

Abstract
Monocyte chemotactic protein-1-induced protein 1 (MCPIP1) plays a important role in ischemia/reperfusion (I/R) injury. Autophagy is involved in activating endothelial cells in response to I/R. However, researchers have not clearly determined whether MCPIP1 mediates I/R injury in endothelial cells via autophagy, and its downstream mechanism remains unclear. Western blotting analyses and immunocytochemistry were applied to detect protein levels were detected in HUVECs. An in vitro scratch assay was used to detect cell migration. Cells were transfected with siRNAs to knockdown MCPIP1 and high mobility group box 1 (HMGB1) expression. The pharmacological activator of autophagy rapamycin and the specific calcium-sensing receptor (CaSR) inhibitor NPS-2143 were used to confirm the roles of autophagy and CaSR in I/R injury. I/R induced HMGB1 and CaSR expression, which subsequently upreguated the migration and apoptosis of HUVECs and coincided with the increase of autophagy. HMGB1 was involved in cell migration, whereas CaSR specifically participated in I/R-induced HUVEC apoptosis. Based on these findings, I/R-induced MCPIP1 expression regulates the migration and apoptosis of HUVECs via HMGB1 and CaSR, respectively, suggesting a new therapeutic targetof I/R injury.
Introduction
Vascular endothelial cell dysfunction plays an crucial role in ischemia/reperfusion (I/R) injury, a common aspect of cardiovascular disease that is characterized by the over-production of inflammatory factors, such as cytokines and chemokines1,2. Various biological events, such as autophagy, endoplasmic reticulum stress (ERS) and ubiquitination, are involved in endothelial cell dysfunction. Autophagy plays an important role in the ability of cells to adapt to changing environmental conditions and in cellular remodeling during angiogenesis3–6. However, the mechanisms underlying I/R-induced endothelial cell dysfunction that are associated with autophagy remain poorly understood.
Based on accumulating evidence, monocyte chemotactic protein-1 (MCP-1) and its downstream molecule MCP-1-induced protein 1 (MCPIP1) facilitate vascular inflammation and endothelial dysfunction in response to I/R1,7–10. For example, MCPIP1 has been shown to induce angiogenesis during placental vasculogenesis, which in turn leads to vascular remodeling11–13. Interestingly, recent studies have linked MCP-1/MCPIP1 with autophagy under different conditions that induce cell activation and apoptosis. For instance, MCP-1 and MCPIP1 contribute to cardiomyoblast death in patients with heart failure and are associated with autophagy resulting from ERS14. Moreover, MCPIP1-induced autophagy is required for angiogenesis in patients with angiogenesis-related cardiovascular diseases15. On the other hand, MCPIP1/p53 expression is induced after SiO2 exposure and promotes macrophage activation and apoptosis, suggesting the presence of a general link between the MCPIP1 signaling pathway and autophagy in different diseases16,17.
Autophagy is an important biological event required to maintain cell homeostasis5,18,19. However, several stimuli may cause irreversible cell injury and cell death via autophagy, which contribute to some pathologies20–23. According to previous data from our lab, I/R induces the expression of inducible nitric oxide synthase (iNOS), which subsequently increases the migration and apoptosis of human umbilical vein endothelial cells (HUVECs) by promoting autophagy3. Moreover, some studies have suggested a direct link between MCP-1 and iNOS, which is consistent with our findings in endothelial cells cultured under I/R conditions24–26. However, the downstream effects of I/R-induced MCPIP1 expression on cell migration and apoptosis associated with autophagy remain unknown.
The present study aimed to determine the mechanism by which MCPIP1 regulated I/R-induced HUVEC migration and apoptosis and the specific roles of autophagy in these processes. The conclusions of current research may help to understand the mechanisms regulating MCPIP1 expression and its functional relevance to I/R injury, providing insight into potential therapeutic targets for myocardial ischemia.
Materials and Methods
Reagents
Fetal bovine serum (FBS), normal goat serum, Dulbecco’s Modified Eagle’s Medium (DMEM; #1200-046), and 10× MEM (11430–030) were all obtained from Life Technologies. Amphotericin B (BP2645) and the supplement GlutaMAX (35050–061) were obtained from Gibco, and Pen/Strep (15140–122) was obtained from Fisher Scientific. Antibodies against the calcium-sensing receptor (CaSR, sc33821, rabbit), MCPIP1 (sc136750, goat) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; sc32233, mouse) were obtained from Santa Cruz Biotechnology, Inc. The antibody against high mobility group box 1 (HMGB1) protein (ab18256, rabbit) was obtained from Abcam Biotechnology, Inc. Control siRNA (sc-37007), a non-targeting 20–25-nt siRNA designed as a negative control, was obtained from Santa Cruz Biotechnology, Inc. The reagent used for siRNA transfection was purchased from Santa Cruz Biotechnology, Inc.
Cell culture
HUVECs were purchased from ScienCell® and maintained as previously described1,3.
Cell-based model simulating ischemia/reperfusion injury
The I/R model used here based upon a version of a described method1,3,27–30.
Lentiviral transduction of HUVECs
HUVECs were transduced with the LV-GFP lentivirus (Hanbio, Inc., Shanghai, CN) as previously described31–34. Briefly, HUVECs (passage (P) 3–5) were cultured in a 24-well plate (1 × 104 cells/well) in DMEM supplemented with 10% FBS for 48 h. The medium was then replaced with 1 mL of fresh medium and 8 µg/mL of polybrene. 15 μl of lentivirus solution (107 IU/mL) were added to each well, and the 24-well plate was incubated at 37 °C with 5% CO2 for 24 h. Following incubation, the medium was replaced with fresh DMEM containing 10% FBS, and the cells were cultured at 37 °C and 5% CO2 until they reached >50% confluence. The transduced cells were selected using puromycin by replacing the medium with DMEM containing 10 µg/mL puromycin and 10% FBS and culturing the cells at 37 °C in a 5% CO2 atmosphere for 24 h. The cells were subsequently washed twice with fresh DMEM containing 10% FBS. Transduced and pure HUVEC cultures were expanded and stored in liquid nitrogen as previously described35.
In vitro scratch assay
Cell migration in a 2D culture system was evaluated using an in vitro scratch assay1. To evaluate the effect of cell division on migration, mitomycin pretreatment was applied for one hour. As shown in Fig. S1, mitomycin did not alter the effect of I/R on cell migration, which ruled out the effect of cell division on migration.
Immunoblotting
Immunoblotting was performed as previously described3.
Immunofluorescence staining
Immunofluorescence staining was performed as previously described35. HUVECs (2 × 105 cell/well) were seeded onto coverslips placed on 24-well plates. After treatment, the coverslips were rinsed twice with PBS, and the cells were fixed with 4% paraformaldehyde overnight. The fixed cells were incubated with 0.3% Triton X-100 in PBS for 30 min and then rinsed twice with PBS. The cells were blocked with 10% normal goat serum (NGS) in 0.3% Triton X-100 for 2 h at room temperature and then incubated with primary antibodies overnight at 4 °C. After three washes with PBS, the cells were incubated with fluorescently-labeled secondary antibodies (Alex Flour® 488 and 594, 1:250), and the cell nuclei were stained with DAPI (4,′6-diamidino-2-phenylindole) and mounted with ProLong Gold antifade reagent (Life Technologies®). Images were captured under a fluorescence microscope (Olympus IX70, Olympus America, Inc., Center Valley, PA).
MTT assay
Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) method31.
CCK8 assay
Cell viability was measured using the CCK8 assay (Dojindo, Tokyo, Japan) following the manufacturer’s protocol. Briefly, 10 μl of CCK8 was added to each culture well, and then, the cells were incubated at 37 °C for 2 h. The absorbance at 450 nm was measured using a spectrophotometer. Cell viability was expressed as the percentage of viable cells in the experimental group relative to the control group.
Hoechst staining
Cells were fixed and stained with 5 μM Hoechst 33324 (Invitrogen) for 15 min at room temperature to quantify the number of apoptotic cells1,36.
Statistics
The data are expressed as the means ± SEM. Unpaired numerical data were compared using either an unpaired t-test (two groups) or an ANOVA (more than two groups). Significance was established at p < 0.05.
Results
I/R-induced MCPIP1 expression is associated with up-regulated HMGB1 expression in HUVECs
Based on previous data from our lab, MCPIP1 expression is induced by I/R in HUVECs and is associated with cell migration and apoptosis, but the detailed mechanism remains unclear1. Interestingly, a recently study suggested a link between MCPIP1 and HMGB1 in macrophage activation/apoptosis and migration, indicating a role for HMGB1 in I/R-induced endothelial cell dysfunction37. HMGB1 induces endothelial cells to increase the expression of adhesion molecules38 and causes dendritic cell maturation39 through a downstream signaling pathway, such as the mitogen-activated protein kinase (MAPK) pathway or the nuclear factor kappa B (NF-κB) pathway, thereby facilitating cellular responses40. Therefore, we first examined the role of HMGB1 in the functional changes observed in HUVECs subjected to I/R. As shown in Fig. 1A,B, the exposure of HUVECs to I/R resulted in significant increases in the levels of the HMGB1 protein in a time-dependent manner, which was confirmed using immunofluorescence staining (Fig. 1C). The expression of both HMGB1 and MCPIP1 was down-regulated with siRNAs to investigate the association between up-regulated HMGB1 and MCPIP1 expression. As shown in Fig. 1D,E, MCPIP1 siRNA significantly inhibited the I/R-induced increase in MCPIP1 expression as expected; the I/R-induced increase in HMGB1 levels was also attenuated. Moreover, HMGB1 siRNA significantly inhibited the I/R-induced increase in HMGB1 expression, but not MCPIP1 expression, indicating that HMGB1 is downstream of MCPIP1 (Fig. 1F,G). The effect of MCPIP1 siRNA on HMGB1 levels was confirmed by immunocytochemistry staining (Fig. 2A). To further understand the mechanism by which MCPIP1 induced HMGB1 expression in I/R, the effect of MCPIP1 siRNA on hmbg1 mRNA expression was measured. Interestingly, MCPIP1 siRNA had no significant effect on hmbg1 mRNA expression (Fig. 2B), indicating that a post-transcriptional mechanism may be involved.
Figure 1.

I/R induced HMGB1 expression in HUVECs. (A) Representative blots showing that I/R induced HMGB1 expression in a time-dependent manner. (B) Densitometric analyses of HMGB1 levels from four independent experiments; *p < 0.05 compared with the 0 h group. (C) Representative images of immunocytochemical staining showing that I/R induced MCPIP1 and HMGB1 expression in HUVECs. Scale bar, 20 μm. (D) Representative blots showing the effects of MCPIP1 siRNA on I/R-induced MCPIP1 and HMGB1 expression. (E) Densitometric analyses of MCPIP1 and HMGB1 levels from four independent experiments; *p < 0.05 compared with the control group. (F) Representative blots showing the effects of HMGB1 siRNA on I/R-induced MCPIP1 and HMGB1 expression. (G) Densitometric analyses of MCPIP1 and HMGB1 levels from four independent experiments; *p < 0.05 compared with the control group, #p < 0.05 compared with the I/R group.
Figure 2.

HMGB1 is involved in I/R-induced HUVEC migration but not apoptosis. (A) Representative images of immunocytochemical staining showing that I/R-induced HMGB1 expression was attenuated by MCPIP1 siRNA in HUVECs. Scale bar, 20 μm. (B) Results from quantitative RT-PCR demonstrating that the expression of hmgb1 mRNA was not affected by MCPIP1 siRNA treatment in HUVEC cells, n = 3. (C) Representative images showing that the I/R-induced migration of HUVECs grown in monolayer cultures was abolished by HMGB1 siRNA. Scale bar, 80 μm. (D) The quantification of the scratch gap distance in six independent experiments is presented. *p < 0.05 compared with the control group. (E) MTT assay showing the effect of HMGB1 siRNA on cell viability following I/R treatment. *p < 0.05 compared with the control group, #p < 0.05 compared with the I/R group. (F) CCK8 assay showing the effect of HMGB1 siRNA on cell viability following I/R treatment. *p < 0.05 compared with the control group, #p < 0.05 compared with the I/R group.
HMGB1 is involved in I/R-mediated HUVEC migration but not apoptosis
As a feature of ischemic heart disease, abnormal angiogenesis is an important aspect of endothelial cell dysfunction, which is is characterized by migratory and proliferative phenotypes and the differentiation of endothelial cells into an angiogenic phenotype1,4,8,41,42. Apoptosis of endothelial cells is the initial step in angiogenesis and the regression of neo-vessels1,3,11–13. MCPIP1 has recently been implicated in the migration and apoptosis of HUVECs in response to I/R and several other stimuli1,35. Functional assays of cell migration and apoptosis were conducted to determine how up-regulated HMGB1 expression is associated with the I/R-induced increase in MCPIP1 expression. As shown in Fig. 2C,D, the results of the scratch assay revealed an increase in HUVEC migration following I/R, which was attenuated by HMGB1 siRNA. Interestingly, although HMGB1 is involved in cell apoptosis under different conditions, the HMGB1 siRNA did not alter the I/R-induced death of HUVECs (Fig. 2E,F), indicating that HMGB1 has a specific role in I/R injury.
The effect of HMGB1 on HUVEC migration is mediated by autophagy
Previous data from our laboratory have suggested a role for autophagy in I/R-induced endothelial cell dysfunction3, whereas HMGB1 has recently been shown to play a role in autophagy43. However, researchers have not determined whether HMGB1-induced cell migration is related to autophagy. We first detected the level of the HMGB1 protein and the autophagy marker LC3B in HUVECs using immunofluorescence staining. HMGB1 and LC3B were co-localized in HUVECs after 6 h of exposure to I/R. Furthermore, HMGB1 siRNA significantly attenuated the I/R-induced increase in LC3B levels (Fig. 3B,C). HMGB1/MCPIP1 siRNAs and the autophagy activator rapamycin (RAPA) were used to investigate the role of autophagy in HMGB1-mediated migration. As shown in Fig. 3D,E, both the HMGB1 and MCPIP1 siRNAs attenuated I/R-induced cell migration, which was restored by pretreatment with RAPA (1 μmol/L) for 1 h. Therefore, MCPIP1 and HMGB1 induced cell migration by promoting autophagy in response to I/R injury.
Figure 3.

Autophagy mediated the effect of HMGB1 on I/R-induced cell migration. (A) Representative images of immunocytochemical staining showing that HMGB1 co-localized with the autophagy marker LC3B in HUVECs in response to I/R injury. Scale bar, 20 μm. (B) Representative blots showing that HMGB1 siRNA attenuated the I/R-induced increase in LC3B expression. (C) Densitometric analyses of LC3B levels from four independent experiments; *p < 0.05 compared with the control group, #p < 0.05 compared with the I/R group. (D) Representative images depicting that the inhibitory effects of HMGB1 and MCPIP1 siRNAs on I/R-induced cell migration were reversed by pretreatment with rapamycin (1 μmol/L, 1 hour). Scale bar, 80 μm. (E) The quantification of the scratch gap distance in six independent experiments is presented. *p < 0.05 compared with the corresponding time point in the control group, #p < 0.05 compared with the corresponding time point in the I/R group.
I/R-induced MCPIP1 expression is associated with increased CaSR expression in HUVECs
Previous data from our laboratory have suggested an important role for CaSR in I/R-induced cardiomyocyte death27,44. As HMGB1 was not involved in apoptosis, we next determined whether CaSR mediated HUVEC apoptosis. First, the CaSR level was detected in HUVECs exposed to the I/R model. As shown in Fig. 4A,B, I/R induced CaSR expression in a time-dependent manner, with a peak at 6 h of reperfusion, which was confirmed by immunofluorescence staining (Fig. 4C). MCPIP1 expression in HUVECs was down-regulated with an siRNA to further examine the link between MCPIP1 and CaSR. As shown in Fig. 4C,D, MCPIP1 siRNA not only significantly inhibited the I/R-induced increase in CaSR expression but also decreased the basal CaSR levels in HUVECs, indicating a strong relationship between MCPIP1 and CaSR. The CaSR-specific inhibitor NPS-2143 was used to clarify the relationship between MCPIP1 and CaSR45. As shown in Fig. 4D,E, inhibition of CaSR with NPS-2143 (5 μmol/L) had no effect on MCPIP1 and CaSR expression. Based on the results shown in Fig. 4C–E, the I/R-induced increase in CaSR expression occurred downstream of MCPIP1 signaling in HUVECs. The effect of MCPIP1 siRNA on CaSR levels was confirmed by immunocytochemistry (Fig. 5A). To further understand the mechanism by which MCPIP1 induced HMGB1 expression in I/R, the effect of MCPIP1 siRNA on casr mRNA expression was measured. Interestingly, MCPIP1 siRNA had no significant effect on casr mRNA expression (Fig. 5B), indicating that a post-transcriptional mechanism may be involved.
Figure 4.

I/R induced CaSR expression in HUVECs. (A) Representative blots showing that I/R induced CaSR expression in a time-dependent manner. (B) Densitometric analyses of CaSR levels from four independent experiments; *p < 0.05 compared with the 0 h group. (C) Representative images of immunocytochemical staining showing that I/R induced MCPIP1 and CaSR expression in HUVECs. Scale bar, 20 μm. (D) Representative blots showing the effects of MCPIP1 siRNA on I/R-induced CaSR expression. (E) Densitometric analyses of CaSR expression from four independent experiments; *p < 0.05 compared with the control group. (F) Representative blots showing the effects of the CaSR-specific inhibitor NPS-2143 (5 μmol/L, 1 h) on I/R-induced MCPIP1 and CaSR expression. (G) Densitometric analyses of MCPIP1 and CaSR levels from four independent experiments; *p < 0.05 compared with the control group, #p < 0.05 compared with the I/R group.
Figure 5.

CaSR was involved in I/R-induced HUVEC cell apoptosis, but not migration. (A) Representative images of immunocytochemical staining showing that I/R-induced CaSR expression was attenuated by MCPIP1 siRNA in HUVECs. Scale bar, 20 μm. (B) Results from quantitative RT-PCR demonstrating that the expression of casr mRNA was not affected by MCPIP1 siRNA treatment in HUVEC cells, n = 3. (C) The quantification of the scratch gap distance in six independent experiments shows the effect of NPS-2143 on I/R-induced cell migration in HUVECs grown in monolayer cultures. *p < 0.05 compared with the corresponding time point in the control group, #p < 0.05 compared with the corresponding time point in the I/R group. (D) MTT assay showing that NPS-2143 rescued cells from I/R-induced apoptosis. *p < 0.05 compared with the control group, #p < 0.05 compared with the I/R group. (E) Representative images of Hoechst 33342 staining showing that I/R-induced HUVEC apoptosis was attenuated by NPS-2143 pretreatment. Scale bar, 10 μm. (F) Percentages of apoptotic cells from six independent experiments are presented. *p < 0.05 compared with the I/R group.
CaSR is involved in I/R-mediated HUVEC apoptosis but not migration
NPS-2143 was used to investigate the role of CaSR in I/R-induced cell apoptosis and migration to further examine the functional role of CaSR in HUVECs. As shown in Fig. 5C, NPS-2143 alone did not alter HUVEC migration, and the I/R-induced increase in migration was not prevented by pretreatment with NPS-2143, excluding the possibility that CaSR participates in I/R-induced endothelial cell migration. Interestingly, although NPS-2143 pretreatment alone did not induce cell proliferation, this pretreatment rescued HUVECs from I/R-induced death as determined using MTT and CCK8 assays (Fig. 5D,E). This finding was confirmed by staining the cells with Hoechst 33342, a dye that specifically stains nuclei1. As shown in Fig. 5F,G, condensation and fragmentation of nuclei, which are characteristic features of apoptotic cells, were observed in HUVECs that had been subjected to I/R for 6 h. The I/R-induced cell death was significantly ameliorated by pretreatment with NPS-2143, as indicated by the appearance of normal cells characterized by regular and round nuclei (Fig. 5F,G).
The effect of CaSR on HUVEC apoptosis is mediated by autophagy
Recent studies have suggested a link between CaSR and autophagy under different conditions46,47. Although the role of autophagy in the apoptosis of HUVECs subjected to I/R injury has been determined, researchers have not determined whether the effect of CaSR on I/R-induced cell death is mediated by autophagy. The levels of the CaSR protein and the autophagy marker LC3B in HUVECs were detected using immunofluorescence staining. CaSR and LC3B were co-localized in HUVECs exposed to 6 h of I/R. Moreover, NPS-2143 pretreatment attenuated the I/R-induced increase in LC3B expression in HUVECs, indicating a direct link between CaSR and autophagy (Fig. 6A), which was confirmed by western blotting (Fig. 6B,C). HUVECs were treated with both NPS-2143 and RAPA and analyzed using the MTT assay to further examine the functional relationship between CaSR and autophagy. As shown in Fig. 6D, NPS-2143-mediated inhibition of cell death was reversed by RAPA treatment, indicating that the effect of CaSR on I/R-induced cell death was mediated by autophagy.
Figure 6.

Autophagy mediated the effect of CaSR on I/R-induced cell apoptosis. (A) Representative images of immunocytochemical staining showing that CaSR co-localized with the autophagy marker LC3B in HUVECs following I/R injury. The I/R-induced increase in the level of LC3B, but not CaSR, was attenuated by NPS-2143 pretreatment. Scale bar, 20 μm. (B) Representative blots showing that NPS-2143 pretreatment attenuated the I/R-induced increase in LC3B expression. (C) Densitometric analyses of LC3B expression from four independent experiments; *p < 0.05 compared with the control group, #p < 0.05 compared with the I/R group. (D) MTT assay showing that the rescue effect of NPS-2143 on HUVECs subjected to I/R injury was reversed by treatment with rapamycin. n = 6, *p < 0.05 compared with the control group, #p < 0.05 compared with the I/R group.
Discussion
Whereas many studies have examined I/R injury in cardiomyocytes, the direct effect of I/R on endothelial cells has received less attention. Apoptosis occurs in endothelial cells in the very early stages of reperfusion following the radial spread of apoptosis to surrounding cardiac myocytes, indicating that soluble pro-apoptotic mediators are released from endothelial cells to promote myocyte death48. Cytokines, chemokines and oxidative stress have been shown to be involved in this process1,3,49. The aim of the current study was to investigate the downstream events mediated by endothelial cell-derived MCPIP1 that lead to cell activation and migration in response to I/R injury.
MCPIP1 plays an important role in inflammatory diseases, such as pneumoconiosis16, neuronal injury50, and I/R injury1. MCPIP1 expression is rapidly increased in HUVECs after I/R, whereas most functional changes occur several hours after reperfusion; therefore, the downstream pathway is of interest. Recently, studies have identified a link between MCPIP1 and HMGB1 under various conditions37,50, indicating a potential role for the interaction between MCPIP1 and HMGB1 in I/R-induced endothelial cell dysfunction. HMGB1 has been reported to exert multiple cardioprotective effects51. Interestingly, although HMGB1 mediated I/R-induced cell migration, which is consistent with previous studies of HMGB1 and cell migration52–54, HMGB1 did not have an effect on cell viability. However, contradictory reports have been published regarding the role of HMGB1 in apoptosis in different cell types. HMGB1 exerts a protective effect on cardiomyocytes, but it also induces endothelial progenitor cell apoptosis through the receptor for the advanced glycation end products (RAGE)-dependent protein kinase R-like ER kinase (PERK)/eukaryotic translation initiation factor 2 alpha (eIF2α) pathway51,55, indicating that HMGB1 has a complex role in cell apoptosis. HMGB1 is associated with autophagy under various conditions43, and our results provided another example of an association between HMGB1 and autophagy.
Apoptosis is an important aspect of endothelial cell dysfunction. Although CaSR is involved in I/R-induced cardiomyocyte death, further studies are required to determine whether the I/R-induced increase in CaSR expression is a general phenomenon. CaSR not only responds to extracellular Ca2+ but is also activated by many ligands, such as divalent and trivalent cations, L-amino acids, and polyamines56. Although most reports indicate a role for CaSR in apoptosis, it also promotes osteoblast proliferation during bone remodeling57. In the current study, NPS-2143, a CaSR-specific inhibitor45, rescued cells stimulated by I/R from apoptosis and increased the levels of LC3B, a marker of autophagy, clearly indicting that CaSR plays a role in apoptosis associated with endothelial cell dysfunction. Furthermore, the preventative effect of NPS-2143 was reversed by treatment with rapamycin, a specific autophagy activator, strongly indicating that CaSR induced apoptosis via autophagy. Interestingly, CaSR was involved in cell apoptosis, but it had no effect on cell migration, suggesting that HMGB1 and CaSR coordinately regulated I/R-induced endothelial cell dysfunction. Moreover, both HMGB1 and CaSR affect downstream functions via autophagy, suggesting that autophagy has a key role in endothelial cell dysfunction, which is consistent with our previous findings of autophagy in endothelial cells subjected to I/R injury3. Interestingly, MCPIP1 siRNA did not affect hmgb1 or casr mRNA expression, indicating that a post-transcriptional mechanism may be involved. In fact, a recent study from our group found that circRNA was involved in the effect of MCPIP1/ZC3H12A on downstream cascades via ubiquitination. Whether MCPIP1 also regulates HMGB1 and CaSR through a post-transcriptional mechanism warrants further investigation.
In summary, our study identified a mechanism by which MCPIP1 regulates endothelial cells via HMGB1 and CaSR in response to I/R. Furthermore, I/R-mediated both HMGB1 and CaSR expression affected cell migration and apoptosis via autophagy, resulting in upregulation of angiogenesis and apoptosis during the late stages of I/R injury (Fig. 7). These findings have implications for I/R injury in heart failure patients. A better understanding of the mechanisms regulating MCPIP1 and autophagy may aid in the development of therapeutic strategies to treat I/R injuries.
Figure 7.
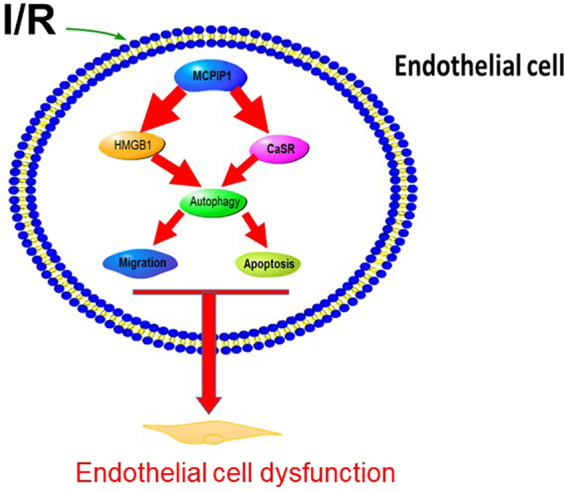
Schematic model of the mechanism by which HMGB1, CaSR, and autophagy promote I/R-induced endothelial cell dysfunction.
Electronic supplementary material
Acknowledgements
This study was partially supported by the resources and facilities of the core laboratory of the Medical School of Southeast University. BTZ was supported by a grant obtained from the Natural Science Foundation of Jiangsu Province, China (No. BK20141497). JC was supported by grants obtained from the National Natural Science Foundation of China (81473263) and the Natural Science Foundation of Jiangsu Province, China (No. BK20141347).
Author Contributions
X.X. and T.Z. performed the experiments, interpreted the data, prepared the figures, and wrote the manuscript. L.C., S.D., H.C., and J.W. performed the experiments and interpreted the data. H.Y. designed the experiments, interpreted the data, and wrote the manuscript. T.Z. and J.C. provided laboratory space and funding, designed the experiments, interpreted the data, wrote the manuscript, and directed the project. All authors read, discussed, and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-20195-6.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Tiebing Zhu, Email: jyfz15117@sina.com.
Jie Chao, Email: chaojie@seu.edu.cn.
References
- 1.Zhu T, et al. The Role of MCPIP1 in Ischemia/Reperfusion Injury-Induced HUVEC Migration and Apoptosis. Cell Physiol Biochem. 2015;37:577–591. doi: 10.1159/000430378. [DOI] [PubMed] [Google Scholar]
- 2.Song MA, Paradis AN, Gay MS, Shin J, Zhang L. Differential expression of microRNAs in ischemic heart disease. Drug Discov Today. 2015;20:223–235. doi: 10.1016/j.drudis.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu T, Yao Q, Wang W, Yao H, Chao J. iNOS Induces Vascular Endothelial Cell Migration and Apoptosis Via Autophagy in Ischemia/Reperfusion Injury. Cell Physiol Biochem. 2016;38:1575–1588. doi: 10.1159/000443098. [DOI] [PubMed] [Google Scholar]
- 4.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 5.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA. 2008;105:20567–20574. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang SJ, et al. Inhibition of endoplasmic reticulum stress by neuregulin-1 protects against myocardial ischemia/reperfusion injury. Peptides. 2017;88:196–207. doi: 10.1016/j.peptides.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 7.Deindl E, Schaper W. The art of arteriogenesis. Cell Biochem Biophys. 2005;43:1–15. doi: 10.1385/CBB:43:1:001. [DOI] [PubMed] [Google Scholar]
- 8.Ito WD, et al. Monocyte chemotactic protein-1 increases collateral and peripheral conductance after femoral artery occlusion. Circ Res. 1997;80:829–837. doi: 10.1161/01.RES.80.6.829. [DOI] [PubMed] [Google Scholar]
- 9.Moldovan NI, Goldschmidt-Clermont PJ, Parker-Thornburg J, Shapiro SD, Kolattukudy PE. Contribution of monocytes/macrophages to compensatory neovascularization: the drilling of metalloelastase-positive tunnels in ischemic myocardium. Circ Res. 2000;87:378–384. doi: 10.1161/01.RES.87.5.378. [DOI] [PubMed] [Google Scholar]
- 10.Voskuil M, et al. Modulation of collateral artery growth in a porcine hindlimb ligation model using MCP-1. Am J Physiol Heart Circ Physiol. 2003;284:H1422–1428. doi: 10.1152/ajpheart.00506.2002. [DOI] [PubMed] [Google Scholar]
- 11.Peters K, Troyer D, Kummer S, Kirkpatrick CJ, Rauterberg J. Apoptosis causes lumen formation during angiogenesis in vitro. Microvasc Res. 2002;64:334–338. doi: 10.1006/mvre.2002.2438. [DOI] [PubMed] [Google Scholar]
- 12.Tertemiz F, Kayisli UA, Arici A, Demir R. Apoptosis contributes to vascular lumen formation and vascular branching in human placental vasculogenesis. Biol Reprod. 2005;72:727–735. doi: 10.1095/biolreprod.104.034975. [DOI] [PubMed] [Google Scholar]
- 13.Duval H, Harris M, Li J, Johnson N, Print C. New insights into the function and regulation of endothelial cell apoptosis. Angiogenesis. 2003;6:171–183. doi: 10.1023/B:AGEN.0000021390.09275.bc. [DOI] [PubMed] [Google Scholar]
- 14.Younce CW, Kolattukudy PE. MCP-1 causes cardiomyoblast death via autophagy resulting from ER stress caused by oxidative stress generated by inducing a novel zinc-finger protein, MCPIP. Biochem J. 2010;426:43–53. doi: 10.1042/BJ20090976. [DOI] [PubMed] [Google Scholar]
- 15.Roy A, Kolattukudy PE. Monocyte chemotactic protein-induced protein (MCPIP) promotes inflammatory angiogenesis via sequential induction of oxidative stress, endoplasmic reticulum stress and autophagy. Cell Signal. 2012;24:2123–2131. doi: 10.1016/j.cellsig.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 16.Liu H, et al. Macrophage-derived MCPIP1 mediates silica-induced pulmonary fibrosis via autophagy. Part Fibre Toxicol. 2016;13:55. doi: 10.1186/s12989-016-0167-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu H, et al. BBC3 in macrophages promoted pulmonary fibrosis development through inducing autophagy during silicosis. Cell Death Dis. 2017;8:e2657. doi: 10.1038/cddis.2017.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwata J, et al. Excess peroxisomes are degraded by autophagic machinery in mammals. J Biol Chem. 2006;281:4035–4041. doi: 10.1074/jbc.M512283200. [DOI] [PubMed] [Google Scholar]
- 20.Azad MB, et al. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy. 2008;4:195–204. doi: 10.4161/auto.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yousefi S, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 22.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Gonzalez J, Alfon J, Berrozpe M, Badimon L. HMG-CoA reductase inhibitors reduce vascular monocyte chemotactic protein-1 expression in early lesions from hypercholesterolemic swine independently of their effect on plasma cholesterol levels. Atherosclerosis. 2001;159:27–33. doi: 10.1016/S0021-9150(01)00469-5. [DOI] [PubMed] [Google Scholar]
- 25.Khanna A, Guo M, Mehra M, Royal W., III Inflammation and oxidative stress induced by cigarette smoke in Lewis rat brains. J Neuroimmunol. 2013;254:69–75. doi: 10.1016/j.jneuroim.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, et al. SCM-198 attenuates early atherosclerotic lesions in hypercholesterolemic rabbits via modulation of the inflammatory and oxidative stress pathways. Atherosclerosis. 2012;224:43–50. doi: 10.1016/j.atherosclerosis.2012.06.066. [DOI] [PubMed] [Google Scholar]
- 27.Yan L, et al. Inhibitory effect of hepatocyte growth factor on cardiomyocytes apoptosis is partly related to reduced calcium sensing receptor expression during a model of simulated ischemia/reperfusion. Mol Biol Rep. 2011;38:2695–2701. doi: 10.1007/s11033-010-0412-8. [DOI] [PubMed] [Google Scholar]
- 28.Van Horn K, Toth C. Evaluation of the AnaeroPack Campylo system for growth of microaerophilic bacteria. J Clin Microbiol. 1999;37:2376–2377. doi: 10.1128/jcm.37.7.2376-2377.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lo LW, Koch CJ, Wilson DF. Calibration of oxygen-dependent quenching of the phosphorescence of Pd-meso-tetra (4-carboxyphenyl) porphine: a phosphor with general application for measuring oxygen concentration in biological systems. Anal Biochem. 1996;236:153–160. doi: 10.1006/abio.1996.0144. [DOI] [PubMed] [Google Scholar]
- 30.Chao J, Wood JG, Blanco VG, Gonzalez NC. The systemic inflammation of alveolar hypoxia is initiated by alveolar macrophage-borne mediator(s) Am J Respir Cell Mol Biol. 2009;41:573–582. doi: 10.1165/rcmb.2008-0417OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X, et al. Role of human pulmonary fibroblast-derived MCP-1 in cell activation and migration in experimental silicosis. Toxicol Appl Pharmacol. 2015;288:152–160. doi: 10.1016/j.taap.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Chao J, et al. Expression of green fluorescent protein in human foreskin fibroblasts for use in 2D and 3D culture models. Wound Repair Regen. 2014;22:134–140. doi: 10.1111/wrr.12121. [DOI] [PubMed] [Google Scholar]
- 33.Liu H, et al. MCPIP1 mediates silica-induced cell migration in human pulmonary fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2015;310:L121–132. doi: 10.1152/ajplung.00278.2015. [DOI] [PubMed] [Google Scholar]
- 34.Chao J, et al. MCPIP1 Regulates Fibroblast Migration in 3-D Collagen Matrices Downstream of MAP Kinases and NF-kappaB. J Invest Dermatol. 2015;135:2944–2954. doi: 10.1038/jid.2015.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chao J, et al. Role of MCPIP1 in the Endothelial-Mesenchymal Transition Induced by Silica. Cell Physiol Biochem. 2016;40:309–325. doi: 10.1159/000452547. [DOI] [PubMed] [Google Scholar]
- 36.Li HY, et al. Pyk2 and Src mediate signaling to CCL18‐induced breast cancer metastasis. J Cell Biochem. 2014;115:596–603. doi: 10.1002/jcb.24697. [DOI] [PubMed] [Google Scholar]
- 37.Zhang W, et al. Neogambogic acid prevents silica-induced fibrosis via inhibition of high-mobility group box 1 and MCP-1-induced protein 1. Toxicol Appl Pharmacol. 2016;309:129–140. doi: 10.1016/j.taap.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Fiuza C, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 39.Messmer D, et al. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J Immunol. 2004;173:307–313. doi: 10.4049/jimmunol.173.1.307. [DOI] [PubMed] [Google Scholar]
- 40.Yang J, et al. High mobility group box-1 induces migration of vascular smooth muscle cells via TLR4-dependent PI3K/Akt pathway activation. Mol. Biol. Rep. 2012;39:3361–3367. doi: 10.1007/s11033-011-1106-6. [DOI] [PubMed] [Google Scholar]
- 41.Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- 42.Niu J, Azfer A, Zhelyabovska O, Fatma S, Kolattukudy PE. Monocyte chemotactic protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein (MCPIP) J Biol Chem. 2008;283:14542–14551. doi: 10.1074/jbc.M802139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao, D., Xiao, Z., Li, H. P., Han, D. H. & Zhang, Y. P. LncRNA MALAT-1 Elevates HMGB1 to Promote Autophagy Resulting in Inhibition of Tumor Cell Apoptosis in Multiple Myeloma. J Cell Biochem, 10.1002/jcb.25987 (2017). [DOI] [PubMed]
- 44.Yan L, et al. Activation of calcium-sensing receptors is associated with apoptosis in a model of simulated cardiomyocytes ischemia/reperfusion. J Biomed Res. 2010;24:301–307. doi: 10.1016/S1674-8301(10)60042-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohsu T, et al. Involvement of the calcium-sensing receptor in human taste perception. J Biol Chem. 2010;285:1016–1022. doi: 10.1074/jbc.M109.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Conigrave AD, Franks AH, Brown EM, Quinn SJ. L-amino acid sensing by the calcium-sensing receptor: a general mechanism for coupling protein and calcium metabolism? Eur J Clin Nutr. 2002;56:1072–1080. doi: 10.1038/sj.ejcn.1601463. [DOI] [PubMed] [Google Scholar]
- 47.Liu L, et al. Suppression of calciumsensing receptor ameliorates cardiac hypertrophy through inhibition of autophagy. Mol Med Rep. 2016;14:111–120. doi: 10.3892/mmr.2016.5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scarabelli T, et al. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischemia/reperfusion injury. Circulation. 2001;104:253–256. doi: 10.1161/01.CIR.104.3.253. [DOI] [PubMed] [Google Scholar]
- 49.Zhu Z, Fang Z. Statin protects endothelial cell against ischemia reperfusion injury through HMGB1/TLR4 pathway. Int J Cardiol. 2016;203:74. doi: 10.1016/j.ijcard.2015.10.095. [DOI] [PubMed] [Google Scholar]
- 50.Liu XX, et al. Regnase-1 in microglia negatively regulates high mobility group box 1-mediated inflammation and neuronal injury. Sci Rep. 2016;6:24073. doi: 10.1038/srep24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foglio E, Puddighinu G, Germani A, Russo MA, Limana F. HMGB1 Inhibits Apoptosis Following MI and Induces Autophagy via mTORC1 Inhibition. J Cell Physiol. 2017;232:1135–1143. doi: 10.1002/jcp.25576. [DOI] [PubMed] [Google Scholar]
- 52.Khoo CP, et al. miR-193a-3p interaction with HMGB1 downregulates human endothelial cell proliferation and migration. Sci Rep. 2017;7:44137. doi: 10.1038/srep44137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin F, Xue D, Xie T, Pan Z. HMGB1 promotes cellular chemokine synthesis and potentiates mesenchymal stromal cell migration via Rap1 activation. Mol Med Rep. 2016;14:1283–1289. doi: 10.3892/mmr.2016.5398. [DOI] [PubMed] [Google Scholar]
- 54.Yang J, et al. MicroRNA-24 inhibits high glucose-induced vascular smooth muscle cell proliferation and migration by targeting HMGB1. Gene. 2016;586:268–273. doi: 10.1016/j.gene.2016.04.027. [DOI] [PubMed] [Google Scholar]
- 55.Huang, Q., Yang, Z., Zhou, J. P. & Luo, Y. HMGB1 induces endothelial progenitor cells apoptosis via RAGE-dependent PERK/eIF2alpha pathway. Mol Cell Biochem, 10.1007/s11010-017-2976-2 (2017). [DOI] [PubMed]
- 56.Canton J, et al. Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat Commun. 2016;7:11284. doi: 10.1038/ncomms11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu F, et al. Elevation of extracellular Ca2 + induces store-operated calcium entry via calcium-sensing receptors: a pathway contributes to the proliferation of osteoblasts. PLoS One. 2014;9:e107217. doi: 10.1371/journal.pone.0107217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.