promote nuclear accumulation of monomeric actin, we examined the cellular mechanisms by which it acts as a potent osteogenic inducer of adipose MSCs. Treatment of MSCs with CytoD affects the actin cytoskeleton and alters cell morphology. MSCs are fibroblastic cells that exhibit a classical spindle‐shaped morphology with a normal actin cytoskeleton. However, CytoD disrupts actin fiber alignment and alters overall cell morphology (Fig. 2A), while not compromising cell viability.
Figure 2.
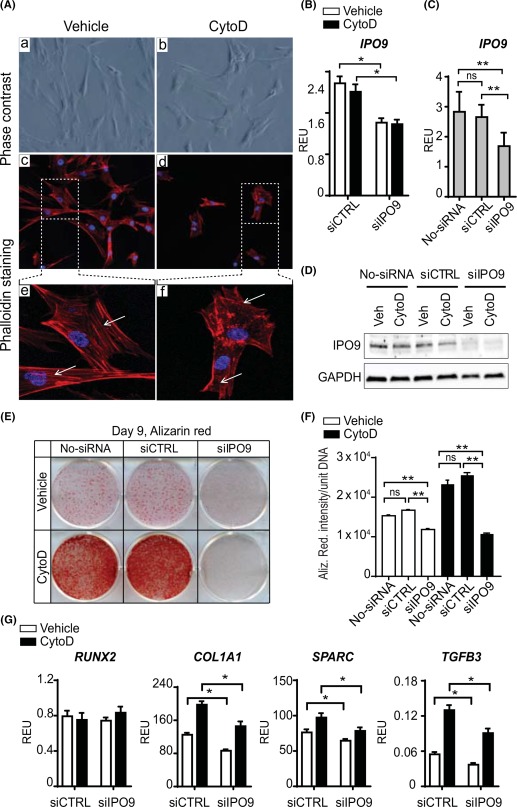
CytoD reorganizes the actin‐cytoskeleton and biological mechanism elucidated using siRNA knockdown of importin‐9 in adipose mesenchymal stem/stromal cells (MSCs). (A): Phase contrast images (Aa, Ab) and immunofluorescence staining of actin fibers (red, phalloidin‐rhodamine, white arrows) and nuclei (blue, DAPI) (Ac, Ad, Ae, Af) showed alterations in actin organization with CytoD treatment (Ad, Af). Magnification: ×10 (Aa, Ab, Ac, Ad); ×40 (Ae, Af). (B, C): Quantitative real time PCR analysis to detect IPO9 mRNA transcripts on un‐transfected (no‐siRNA) and transfected adipose MSCs with control siRNA (siCTRL) or importin‐9 siRNA (siIPO9) treated with or without CytoD for 72 hours. Data show that silencing of human importin‐9 was successful as seen by the reduction in mRNA levels of IPO9. *, p < .05; **, p < .01. (D): Western blotting analysis of siRNA‐induced knockdown of importin‐9. (E): Representative 6‐well plate stained with Alizarin red at day 9 showing differences in mineralization between un‐transfected (no‐siRNA), nonsilencing (siCTRL) and silencing (siIPO9) groups treated with or without CytoD. The knocking down of importin‐9 (siIPO9 group) showed significant suppression of mineralization. (F): Quantification of Alizarin red staining by densitometry; **, p < .01. (G): Importin‐9 silencing analyzed by quantitative PCR on cells treated with or without CytoD for expression levels of RUNX2, COL1A1, SPARC, and TGFB3. Data show significant downregulation in mRNA levels of bone matrix genes COL1A1, SPARC, as well as TGFB3 in importin‐9 silenced cells, confirming that actin import is required for osteogenesis. y‐axis: REU normalized to GAPDH; *, p < .05. Abbreviations: CytoD, cytochalasin D; ns, not significant; REU, relative expression units; siCTRL, control siRNA; siIPO9, importin‐9 siRNA.
Because IPO9 is an important regulator of osteogenesis in mouse bone‐marrow derived BMSCs 28, we performed siRNA knockdown studies in which we silenced the actin co‐transporter IPO9. Comparison of un‐transfected and scrambled siRNA groups (siCTRL) showed no significant differences in IPO9 mRNA and protein expression (Fig. 2C, 2D). The short‐term knockdown effects of RNA silencing were validated by qPCR (Fig. 2B, 2C) and Western blot (Fig. 2D) at day 3 post‐transfection. These short‐term downregulation of IPO9 had prolonged effects on osteoblast differentiation at day 9 after the initial silencing event, because silencing importin‐9 significantly affected MSC mineralization compared with control (siCTRL) and un‐transfected (no siRNA) cells based on Alizarin Red staining (Fig. 2E, 2F). These data suggest that actin may need to be transported to the nucleus for osteogenesis to proceed, as we have proposed previously 28. Further analysis of day 3 mRNA levels of bone‐related markers in adipose MSCs treated with importin‐9 silencing showed significant downregulation in matrix genes COL1A1 and SPARC (osteonectin). Interestingly, RUNX2 expression remained robust and stable during early stages of differentiation, while TGFβ3 mRNA was significantly downregulated with importin‐9 silencing (Fig. 2G). These experiments suggest that MSC matrix production and mineralization is regulated by actin import to the nucleus. Taken together, the biological mechanism by which CytoD acts involves disruption of the actin cytoskeleton concomitant with accumulation of nuclear actin which drives osteogenic differentiation, in accordance with our previous studies 24, 28.
RNA‐Sequencing Reveals Global Changes in MSC Gene Expression with CytoD
To characterize regulatory changes in gene expression caused by CytoD at a very early stage of osteogenic differentiation, we performed RNA‐sequencing on adipose MSC cultures (passage 6) from three different donors using RNA isolated within 24 hours after CytoD‐treatment in the absence of standard osteogenic supplements (dexamethasone, ascorbic acid and beta‐glycerol phosphate). Gene counts for mapped reads were initially conservatively filtered for mRNAs that (a) are detected at a robust level (RPKM > 0.3) and (b) show a biologically relevant effect (fold‐change > 1.4 between control and CytoD treated cells). Hierarchical clustering of this gene set (n = 4,796) resulted in a heat map in which samples cluster according to donor rather than treatment (CytoD treatment) (Fig. 3A). The heat map shows significant differences in gene expression between control and treated cells for all three donors. This result indicates that CytoD treatment has marked donor‐specific effects on gene expression. Corroborating the latter conclusion, Venn diagram analysis reveals that there is only a limited percentage of genes that is either consistently upregulated (11.8%) or downregulated (14.6%) upon CytoD treatment (Fig. 3B). Furthermore, cross‐table analysis which compares the percentages of genes that are commonly upregulated or downregulated in MSCs for each possible donor pair, shows that only between 19.8% to 36.2% of all upregulated genes and between 23.5% to 39.4% of all downregulated genes respond to CytoD (Fig. 3C). These findings indicate that CytoD has a potent biological activity that is capable of dominantly altering the diverse biological ground‐states of undifferentiated MSCs from different donors to promote osteogenic lineage differentiation.
Figure 3.
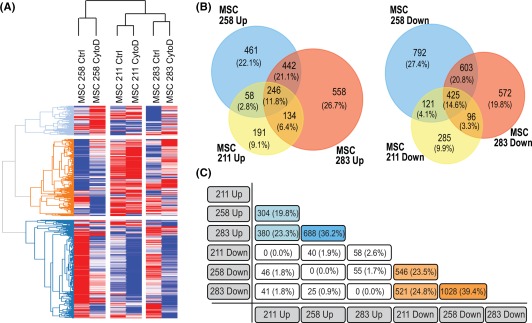
RNA sequencing reveals global changes in gene expression with CytoD treatment. (A): Heat map analysis of n = 4,796 genes sorted for reads per kilobase pair per million mapped reads RPKM > 0.3 and FC > 1.4 showed that the RNA samples from control‐ and CytoD‐treated MSCs from three donors clustered according to donors, indicating the donor‐specific responses to CytoD. (B): Venn diagram analysis of genes upregulated and downregulated with CytoD treatment. (C): Cross‐table analysis indicating percentages of expression of commonly upregulated genes (blue‐shaded boxes) and commonly downregulated genes (orange‐shaded boxes). Abbreviations: ctrl, control; CytoD, cytochalasin D; MSCs, mesenchymal stem/stromal cells.
Identification of Novel CytoD Responsive Genes During Osteogenic Lineage Commitment
To identify the subset of genes that are commonly activated or suppressed during in MSCs by CytoD, we filtered genes for statistically relevant differences (p < .05) based on pair‐wise t‐testing. As expected, heat map analysis reveals clear distinctions between control and CytoD‐treated MSCs, as well as identifies groups of genes that are either upregulated or downregulated with CytoD (Fig. 4A). Gene ontology analysis revealed that CytoD decreases expression of genes involved in nuclear processes (e.g., chromatin assembly, DNA binding and DNA repair) and mitosis (e.g., cell cycle regulation, mitotic spindle assembly). These changes in gene expression indicate that CytoD has cytostatic effects on MSCs. Because CytoD induces osteogenic differentiation, genes that are upregulated are of greater interest, because their expression positively correlates with acquisition of the early osteoblastic phenotype. These genes mostly have molecular functions linked to ECM proteins, plasma‐membrane associated proteins, glycoproteins, and cell surface receptors, as well as the corresponding biological processes associated with cell communication and signal transduction (Fig. 4B). Representative CytoD responsive genes are presented in Figure 4C and 4D. One of the more prominent CytoD stimulated genes is HSD11B1, which encodes the glucocorticoid activating enzyme hydroxysteroid 11‐beta dehydrogenase; remarkably, the corticosteroid dexamethasone is a critical additive for differentiation of cocktails for human osteoblasts. Furthermore, expression of the NPR1 gene increases sixfold with CytoD; this gene is related to the NPR2 gene that is required for normal endochondral bone formation and also associated with osteocrin (OSTN), a ligand that has been linked in osteoblasts to nuclear export of YAP1; the latter is a co‐factor that represses the activating function of RUNX2 and its nuclear localization decreases with CytoD 28. Additionally, in order to explore the commitment of alternative cell lineages, we examined biomarkers specific to a range of cell and tissue types. These analyses included epithelium (cytokeratin 18 [KRT18], E‐cadherin [CDH1], claudin 11 [CLDN11], perlecan [HSPG2], tropomyosin 1 [TPM1], α‐fetoprotein [AFP]), muscle (α‐smooth muscle actin [ACTA2], fatty acid binding protein 3 [FABP4], desmin [DES], creatine kinase [CKM], calponin [CNN], vimentin [VIM]) and hepatocytes (α‐fetoprotein [AFP], cytokeratin 18 [KRT18], cytokeratin 19 [KRT19], albumin [ALB], plasminogen activator inhibitor‐1 [SERPINE1], α‐2‐macroglobulin [A2M]). Our data show that CytoD does not promote significant changes in biomarker gene expression of these lineages (Supporting Information Fig. 3A). Furthermore, key muscle markers such as MyoD, Myogenin, VE cadherin, Myostatin, and Troponin I (cardiac) were not expressed in adipose MSCs regardless of CytoD treatment (Supporting Information Fig. 3B). Network analysis of CytoD upregulated genes revealed that these genes form circuits of genes with related function and representative nodes in these networks are PDGFRB, MAPK10, HDAC5, and HDAC9 (Fig. 4E). Thus, CytoD stimulates genes involved in growth factor dependent signaling pathways and epigenetic regulators, including HDAC5 which is required for parathyroid hormone dependent suppression of the WNT inhibitor SOST 29, 30.
Figure 4.
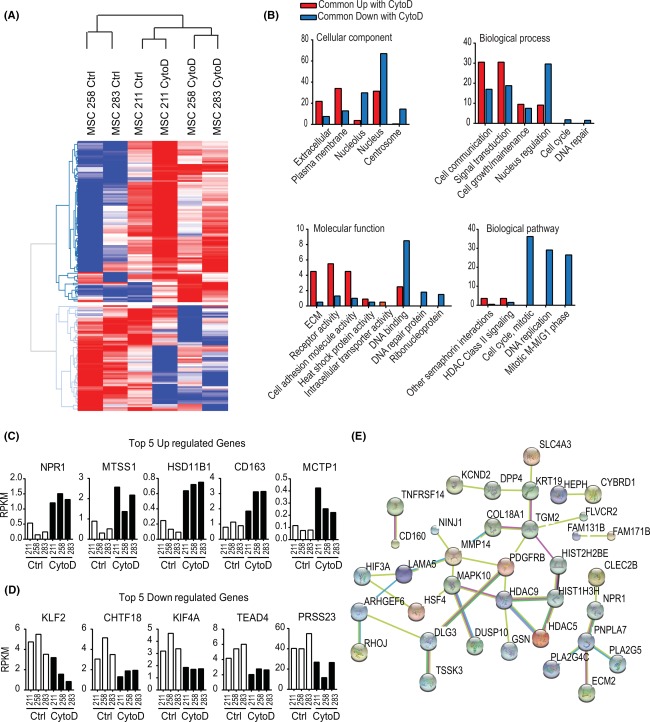
Mathematical enrichment of genes revealed distinct clustering based on CytoD treatment. (A): Heat map analysis of selected genes detected at RPKM > 0.3 and exhibiting statistically significant difference (p < .05) and fold change > 1.4 in pairwise comparisons between control‐ and CytoD‐treated samples from three MSC donors. (B): Functional analysis comparison using functional enrichment analysis tool (FunRich) showed upregulation of extracellular and plasma membrane cellular components and downregulation of nuclear and mitosis‐related components with CytoD. (C, D): Top five genes commonly upregulated and downregulated with CytoD; y‐axis = RPKM. (E): STRING v10.0 network analysis of CytoD‐upregulated genes revealed important nodes such as PDGFRB, MAPK10, HDAC 5 and 9, and PNPLA7. Abbreviations: ctrl, control; CytoD, cytochalasin D; MSCs, mesenchymal stem/stromal cells; RPKM, reads per kilobase pair per million mapped reads.
CytoD Suppresses Expression of Polycomb Group 2 Protein EZH2 and Decreases the Gene Repressive Trimethylation of Lysine 27 in Histone H3 (H3K27me3)
Based on the observation that expression of epigenetic regulators is modulated by CytoD, we further focused our expression analyses on >300 annotated epigenetic regulators, many of which are robustly expressed in adipose MSCs 18. This screen identified 17 epigenetic regulators including HDAC5, which is upregulated (as discussed above), as well as UHRF1 (Ubiquitin‐like with PHD and Ring Finger Domains 1) and the Polycomb Group 2 (PcG2) protein EZH2 (Enhancer of Zeste 2) that are both downregulated within 24 hours after CytoD treatment (Fig. 5A, 5B). UHRF1 is a primary regulator of cell senescence, while EZH2 is a critical suppressor of osteoblastogenesis 18, 19. Network analysis revealed that EZH2 is a principal node of a set of epigenetic regulators genes that are functionally interlinked and exhibit a correlating gene expression pattern (Fig. 5C). Our findings suggest that CytoD may modulate cell fate determination of adipose MSCs by modulating the expression of key epigenetic regulators, including EZH2.
Figure 5.
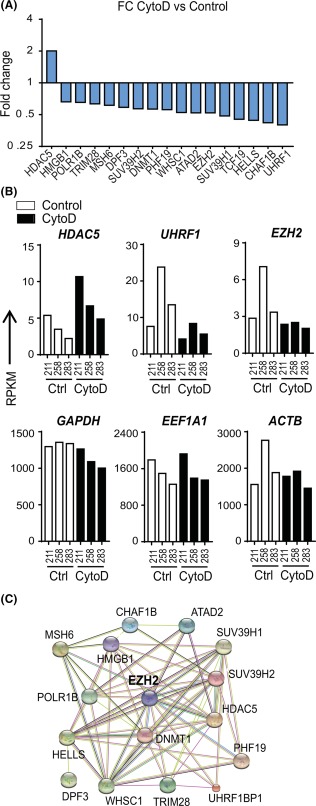
CytoD‐dependent modulations of epigenetic regulators. (A): Analysis on 326 epigenetic regulators expressed at detectable levels (RPKM > 0.3) and filtered for expression levels >1 and fold change >1.5 enabled identification of 17 key epigenetic regulators with CytoD treatment. (B): HDAC5 was found to be highly upregulated with CytoD, and EZH2 and UHRF1 highly downregulated 24 hours post‐treatment. Also shown are RPKM values for housekeeping genes GAPDH, EEF1A1, and ACTB. (C): STRING v10.0 network analysis of the identified epigenetic regulator panel revealed EZH2, an important inhibitor of osteoblastogenesis, as a central node in the network. Abbreviations: ACTB, beta‐actin; CytoD, cytochalasin D; EEF1A1, Elongation factor 1‐alpha 1; EZH2, Enhancer of Zeste Homolog 2; FC, fold change; HDAC5, Histone deacetylase 5; RPKM, reads per kilobase pair per million mapped reads; UHRF1, Ubiquitin‐like with PHD and Ring Finger Domains 1.
We used qPCR and Western blot to validate the RNA‐sequencing results for the chromatin‐structure related protein EZH2. Because CytoD promotes osteogenesis in bone marrow‐derived MSCs by increasing the levels of nuclear actin, we also assessed the mRNA levels of genes associated with nuclear architecture, such as RUNX2 (nuclear matrix) 31, SUN1 and SUN2 (nuclear envelope), as well as LMNA (encoding for lamins A and C) and LMNB1 (nuclear lamina). CytoD significantly accelerates the decrease in the mRNA levels of EZH2, which is an epigenetic suppressor of osteogenic differentiation, while RUNX2 expression increases (Fig. 6). Additionally, CytoD suppresses expression of SUN1 and SUN2, as well as LMNA and LMNB1, which all encode for nuclear envelope proteins. Because EZH2 and LMNB1 have dual functions in cellular differentiation and normal cell division, the downregulation of these genes could perhaps in part be due to the cytostatic effects of CytoD. We performed Western blot analysis to examine how EZH2 protein levels are modulated by CytoD in different experimental conditions (Fig. 7A–7D). CytoD consistently decreases EZH2 protein levels and increases RUNX2 levels in adipose MSCs over a 6‐day culturing period (Fig. 7A), while EZH1 levels are not significantly affected with CytoD treatment. Consistent with the cytostatic properties of CytoD in adipose MSCs revealed by RNA‐seq analysis, we found that CytoD reduces the levels of cyclin A and cyclin B1, as well as the levels of Lamin B within the first two days in culture (Fig. 7A).
Figure 6.
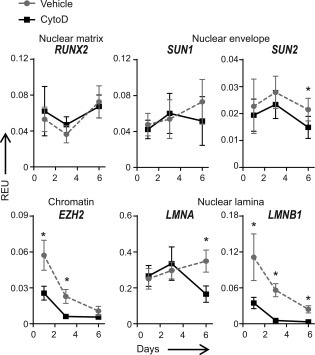
CytoD affects genes linked to nuclear architecture. Quantitative PCR analysis on MSCs treated with CytoD over a 6‐day period showed downregulation of nuclear envelope genes SUN1 and SUN2, as well as lamin genes LMNA and LMNB1, associated with a decrease in mRNA levels of nuclear chromatin‐related EZH2; *, p < .05. Abbreviations: CytoD, cytochalasin D; EZH2, Enhancer of Zeste Homolog 2; LMNA, Lamin A; LMNB1, Lamin B1; REU, relative expression units.
Figure 7.
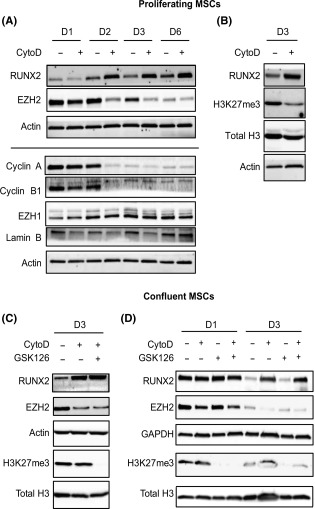
CytoD suppresses expression of epigenetic regulator EZH2. (A): Western blotting on proliferating MSCs treated with or without CytoD over a 6‐day period. CytoD increases RUNX2 levels while suppressing EZH2. Cell cycle proteins, Cyclin A and Cyclin B, as well as lamin B levels were reduced with CytoD. (B): Day 3 (72 hours) lysates of proliferating MSCs treated with CytoD showed decreased expression of lysine‐23 methylation on histone‐3 (H3K27me3). (C): In confluent cultures analyzed at day 3, CytoD reduced EZH2 expression while increasing RUNX2, similar to proliferating conditions. No change was seen in the levels of H3K27me3 in confluent cultures treated with CytoD, whereas GSK126, a known EZH2 inhibitor, blocked H3K27me3. Note that in confluent cultures, CytoD inhibited EZH2 without having an effect on H3K27me3. (D): Similarly, confluent MSCs at 24 hours and 72 hours showed reduction in EZH2 levels with no direct effects on methylation of lysine‐27 on histone‐3. Abbreviations: CytoD, cytochalasin D; EZH2, Enhancer of Zeste Homolog 2; MSCs, mesenchymal stem/stromal cells.
Because EZH2 regulates trimethylation of lysine 27 in histone H3, we examined H3K27me3 levels in CytoD treated cells at 72 hours post‐induction. In proliferating cells (∼50% confluency), CytoD decreases the protein levels of both EZH2 and H3K27me3, while increasing the levels of RUNX2 (Fig. 7A, 7B). However, in confluent cultures, CytoD did not suppress H3K27me3 levels regardless of decreased levels of EZH2, unless we co‐administer the EZH2 inhibitor GSK126 (Fig. 7C, 7D). The latter results indicate that loss of EZH2 does not immediately result in loss of H3K27me3 even though presumably the cognate demethylases remain active in the presence of CytoD and/or GSK126.
Discussion
Adult stem cells, in particular, mesenchymal stem or stromal cells (MSCs) represent a versatile cellular source for cell therapy. To improve the utility of adipose MSCs for bone regenerative therapy that are generally considered to be less osteogenic than bone marrow MSCs, we have shown here that the fungal metabolite Cytochalasin D (CytoD) enhances the osteogenic capacity adipose‐derived MSCs. Specifically, as early as day 6 post‐treatment, CytoD‐treated cells exhibit high ALPL activity and extensive mineralization within just 2 weeks. Mechanistically, these effects of CytoD are due to genome‐wide molecular enhancement of osteoblast differentiation‐related expression programs that render these cells competent to support ECM mineralization.
Previous studies that sought to decipher the molecular mechanisms underlying the mechanical control of stem cell lineage specification have shown that biomechanical signals transmitted through the actin cytoskeleton can modulate stem cell differentiation 32, 33 potentially through forces transmitted to the nuclear envelope and communicated to the internal nucleoskeleton 34. External force not only changes nuclear shape 35, 36, but also alters chromatin structure and gene silencing within the nucleus 36, 37, 38. While these findings collectively suggest that cellular tension is important for mediating cell fate decisions, studies by Sen et al. recently showed cellular tension transmitted through the actin cytoskeleton (and subsequent bone formation in vitro and in vivo) was not the sole determining factor of deciding cell fate but that the specific control of gene expression was primarily dependent on intranuclear actin transport 24. In other studies by Shi et al. and Chen et al., CytoD has been shown to have reduced the viability and osteogenic differentiation of calvarial osteoblasts and tendon stem cells, in vitro 39, 40. These inhibitory responses could be attributed to cell shape status and confluency conditions before CytoD treatment. From our previous and current studies, we observe that CytoD induces similar osteogenic differentiation in MSCs across tissue types (bone marrow MSCs vs. adipose MSCs) as well as among species (human MSCs vs. mouse MSCs) (unpublished data). Our present work included siRNA knockdown of IPO9 that subsequently blocked mineralization completely, with downregulation of bone matrix genes, collagen‐1, and osteonectin, indicating that nuclear actin transport is required for matrix production and osteogenesis to occur (Fig. 2E, 2F, 2G), further supporting our observations and consistent with our recent findings 28. Future studies are warranted to fully unravel this unique and intriguing interaction between cytoskeletal actin reorganization and intranuclear actin transport.
We also investigated changes brought about by CytoD with high‐resolution RNA‐sequencing and identified that that CytoD‐treatment had donor‐specific effects resulting in strong upregulated or downregulated genes as early as 24 hours post‐treatment without osteogenic supplements. Based on our previous study, we note that prolonged treatment (>48 hours) with CytoD may trigger secondary effects that causes cells to differentiate into both osteogenic and adipogenic pathways 28. In the current study, we note that short‐term (24 hours) treatment results in activation of the osteogenic program in adipose MSCs. The most prominently upregulated gene is HSD11B1 which facilitates glucocorticoid synthesis, subsequently affecting their proliferation and differentiation 41. Also, polymorphisms in HSD11B11 have been associated with age‐related osteoporosis 42, 43. CytoD‐induced increase in HSD11B1 expression implies that CytoD is indeed upregulating genes linked to bone metabolism, thus validating the observed in vitro phenotypic effects on MSCs. Other genes involved in the CytoD response in adipose MSCs are NPR1, which is a cell surface binding site for VEGF 44, 45, as well as MTSS1 (missing in metastasis) protein, which is implicated in actin reorganization 46 and regulation of cell motility by modulating different Arp2/3 activators 47. The observation that CytoD upregulates MTSS1 supports the concept that CytoD‐induced osteogenesis proceeds by modulation of the Arp2/3 complex 28. Our data also revealed downregulation of TEAD4 with CytoD. Importantly, TEAD transcriptional factors interact with transcriptional cofactors YAP and TAZ that are critical for controlling skeletal stem cell renewal and differentiation 48, 49, 50.
We note that CytoD‐induced osteogenesis is not accompanied by a significant increase in the mRNA levels of RUNX2 at early time points (<3 days) based on both qPCR and RNA‐seq data (Fig. 1D; Supporting Information Fig. 2). However, the protein levels of RUNX2 are increased during early osteogenesis (Fig. 7). The differences in mRNA and protein levels may be related to other gene regulatory mechanisms (e.g., RUNX2 protein stability and miRNA mediated control), as well as perhaps additional changes in the intrinsic DNA binding activity and transcriptional activity of RUNX2 51, 52. Importantly, the mechanism by which CytoD‐mediated osteogenic pathway proceeds initially is mechanistically different. This is supported by our RNA‐sequencing data which revealed that the expression of bone‐related genes such as RUNX2, ALP, IBSP, COL2A1, and DCN do not exhibit major changes at earlier time points, similar to the mRNA levels assayed by qPCR (Fig. 1D).
Epigenetic enzymes and chromatin binding proteins control lineage determination in MSCs. Using our recently reported hybrid expression screening platform 53, we identified epigenetic regulators modified in adipose MSCs following CytoD treatment. CytoD‐induced osteogenic differentiation was accompanied by alterations in the expression of EZH2 and HDAC5, factors shown by us and others to regulate bone formation 9, 10, 18, 19, 29. Collectively, our RNA‐sequencing data validates CytoD‐induced phenotypic changes in adipose MSCs presented in this study and confirms that CytoD improves adipose MSC osteogenesis by modulating epigenetic regulators.
To understand the downstream components of CytoD‐mediated osteogenesis, we established an inverse relationship between the epigenetic regulator EZH2 and cell‐fate‐determining transcription factor RUNX2 at both mRNA and protein levels. This finding is consistent with a model wherein CytoD‐driven osteogenesis via upregulation of osteogenic master regulator RUNX2 51 is associated with a concomitant repression of EZH2. This finding suggests that RUNX2 and EZH2 may function in a reciprocal and interlocked regulatory circuit. Previous studies from our group have identified that EZH2 has effects on skeletal development and this has been shown both in vitro and in vivo using EZH2 inhibitor drugs and genetic models respectively. The major novelty of the current findings is that osteogenic induction by Cytochalasin D is accompanied by a reduction of the epigenetic Polycomb Group protein EZH2, which is known to inhibit osteogenic differentiation, whereas is less critical for adipogenesis 18, 19.
Given the translational potential of adipose‐MSCs cells for orthopedic repair, it is important to understand how best to sensitize these cells to initiate a robust differentiation program and enable them into becoming potent bone‐forming or mineralizing cells without perturbing normal cellular functions. Our current findings favor the conclusion that osteogenic differentiation of adipose derived MSCs is improved upon treatment with CytoD, because there is increased expression of histochemical markers (after 1 week) and osteogenic mRNA markers (at 24 hours) after CytoD treatment (based on our RNA‐sequencing data) even in the absence of standard osteogenic cocktail (dexamethasone, ascorbic acid, beta‐glycerol phosphate). It remains to be established whether CytoD alone is superior over the use of standard osteogenic medium, and what the precise sequela of temporal and mechanistic events are that ensue upon induction by CytoD. Another limitation with implementing CytoD as osteogenic agent is that CytoD has pleiotropic activity and distinct donor‐specific effects. Yet, we are optimistic that future studies may validate the concept that CytoD can improve the osteogenic potential for adipose MSCs through specific mechanisms to complement standard osteogenic cocktails. In addition, it will be important to compare the effects on MSCs between CytoD‐only treatment versus CytoD‐supplemented osteogenic medium.
Conclusion
Our study has established that CytoD treatment represent a novel approach for improving osteogenesis in adipose MSCs and that CytoD functions at least in part by modulating the reciprocal functional interplay between EZH2 and the bone master regulator RUNX2 program and nuclear actin‐controlled stem cell commitment/differentiation. In a clinical context, because of the observed stimulatory effects of CytoD, pre‐conditioning adipose MSCs with CytoD should permit or enhance the osteogenic capacity of adipose MSCs for bone formation. Our work suggests that osteogenic differentiation is associated with rearrangements in cytoskeletal and nuclear architecture via actin relocalization, as well as alterations in epigenetic mechanisms that normally suppress osteoblastogenesis.
Author Contributions
R.M.S.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; A.D.: collection and assembly of data, data analysis and interpretation, manuscript review; B.M.: collection and assembly of data; B.S. and S.M.C.: data analysis and interpretation, manuscript review; A.B.D.: data analysis and interpretation, provision of study material, manuscript review; J.R.: conception and design, data analysis and interpretation, manuscript review. A.J.v.W.: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
A.B.D. is an employee of and has intellectual property rights with Mill Creek Life Sciences The other authors indicated no potential conflicts of interest.
Note Added in Proof
This article was published online on 27 December 2017. Minor edits have been made that do not affect data. This notice is included in the online and print versions to indicate that both have been corrected 29 January 2018.
Supporting information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Table 1
Acknowledgments
These studies were supported by funding from NIH (R01 AR049069 to A.Jv.W., R01 AR056655 and AR066616 to J.R.), the Mayo Clinic Center of Regenerative Medicine (to R.M.S.) and Kogod Center on Aging (to R.M.S.), NIH F32 AR066508 (to A.D.), and the generous philanthropic support of William and Karen Eby. We appreciate our institutional colleagues in Mayo Clinic for stimulating discussions, including Drs. Eric Lewallen, Janet Denbeigh and Roman Thaler, as well as Gunes Uzer (UNC). We also acknowledge the support of the Bioinformatics Core and the Medical Genome Facility at Mayo Clinic.
References
- 1. Squillaro T, Peluso G, Galderisi U. Clinical trials with mesenchymal stem cells: An update. Cell Transplant 2016;25:829–848. [DOI] [PubMed] [Google Scholar]
- 2. Riester SM, Denbeigh JM, Lin Y et al. Safety studies for use of adipose tissue‐derived mesenchymal stromal/stem cells in a rabbit model for osteoarthritis to support a phase i clinical trial. Stem Cells Translational Medicine 2017;6:910–922., [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen BK, Staff NP, Knight AM et al. A safety study on intrathecal delivery of autologous mesenchymal stromal cells in rabbits directly supporting Phase I human trials. Transfusion 2015;55:1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Camilleri ET, Gustafson MP, Dudakovic A et al. Identification and validation of multiple cell surface markers of clinical‐grade adipose‐derived mesenchymal stromal cells as novel release criteria for good manufacturing practice‐compliant production. Stem Cell Res Ther 2016;7:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Samsonraj RM, Rai B, Sathiyanathan P et al. Establishing criteria for human mesenchymal stem cell potency. Stem Cells 2015;33:1878–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pollock K, Samsonraj RM, Dudakovic A et al. Improved post‐thaw function and epigenetic changes in mesenchymal stromal cells cryopreserved using multicomponent osmolyte solutions. Stem Cells Dev 2017;26:828–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ling L, Camilleri ET, Helledie T et al. Effect of heparin on the biological properties and molecular signature of human mesenchymal stem cells. Gene 2016;576:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wagner ER, Bravo D, Dadsetan M et al. Ligament tissue engineering using a novel porous polycaprolactone fumarate scaffold and adipose tissue‐derived mesenchymal stem cells grown in platelet lysate. Tissue Eng Part A 2015;21:2703–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dudakovic A, Camilleri E, Riester SM et al. High‐resolution molecular validation of self‐renewal and spontaneous differentiation in clinical‐grade adipose‐tissue derived human mesenchymal stem cells. J Cell Biochem 2014;115:1816–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dudakovic A, Camilleri ET, Lewallen EA et al. Histone deacetylase inhibition destabilizes the multi‐potent state of uncommitted adipose‐derived mesenchymal stromal cells. J Cell Physiol 2015;230:52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lewallen EA, Jones DL, Dudakovic A et al. Osteogenic potential of human adipose‐tissue‐derived mesenchymal stromal cells cultured on 3D‐printed porous structured titanium. Gene 2016;581:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marmotti A, de Girolamo L, Bonasia DE et al. Bone marrow derived stem cells in joint and bone diseases: A concise review. Int Orthop 2014;38:1787–1801. [DOI] [PubMed] [Google Scholar]
- 13. Hermida‐Gómez T, Fuentes‐Boquete I, Gimeno‐Longas MJ et al. Bone marrow cells immunomagnetically selected for CD271+ antigen promote in vitro the repair of articular cartilage defects. Tissue Eng Part A 2011;17:1169–1179. [DOI] [PubMed] [Google Scholar]
- 14. Chen G, Deng C, Li Y‐P. TGF‐β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 2012;8:272–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lombardi G, Di Somma C, Rubino M et al. The roles of parathyroid hormone in bone remodeling: Prospects for novel therapeutics. J Endocrinol Invest 2011;34:18–22. [PubMed] [Google Scholar]
- 16. Langenbach F, Handschel J. Effects of dexamethasone, ascorbic acid and beta‐glycerophosphate on the osteogenic differentiation of stem cells in vitro. Stem Cell Res Ther 2013;4:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maria S, Swanson MH, Enderby LT et al. Melatonin‐micronutrients Osteopenia Treatment Study (MOTS): A translational study assessing melatonin, strontium (citrate), vitamin D3 and vitamin K2 (MK7) on bone density, bone marker turnover and health related quality of life in postmenopausal osteopenic women following a one‐year double‐blind RCT and on osteoblast‐osteoclast co‐cultures. Aging (Albany NY) 2017;9:256–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dudakovic A, Camilleri ET, Riester SM et al. Enhancer of Zeste Homolog 2 inhibition stimulates bone formation and mitigates bone loss caused by ovariectomy in skeletally mature mice. J Biol Chem 2016;291:24594–24606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dudakovic A, Camilleri ET, Xu F et al. Epigenetic control of skeletal development by the histone methyltransferase Ezh2. J Biol Chem 2015;290:27604–27617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thaler R, Maurizi A, Roschger P et al. Anabolic and antiresorptive modulation of bone homeostasis by the epigenetic modulator sulforaphane, a naturally occurring isothiocyanate. J Biol Chem 2016;291:6754–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hemming S, Cakouros D, Isenmann S et al. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells 2014;32:802–815. [DOI] [PubMed] [Google Scholar]
- 22. Hemming S, Cakouros D, Codrington J et al. EZH2 deletion in early mesenchyme compromises postnatal bone microarchitecture and structural integrity and accelerates remodeling. FASEB J 2017;31:1011–1027. [DOI] [PubMed] [Google Scholar]
- 23. Sen B, Guilluy C, Xie Z et al. Mechanically induced focal adhesion assembly amplifies anti‐adipogenic pathways in mesenchymal stem cells. Stem Cells 2011;29:1829–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sen B, Xie Z, Uzer G et al. Intranuclear actin regulates osteogenesis. Stem Cells 2015;33:3065–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dopie J, Skarp K‐P, Kaisa Rajakyla E et al. Active maintenance of nuclear actin by importin 9 supports transcription. Proc Natl Acad Sci USA 2012;109:E544–E552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crespo‐Diaz R, Behfar A, Butler GW et al. Platelet lysate consisting of a natural repair proteome supports human mesenchymal stem cell proliferation and chromosomal stability. Cell Transplant 2011;20:797–811. [DOI] [PubMed] [Google Scholar]
- 27. Samsonraj RM, Raghunath M, Hui JH et al. Telomere length analysis of human mesenchymal stem cells by quantitative PCR. Gene 2013;519:348–355. [DOI] [PubMed] [Google Scholar]
- 28. Sen B, Uzer G, Samsonraj RM et al. Intranuclear actin structure modulates mesenchymal stem cell differentiation. Stem Cells 2017;35:1624–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wein MN, Spatz J, Nishimori S et al. HDAC5 controls MEF2C‐driven sclerostin expression in osteocytes. J Bone Miner Res 2015;30:400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wein MN, Liang Y, Goransson O et al. SIKs control osteocyte responses to parathyroid hormone. Nat Commun 2016;7:13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Merriman HL, van Wijnen AJ, Hiebert S et al. The tissue‐specific nuclear matrix protein, NMP‐2, is a member of the AML/CBF/PEBP2/runt domain transcription factor family: Interactions with the osteocalcin gene promoter. Biochemistry 1995;34:13125–13132. [DOI] [PubMed] [Google Scholar]
- 32. McBeath R, Pirone DM, Nelson CM et al. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell 2004;6:483–495. [DOI] [PubMed] [Google Scholar]
- 33. Treiser MD, Yang EH, Gordonov S et al. Cytoskeleton‐based forecasting of stem cell lineage fates. Proc Natl Acad Sci USA 2010;107:610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Driscoll TP, Cosgrove BD, Heo S‐J et al. Cytoskeletal to nuclear strain transfer regulates YAP signaling in mesenchymal stem cells. Biophys J 2015;108:2783–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martins RP, Finan JD, Guilak F et al. Mechanical regulation of nuclear structure and function. Annu Rev Biomed Eng 2012;14:431–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haase K, Macadangdang JK, Edrington CH et al. Extracellular forces cause the nucleus to deform in a highly controlled anisotropic manner. Sci Rep 2016;6:21300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Le HQ, Ghatak S, Yeung CY et al. Mechanical regulation of transcription controls Polycomb‐mediated gene silencing during lineage commitment. Nat Cell Biol 2016;18:864–875. [DOI] [PubMed] [Google Scholar]
- 38. Heo S‐J, Thorpe SD, Driscoll TP et al. Biophysical regulation of chromatin architecture instills a mechanical memory in mesenchymal stem cells. Sci Rep 2015;5:16895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen B et al. Influence of inhibition of actin polymerization on adipogenic differentiation of rat Achilles‐derived tendon stem cells in vitro. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 2015;29:206–212. [PubMed] [Google Scholar]
- 40. Shi W, Xie Y, He J et al. Microgravity induces inhibition of osteoblastic differentiation and mineralization through abrogating primary cilia. Sci Rep 2017;7:1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cooper MS, Rabbitt EH, Goddard PE et al. Osteoblastic 11beta‐hydroxysteroid dehydrogenase type 1 activity increases with age and glucocorticoid exposure. J Bone Miner Res 2002;17:979–986. [DOI] [PubMed] [Google Scholar]
- 42. Siggelkow H, Etmanski M, Bozkurt S et al. Genetic polymorphisms in 11beta‐hydroxysteroid dehydrogenase type 1 correlate with the postdexamethasone cortisol levels and bone mineral density in patients evaluated for osteoporosis. J Clin Endocrinol Metab 2014;99:E293–E302. [DOI] [PubMed] [Google Scholar]
- 43. Hwang J‐Y, Lee SH, Kim GS et al. HSD11B1 polymorphisms predicted bone mineral density and fracture risk in postmenopausal women without a clinically apparent hypercortisolemia. Bone 2009;45:1098–1103. [DOI] [PubMed] [Google Scholar]
- 44. Zelzer E, Mamluk R, Ferrara N et al. VEGFA is necessary for chondrocyte survival during bone development. Development 2004;131:2161–2171. [DOI] [PubMed] [Google Scholar]
- 45. Chiba A, Watanabe‐Takano H, Terai K et al. Osteocrin, a peptide secreted from the heart and other tissues, contributes to cranial osteogenesis and chondrogenesis in zebrafish. Development 2016;144:334–344. [DOI] [PubMed] [Google Scholar]
- 46. Wang Y, Zhou K, Zeng X et al. Tyrosine phosphorylation of missing in metastasis protein is implicated in platelet‐derived growth factor‐mediated cell shape changes. J Biol Chem 2007;282:7624–7631. [DOI] [PubMed] [Google Scholar]
- 47. Lin J, Liu J, Wang Y et al. Differential regulation of cortactin and N‐WASP‐mediated actin polymerization by missing in metastasis (MIM) protein. Oncogene 2005;24:2059–2066. [DOI] [PubMed] [Google Scholar]
- 48. Tang Y, Rowe RG, Botvinick EL et al. MT1‐MMP‐dependent control of skeletal stem cell commitment via a beta1‐integrin/YAP/TAZ signaling axis. Dev Cell 2013;25:402–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tang Y, Feinberg T, Keller ET et al. Snail/Slug binding interactions with YAP/TAZ control skeletal stem cell self‐renewal and differentiation. Nat Cell Biol 2016;18:917–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hau JC, Erdmann D, Mesrouze Y et al. The TEAD4‐YAP/TAZ protein‐protein interaction: Expected similarities and unexpected differences. Chembiochem 2013;14:1218–1225. [DOI] [PubMed] [Google Scholar]
- 51. Zhang Y, Xie R‐L, Croce CM et al. A program of microRNAs controls osteogenic lineage progression by targeting transcription factor Runx2. Proc Natl Acad Sci USA 2011;108:9863–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Y, Xie R‐L, Gordon J et al. Control of mesenchymal lineage progression by microRNAs targeting skeletal gene regulators Trps1 and Runx2. J Biol Chem 2012;287:21926–21935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dudakovic A, Gluscevic M, Paradise CR et al. Profiling of human epigenetic regulators using a semi‐automated real‐time qPCR platform validated by next generation sequencing. Gene 2017;609:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Table 1