
Extended Data Figure 8. General features of the cancer methylome and of CGI DMRs.
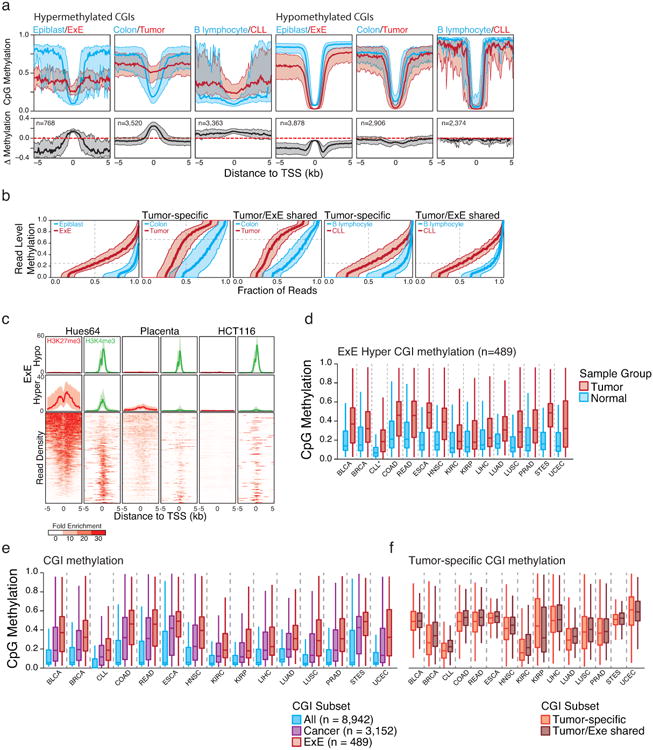
a. Median methylation of differentially regulated CGI-containing promoters in a primary colon tumor isolate and Chronic Lymphocytic Leukemia (CLL) compared to colon and B lymphocytes, respectively. E×E Hyper CGIs as identified in this study and shown in Figure 1 are included for reference. The median methylation difference between extraembryonic or cancerous tissue compared to Epiblast or normal tissue is also included. The general features of both cancer methylomes are similar to that of the E×E, with a maximal increase in DNA methylation centered at the TSS that steadily diminishes within the periphery. Alternatively, hypomethylated CGIs in extraembryonic or tumorigenic contexts are maximally different a distance away from the TSS, within the boundary or “CpG island shore,” as previously reported for cancer61. Shaded area represents the 25th and 75th percentiles per 100 bp bin.
b. Read level methylation of hypermethylated CGIs in E×E vs Epiblast, Colon Tumor vs Colon, and CLL vs B lymphocyte, with those islands that share differential methylation status between the cancer and extraembryonic development included as a subset. The methylation status for every sequencing read within a given hypermethylated CGI was ranked and binned into percentiles. Plotted are the median and 25th/75th percentile for these ranks across CGIs called as hypermethylated in each pairwise comparison. The E×E-Epiblast and CLL-B lymphocyte comparisons exhibit very similar distributions that indicate general discordance, meaning similar aggregate methylation across the feature as is observed in phase, which can only be obtained by dispersive de novo methylation across the majority of alleles within the population. Alternatively, Colon Tumor exhibits substantially higher read level methylation, with a median per read methylation of ∼0.7. However, the per-read methylation level of the non-tumorous, matched colon tissue is also quite high, with >50% of reads exhibiting some methylation. This could indicate a transition in the epigenetic status of these loci within colon tissue that precedes tumorigenesis, as has been noted for several other tissues in Extended Data Figure 9. Moreover, the extent to which E×E Hyper CGIs are methylated within each tumor reflects the read-level methylation distribution for the tumor. As such, the targeting to E×E Hyper CGIs is a conserved feature of human cancer types, but the extent to which they are methylated can be specific to the system.
c. Data taken from ENCODE samples that reflect embryonic and extraembryonic identities in human in comparison to the well-characterized human cancer cell line HCT116. The human embryonic stem cell line HUES64, a proxy for the pluripotent epiblast, displays notable enrichment for both repressive, PRC2 deposited H3K27me3 and activating H3K4me3 modifications at orthologous E×E Hyper CGIs. Alternatively, human placenta exhibits diminished enrichment for both modifications at these regions, as does HCT116. Both systems display substantial methylation over these islands as presented in Figure 4, Extended Data Figure 9, and Supplementary Table 7. As a control, “E×E hypo” demonstrates uniformly high H3K4me3 levels across all three tissues. Enrichment density heatmaps are provided for the full E×E-hyper set and are ranked across plots according to their enrichment for H3K27me3 in HUES64. Normalized enrichment represents the fold ChIP-enrichment against sample matched Whole Cell Extract (WCE).
d. Boxplots of mean methylation for 489 E×E-methylated, orthologous CGIs (E×E Hyper CGIs) across the 14 tissue-matched TCGA tumor types that display disregulated DNA methylation landscapes and for CLL. Note: CLL samples were measured by RRBS (n=119) and represent a comparison between age matched healthy B lymphocytes (n=24). Edges refer to the 25th and 75th percentiles, whiskers the 2.5th and 97.5th percentiles, respectively.
e. Boxplots for TCGA data sets and CLL for the absolute methylation values of All orthologously mapped CGIs, those methylated across Cancer, and those that are specifically methylated in mouse E×E. In all 15 cancer types that exhibit general global hypomethylation and CGI methylation as part of their departure from somatic cells, E×E Hyper CGIs are specifically enriched, more so than for CGIs that are observed as hypermethylated in any tumor.
f. Boxplots for the same TCGA data for tumor-specific CGIs and those that are also methylated in mouse E×E. Notably, the extent to which mouse E×E Hyper CGIs are methylated reflect the tumor, with some cancer types exhibiting higher absolute methylation values than others. However, in 14 out of 15 cases, the absolute methylation status of tumor-specific CGI DMRs and those that are also methylated in E×E are nearly identical, and often slightly greater. Absolute methylation values therefore appear to be determined by the cancer type, while targeting of extraembryonically methylated CGIs is a general feature.