

ABSTRACT
AKT3 is one of the major therapeutic targets in melanoma but clinically targeting AKT3 alone seems to be an ineffective therapeutic approach. To identify unique strategies to enhance the efficacy of targeting AKT3, a screen was undertaken where AKT3 was co-targeted with a panel of kinases important in melanoma development. The screen identified WEE1 as the most potent target that when inhibited along with AKT3 would enhance the efficacy of targeting AKT3 in melanoma. RNAi mediated inhibition of AKT3 and WEE1 synergistically inhibited the viability of melanoma cells leading to a 65–75% decrease in tumor development. This approach was effective by mechanistically modulating pathways associated with the transcription factors p53 and FOXM1. Simultaneously regulating the activity of these two transcriptionally driven pathways, cooperatively deregulated cell cycle control and DNA damage repair to synergistically kill melanoma cells. This study uniquely identifies a potential approach to improve the efficacy of targeting AKT3 in melanoma.
KEYWORDS: AKT3, Melanoma, synergy, WEE1
Introduction
Incidence and mortality rates of malignant melanoma continue to rise annually.1 Advanced stage metastatic melanoma carries a poor prognosis, with a median overall survival of 2 to 8 months, and with only 5% of patients surviving beyond 5 y.1 AKT3, a member of the AKT serine/threonine protein kinase family, is activated in up to 70% of sporadic melanomas to promote cell survival by deregulating apoptotic signaling.2 Preclinical targeting of AKT3 in melanoma cells is effective in killing melanoma cells and retarding tumor development.2 Regretfully, these observations have not translated clinically, and therapies targeting AKT in melanoma have not been effective.3 Hence, novel approaches are needed to improve the efficacy of agents targeting AKT signaling in melanoma.
Another major target in melanoma is the MAP (mitogen-activated protein) kinase signaling pathway that is frequently activated by V600EBRAF, which is the most frequent genetic alteration occurring in 50% of sporadic melanomas.4 Small molecule inhibitors targeting the over-activated MAPK pathway showed promise as effective therapeutic approaches for patients with advanced stage or unresectable melanoma.5-10 Unfortunately, these drugs have only a modest effect on median survival, with recurrent resistant disease developing by circumventing the point of drug inhibition.11,12 Activation of MAPK signaling alone is unable to drive melanoma development but requires co-operation with additional genetic or epigenetic alterations.2 This observation is supported by the fact that, 70 to 80% of melanocytic nevi carry the V600EBRAF mutation but seldom progress into melanoma.4 Activation of AKT signaling is a key event in BRAF mediated tumor progression. AKT promotes melanoma development by phosphorylating the V600EBRAF protein to decrease its activity to the levels that promote rather than inhibit melanocytic cell growth.2 Moreover, activation of AKT signaling has also been shown to play a role in resistance development to MAPK inhibitors.13-16 Hence, efficacy of the combination of MAPK and AKT inhibitors are currently under investigation.17,18 Unfortunately, recent studies suggested that targeting AKT signaling alone or in combination with MAPK is not clinically effective.19,20,21 AZD5363, a new generation pan AKT inhibitor, although well tolerated, yielded a partial response in only 2 of the 92 patients with advanced solid tumors.14 Co-treatment of MEK inhibitor, trametinib, with orally bioavailable pan Akt inhibitor, GSK2141795, led to stable disease in 65% of the melanoma patients, without any partial or complete responses.21 Based on this background and the need to identify targets to inhibit in combination with AKT that could synergize, a set of kinases were screened to identify those that could be targeted in combination with AKT3 to synergistically inhibit melanoma tumor development. WEE1 kinase was identified as a potential target that could accomplish this objective.
WEE1 is involved in the regulation of the cell cycle by phosphorylating and inactivating cyclin-dependent kinase-1 (CDK1).22 As a component of the G2/M checkpoint, it determines the time point for entry into mitosis and inhibits early progression through the cell cycle. It is also involved in the coordination of cellular response to DNA damage. Furthermore, WEE1 was also identified as a key signaling molecule lying downstream of V600EBRAF in the MAPK signaling cascade.23 WEE1 levels were decreased upon genetic or pharmacological inhibition of V600EBRAF, MEK or ERK activity.23 Genetic knockdown of WEE1 reduced tumor development in melanoma xenograft models with similar signaling alterations observed following the inhibition of V600EBRAF.23 In this study, we show that RNAi mediated co-targeting of WEE1 with AKT3 can synergistically inhibit melanoma in culture as well as in tumors, and identified the unique mechanism through which it occurs.
Materials and methods
Cell lines and culture conditions
Metastatic melanoma cell lines, UACC 903 was provided by Dr. Mark Nelson (between 1995 and 1999), University of Arizona, (Tucson, AZ) and the 1205 Lu cell line (between 2003 and 2005) from Dr. Herlyn, Wistar Institute (Philadelphia, PA), both the cell lines harbor V600EB-Raf. All cell lines were maintained in DMEM (Life Technologies, Grand Island, NY) supplemented with 1% GlutaMAX from Gibco (Life Technologies) and 10% FBS (HyClone, Logan, UT) in a 37°C humidified 5% CO2 atmosphere incubator and periodically monitored for genotypic characteristics, phenotypic behavior and tumorigenic potential.
Small interfering RNA (siRNA) transfection
siRNA was introduced into melanoma cells via nucleofection using an Amaxa nucleofector with solution R / program K-17 for UACC 903 and 1205 Lu cells.23,24 Nucleofection efficiency was >90% with 80–90% cell viability. Following siRNA transfection, cells were plated and allowed to recover for 2 d and then replated in 96-well plates to assess viability or harvested for protein knockdown studies.25 Duplexed Stealth siRNA (Invitrogen) sequences for scrambled, V600EBRAF, WEE1, AURKB, GSK3A and TPK1 were as reported previously.23,26
siRNA screening and synergy analysis of cultured cells to identify kinases synergizing with AKT3
siRNA screening was performed as described previously.23,26 For synergy studies, 6.25–100 picomoles of siRNA targeting AKT3 and 5 kinases (WEE1, AURKB, GSK3A, TPK1 or mutant V600EBRAF) that were identified from the screen were introduced into 1 × 106 melanoma cells alone or in combination using an Amaxa nucleofector. 2 d post transfection, cells were trypsinised and 1 × 104 cells / well in 100 uL of serum-free media were plated into 96-well plates with 6 to 8 replicates for each siRNA and for each scrambled siRNA control. Cells were grown for 72 hours and viability of cells was measured using an MTS assay and the decrease in viability was compared with the scrambled siRNA control. The data were subjected to Chou-Talalay analysis for determining the combination index using CalcuSyn software.
RNA-sequencing experiments
Twenty-four hours following treatment with tested agents, total RNA was extracted using the mirVana RNA isolation kit (Life Technologies). Next, using the SureSelect Strand-Specific RNA Library Preparation Kit (Agilent), cDNA libraries were constructed. For multiplexed high-throughput sequencing, unique barcode sequences were incorporated. Libraries were then denatured using the Illumina protocol, diluted with pre-chilled hybridization buffer and loaded onto TruSeq SR v3 flow cells on an Illumina HiSeq 2500 (50 cycles, single-read). Sequencing reads were extracted using Illumina CASAVA pipeline Version 1.8 and aligned to the human reference genome (hg38) using Tophat (version 2.0.9).27 SeqMonk (version 0.32.0) was used for identification of differentially expressed genes. Following RNA-Seq quantitation, 75% percentile normalization was performed, and significant alterations were identified using the intensity difference test with Benjamini and Hochberg multiple testing correction (p-value < 0.05).
RPPA array experiment
UACC 903 cells were transfected with siRNAs targeting AKT3 (50 pmole), WEE1 (25 pmole) or their combination. siScramble (50 pmole) was used as a control. 48 hours after transfections, cell lysates were collected using protein extraction reagent (T-PER, Thermo Scientific) supplemented with 1 mM EDTA, 5 mM NaF, 2 µM staurosporine, PhosSTOP Phosphatase Inhibitor Cocktail (Roche), and Complete Mini Protease Inhibitor Cocktail (Roche). Total protein concentration was determined by bicinchoninic acid assay (Thermo Scientific) and submitted to the Functional Proteomics Core Facility at MD Anderson for the RPPA analysis. The array consisted of 287 antibodies including 64 phospho-specific antibodies.
Western Blotting
Cell lysates were collected 48 hours after siRNA transfection or drug treatment by washing plates with PBS followed by addition of RIPA lysis buffer containing Halt Protease & Phosphatase Inhibitor Cocktail (Thermo Scientific, Rockford, IL, USA).28-33 Lysates were centrifuged (10,000 X g) for 10 minutes at 4°C to remove cell debris. Protein concentration was measured using the bicinchoninic acid assay kit (Thermo). 25 µg of respective lysates were loaded onto 4–12% Bis-Tris NuPAGE gels (Life Technologies) and run in an XCell SureLock Mini-Cell gel apparatus (Life Technologies), transferred to polyvinylidene difluoride membranes (Pall Corporation, Port Washington, NY), probed with respective primary antibody followed by horseradish peroxidase-conjugated secondary antibody, and developed using ECL Western Blotting Substrate (Thermo Scientific) or Supersignal West Femto Chemiluminescent Substrate (Thermo Scientific). Primary antibodies used: p21 (sc-756), p27 (sc-528), p53 (sc-6243) and ERK2 (sc-1647) from Santa Cruz Biotechnology (Dallas, TX), pAKT (9271), AKT3 (3788), Total AKT (4685), pWEE1 (4910), WEE1 (4936), p-CDC2 (Y15) (9111), pRB (S807/811) (9308), pRB (S795) (9301), pRB (S780) (9307), (9309) and p-Histone H2AX (S139) (2577) from Cell Signaling (Danvers, MA). Secondary antibodies goat anti-rabbit IgG-HRP (sc-2004) and goat anti-mouse IgG-HRP (sc-2005) were purchased from Santa Cruz Biotechnology.
In vivo analysis of synergy, cell proliferation and apoptosis in time and size matched tumors
Animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee at the Pennsylvania State University. Tumor kinetics studies were undertaken in athymic-Foxn1nu nude mice (Harlan Laboratories, Indianapolis, IN, USA). siRNA targeting AKT3 (100 picomoles) and WEE1 (6.25 picomoles) alone or in combination was nucleofected into 1 × 106 UACC 903 or 1205 Lu cells. Cells transfected with scrambled siRNA (106.25 picomoles) were used as a control. Since total siRNA was 106.25 picomoles for the combination group, scrambled siRNA was used to compensate for siRNA in AKT3 or WEE1 alone groups. After plating and allowing the cells to recover for 48 hours, 1 × 106 cells were aliquoted in 0.2 ml of 10% FBS-DMEM and then injected subcutaneously above both the left and right rib cages of 4–6 week old female mice (3–4 mice/ group). Dimensions of developing tumors were measured on alternate days up to day 17.5, using calipers by multiplying length, width and depth in mm3.
To generate tumors of the same size developing at parallel time points, 1 × 106 or 10 × 106 of UACC 903 melanoma cells were nucleofected with siScrambled, siAKT3, siWEE1 or siAKT3+siWEE1, respectively, as detailed above and injected into nude mice. Tumors were removed from euthanized mice at days 9 and 11, flash frozen in liquid nitrogen, pulverized and protein lysates collected by the addition of 600 to 800 uL of RIPA protein lysis buffer containing Halt Protease & Phosphatase Inhibitor Cocktail (Thermo Scientific, Rockford, IL, USA). Protein concentration was measured using the bicinchoninic acid assay kit (Thermo) followed by Western blotting to measure the levels of AKT3, and WEE1 proteins in tumors. The band intensity was quantified by scanning the optical density of each band using ImageJ as described previously.23,24 Cell proliferation rates were measured using a mouse anti-human Ki-67 antibody from BD Pharmigen (BD Biosciences, San Diego, CA). Apoptosis rate was measured using the “terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL)” TMR Red Apoptosis kit (Roche, Mannheim, Germany) as described previously.23,26 Number of Ki-67 or TUNEL stained cells were quantified as the percentage of total cells in tumors. Sections were imaged using a Nikon Eclipse 600 and quantified using IP laboratory imaging software (Scanalytics, Fairfax, VA). A minimum of 6 different tumors with 4 to 6 fields per tumor was analyzed.
Statistical analysis
Statistical analysis was performed using Prism 4.0 GraphPad Software. Data were subjected to the Chou-Talalay method for determining the combination index using CalcuSyn software and combination index (CI) values plotted against fraction affected.34 Using this approach, combination index values of < 0.9 are synergistic, >1.1 are antagonistic, and values 0.9–1.1 are nearly additive. For comparison between 2 groups, Student t test (2 tailed) was used. One-way Analysis Of Variance (ANOVA) was used for group wise comparisons, followed by the Tukey's post hoc test. Results represent at least 2 to 3 independent experiments and are shown as averages ± SEM. Results with a P value less than 0.05 (95% CI) were considered significant. Sample sizes and number of times experiments were repeated are indicated in the figure legends. Number of asterisks in the figures indicates the level of statistical significance as follows: *P < 0.05, **P < 0.01, ***P < 0.001.
Results
Identifying WEE1 to target in combination with AKT3 to synergistically inhibit melanoma cell survival
To identify druggable targets that enhance the efficacy of AKT3 inhibition, a screen was undertaken to identify kinases that could retard melanoma development.23 The screen was undertaken using a pool of 3 siRNAs targeting each of the 636 kinases (totaling 1908 individual siRNAs). siRNAs were nucleofected into the UACC 903 melanoma cell line using the AMAXA 96-well shuttle transfection system and 5 d later, the viability of cells was measured by MTS assay. Results were compared with the average of high-, medium-, and low-GC content scrambled siRNA transfected cells. In this primary screen, 34 kinases reduced the viability of UACC 903 cells more significantly than the set experimental cut-off, i.e. 15% growth inhibition. The secondary screen retested the 34 kinases and validated 14 of these hits (Supplementary Table 1). A tertiary screen required that at least 2 of the 3 siRNA targeting different regions of respective mRNAs decrease UACC 903 viability. Based on these criteria, AURKB, WEE1, GSK3A, TPK1, and BRAF were identified as potential targets able to reduce the proliferative potential of UACC 903 melanoma cells. The potential of these targets was then confirmed in 2 additional melanoma cell lines (A375M and 1205 Lu) to show similar growth inhibitory effects. Subsequently, siRNA-mediated knockdown of these kinases was performed to measure the inhibitory effect on xenografted tumor development23,26 (Supplementary Table 1).
Increased AKT3 activity has been reported in up to 70% of melanomas, but inhibition of AKT3 alone in the clinic does not appear to be an effective therapeutic approach.35 Thus, to achieve high-level synergistic melanoma inhibition, AKT3 was co-targeted with the kinases identified from the screen. The viability of UACC 903 cells following siRNA-mediated knockdown of AKT3 alone or in combination with WEE1, GSK3A, AURKB, TPK1, and BRAF was examined by MTS assay. The combination index (CI) values suggested strong synergism between knockdowns of AKT3 and WEE1 (CI value 0.095) or GSK3A (CI value 0.267) (Supplementary Table 2). Since synergism between targeting AKT3 and GSK3A has been previously reported, it is not discussed further in this manuscript.26
The approach in this study to identify synergy between gene targets has been reported previously.26 It involved nucleofection of 50 or 100 picomoles of siRNA targeting AKT3 with increasing concentrations of WEE1 siRNA (6.25, 12.5, 25, 50 and 100 picomoles) into UACC 903 and 1205 Lu cells, and measuring the effect on cell viability (Figs. 1A and B). Analyses of the combination index for the knockdown of AKT3 and WEE1 showed a synergistic relationship in both UACC 903 and 1205 Lu cell lines (Figs. 1C and D). Protein knockdown efficiencies following nucleofection is demonstrated by western blotting (Figs. 1E and F). Both AKT3 and WEE1 siRNAs effectively decreased total and phosphorylated (active) levels of their respective targets.
Figure 1.

Targeting AKT3 and WEE1 synergistically inhibited melanoma cell growth. (A) and B, Dose-response curves of siRNA targeting AKT3 or WEE1 were undertaken by nucleofecting increasing amounts of siRNA targeting WEE1 and AKT3 followed by measurement of cellular viability after 3 d of growth in serum-free medium. For combination analysis, 50 or 100 picomoles of AKT3 were combined with increasing amounts of WEE1 siRNA. (C) and (D), The combination index (CI) analysis showed synergism between AKT3 and WEE1 when targeted together. Columns; mean (n = 6–8); error bars, SEM. (E) and (F), siRNAs targeting AKT3 alone or in combination with increasing amounts of WEE1 siRNA were introduced into melanoma cells via nucleofection and protein levels were measured 2 d later. ERK2 served as a control for equal protein loading. Western blot analysis was reproduced at least twice.
Targeting AKT3 and WEE1 inhibited xenografted melanoma tumor growth by decreasing proliferation and inducing apoptosis
Since targeted inhibition of AKT3 and WEE1 synergistically reduced survival of cultured human melanoma cells, the efficacy of this combination was next examined in human melanoma xenografts. 1 × 106 1205 Lu or UACC 903 cells were transfected with siRNAs targeting AKT3 (100 picomoles) and WEE1 (6.25 picomoles) alone or in combination, and cells transfected with scrambled siRNA (106.25 picomoles) were used as a control. The concentration of WEE1 siRNA for these studies was selected based on the titration for tumor inhibition to occur in a similar range as with 100 picomoles of AKT3 knockdown, using a published approach.2 This amounted to 6.25 picomoles for WEE1 siRNA and 100 picomoles for AKT3 siRNA. Use of more than 6.25 pmoles of WEE1 siRNA inhibited tumor growth into levels that would not allow synergy assessment. Scrambled siRNA was used to compensate for siRNA in AKT3 or WEE1 only groups. Two days after transfections, cells were subcutaneously injected into 4 to 6-week old nude mice, and volumes of tumors were measured on alternate days.23,25 In both melanoma cell lines, combinatorial knockdown of AKT3 and WEE1 was significantly more effective at inhibiting tumor development in contrast to single knockdowns (Figs. 2 A and B). 60 to 80% decrease in tumor volume was observed when AKT3 and WEE1 were cotargeted compared with a 25% decrease when each kinase was targeted alone. Protein knockdown efficiency of siRNAs was measured by western blotting of size and time matched xenografted tumor lysates using an established protocol.25,36 Decreased levels of AKT3 and WEE1 protein expression were also observed in tumor lysates (Fig. 2C).
Figure 2.

Targeting of AKT3 and WEE1 inhibited melanoma tumor growth. Targeting AKT3 and WEE1 synergistically inhibited melanoma tumor growth in vivo. UACC 903 (2A) and 1205 Lu (2B) cells were nucleofected with AKT3 and WEE1 siRNA alone or in combination and 48 hours later, viable cells were s.c. injected into left and right flanks of nude mice. Cells transfected with scrambled siRNA were used as a control. Developing tumors were measured on alternate days for 3 weeks. Significance was measured by one-way analysis of variance, followed by the post hoc test, *P < 0.05, **P < 0.01, ***P < 0.01. Error bars show SEM. Data were obtained from duplicate experiments with 3 mice per group, containing 2 tumors per mouse. C. Western blot analysis showing knockdown of AKT3 and WEE1 in tumor lysates of UACC 903 xenografts harvested at day 9. ERK2 served as a control for equal protein loading. (D) and E, Analysis of size and time matched tumors from animals injected with UACC 903 melanoma cells transfected with scrambled siRNA controls or siRNA to AKT3, WEE1 or AKT3, and WEE1. Tumor sections were immunostained for Ki-67 (2D) or TUNEL (2E) to measure the proliferation and apoptosis, respectively. Bar graphs show the fold change in Ki-67 or TUNEL-positive cells compared with the scrambled siRNA control. Data were obtained from 3 to 4 tumors with 4 to 5 fields averaged per tumor. Significance was measured by one-way analysis of variance, followed by the post hoc test, **P < 0.01, ***P < 0.001, NS; Non-significant. Columns, mean; error bars, SEM.
To dissect the cellular processes mediating enhanced tumor inhibition when targeting AKT3 and WEE1, rates of cellular proliferation and apoptosis were measured in size and time matched human melanoma xenografts.25,36 Formaldehyde-fixed, paraffin-embedded sections of tumors were stained with Ki-67 to measure tumor cell proliferation rates, and TUNEL to detect apoptotic cell death. Cotargeting AKT3 and WEE1 led to 4-fold inhibition in melanoma tumor cell proliferation (Fig. 2D) and 3.5-fold increase in apoptosis (Fig. 2F), compared with 1.3 and 2-fold decrease in proliferating cells and 2.5 to 1.25-fold increase in apoptotic cells, respectively when targeting each gene alone.
Targeting AKT3 and WEE1 leads to deregulation of cell cycle and DNA damage response pathways
Targeting 2 proteins together often leads to a new and novel mechanism of inhibiting tumor development, which is also hypothesized to be occurring when simultaneously targeting AKT3 and WEE1. Therefore, establishing the mechanism through which tumor inhibition occurs when targeting 2 proteins is complex and in recent years has involved the use of an “omics” approach.37 This contemporary approach was used to identify the synergistic mechanisms, through which targeting AKT3 and WEE1 inhibits melanoma tumor development. First, alterations in gene expression levels were identified using genome-wide RNA sequencing analysis of cells following genetic targeting of AKT and WEE1 kinases. Next, RPPA analysis was used to validate alterations at the protein level, which was finally confirmed by Western blotting.
RNA sequencing identified 41 significantly deregulated genes following siRNA-mediated knockdown of WEE1 in UACC 903 cells (Fig. 3A). Enrichment analyses of the 22 upregulated genes showed the p53 protein as a significantly enriched transcriptional regulator (Table 1). As a matter of fact, 14 of the 22 genes were modulated by the p53 gene family transcription factors. Analysis of the remaining 19 downregulated genes identified transcription factors FOXM1 and E2F4 as prominent modulators of this effect. 13 of the 19 (70%) downregulated genes were modulated by these 2 transcription factors. Enrichment analysis of all 41 deregulated genes implicated induction of DNA damage and cell cycle deregulation (Table 1). These observations were consistent with the function of WEE1, since, as a regulator of the G2/M phase checkpoint, inhibition of WEE1 has been shown to induce DNA damage through perturbation of the cell cycle.38
Figure 3.
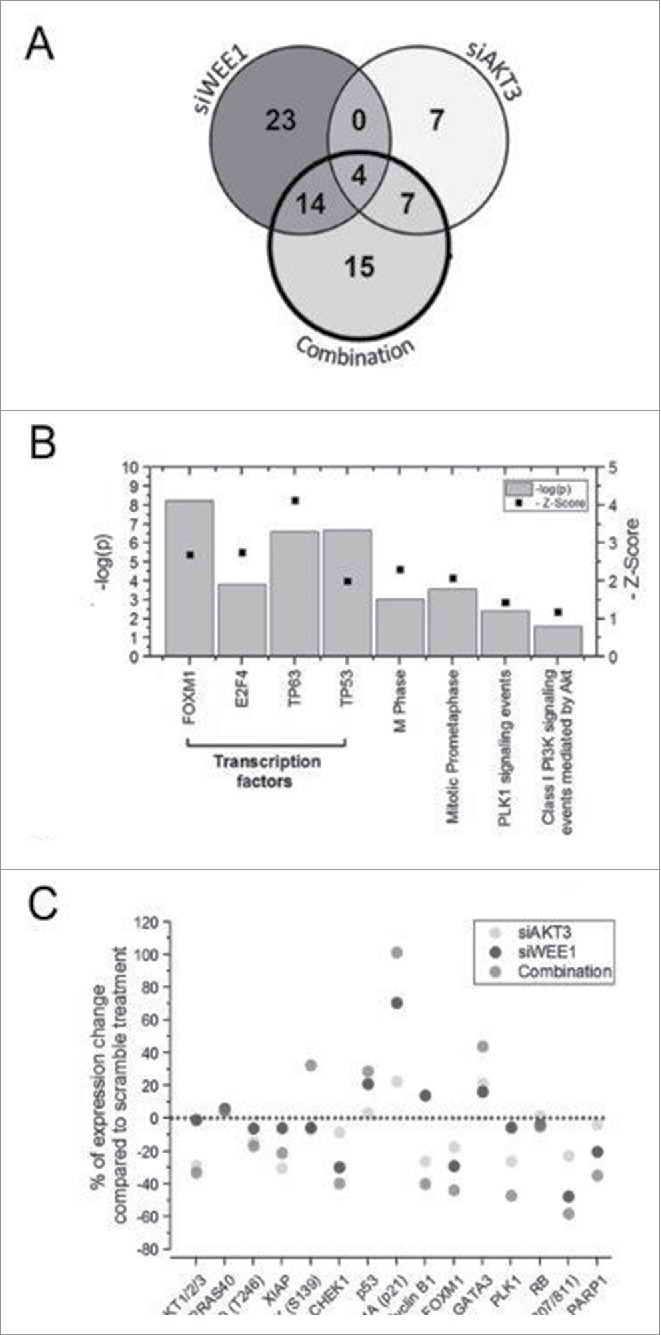
Alteration in gene expression and protein levels through genome-wide RNA sequence and RPPA analysis respectively. A, Venn diagram showing significantly altered genes identified following RNA-Seq analysis of UACC 903 cells transfected with siRNAs targeting AKT3, WEE1 or both. B, Enrichment analysis of 40 genes significantly altered by the combination. C, RPPA array analysis showing some of the altered proteins in UACC 903 melanoma cells transfected with siAKT3, siWEE1 or the combination. In RNA-Seq experiments, significant genes were determined using the intensity difference test with a p-value threshold of 0.05 and Benjamini and Hochberg multiple testing correction.
Table 1.
Gene set enrichment analysis. Enrichment analysis of gene sets identified by RNA sequencing of genetically targeting of AKT and WEE1 pathways in UACC 903 cells. For the full list, please see Supplementary Table 4.
| Treatment | Subgroup | Pathway/Transcription Factor | p-Value | Z Score | Combined Score | Tool | |
|---|---|---|---|---|---|---|---|
| 1 | siWEE1 | DOWN | FOXM1 | 5.00E-14 | −2.70 | 68.11 | CHEA |
| 2 | siWEE1 | DOWN | E2F4 | 1.40E-06 | −2.84 | 26.12 | CHEA |
| 3 | siWEE1 | UP | TP63 | 3.89E-09 | −4.14 | 61.69 | CHEA |
| 4 | siWEE1 | UP | TP53 | 4.30E-12 | −2.00 | 41.32 | CHEA |
| 5 | siWEE1 | ALL | M Phase | 1.82E-03 | −2.30 | 5.61 | REACTOME |
| 6 | siWEE1 | ALL | DNA Damage Response | 2.00E-03 | −1.83 | 6.80 | WIKIPATHWAYS |
| 7 | siWEE1 | ALL | TP53 Network | 2.93E-03 | −1.45 | 5.38 | WIKIPATHWAYS |
| 8 | siWEE1+siAKT3 | DOWN | FOXM1 | 5.73E-09 | −2.70 | 36.85 | CHEA |
| 9 | siWEE1+siAKT3 | DOWN | E2F4 | 1.63E-04 | −2.76 | 15.13 | CHEA |
| 10 | siWEE1+siAKT3 | UP | GATA3 | 2.19E-03 | −1.85 | 4.05 | ENCODE |
| 11 | siWEE1+siAKT3 | ALL | RB in Cancer | 1.38E-03 | −1.92 | 5.44 | WIKIPATHWAYS |
| 12 | siWEE1+siAKT3 | ALL | M Phase | 9.25E-04 | −2.30 | 6.86 | REACTOME |
| 13 | siWEE1+siAKT3 | ALL | Validated transcriptional targets of TAp63 isoforms | 2.89E-05 | −1.70 | 11.82 | NCI NATURE |
| 14 | siWEE1+siAKT3 | ALL | Direct p53 effectors | 2.19E-03 | −1.59 | 5.94 | NCI NATURE |
| 15 | siWEE1+siAKT3 | ALL | PLK1 signaling events | 3.91E-03 | −1.44 | 4.95 | NCI NATURE |
| 16 | siWEE1+siAKT3 | ALL | class I PI3K signaling events mediated by Akt | 2.56E-02 | −1.19 | 2.34 | NCI NATURE |
siRNA-mediated knockdown of AKT3 only altered 18 genes limiting the functionality of the enrichment analysis. Among these 18 genes, the most notable alteration was downregulation of cyclin-dependent kinase 6 (CDK6), which is an important effector of AKT signaling.39 In contrast to targeting AKT3 alone, siRNA-mediated cotargeting of AKT3 with WEE1 deregulated 40 genes, of which 14 were upregulated and 26 were downregulated (Fig. 3A). PLK1 signaling modulating the FOXM1 transcription factor was enriched only during the combination treatment (Fig. 3B and Table 1). FOXM1 and E2F transcription factors were enriched among the downregulated genes while GATA3 was enriched among the upregulated ones (Table 1).
To validate the signaling alterations at the protein level, RPPA analysis was used (Fig. 3C). Confirming the RNA sequencing results, knockdown of WEE1 enhanced the expression of DNA damage marker phospho H2AX, as well as p53 and downstream p21 levels. Furthermore, FOXM1 levels were decreased concomitantly with PLK1 levels (Fig. 3C).40 Knockdown of AKT3 increased p21 and phospho H2AX levels while reducing total AKT, pPRAS40, XIAP, PLK1, FOXM1 and phospho-Rb levels (Fig. 3C). Targeting the 2 kinases synergistically increased GATA3, phospho H2AX, p53 and downstream p21 levels while leading to a significant reduction in PLK1, FOXM1, Cyclin B1 and phospho-Rb levels. These observations were in concordance with the RNA sequencing analysis suggesting that, WEE1 inhibition leads to DNA damage that activates p53/p21 signaling, while AKT inhibition modulates this process through the PLK1/FOXM1 axes (Fig. 3C).
The results obtained from RPPA analyses were confirmed by Western blotting studies. Knockdown of WEE1 kinase led to a dose-dependent decrease in the phosphorylation of its substrate CDK1 (Figs. 4A and B). Decreased phosphorylation of CDK1 was predicted to increase its activity leading to the bypass of the G2/M checkpoint and induction of DNA damage.38 As expected, a dose-dependent increase in the phosphorylation of H2AX was observed. Consequently, this resulted in increased p53, p21 as well as p27 levels, which are known to be inhibitory to cell proliferation41 (Figs. 4A and B). FOXM1 and phosphorylated RB levels were also dose-dependently decreased by WEE1 knockdown. Decreased phosphorylation of RB and enhanced expression of p21 were observed with the AKT3 knockdown. Co-targeting of AKT3 and WEE1 was more effective at reducing FOXM1 and phosphorylated RB1 levels while enhancing cellular levels of p53 (Figs. 4A and B).
Figure 4.
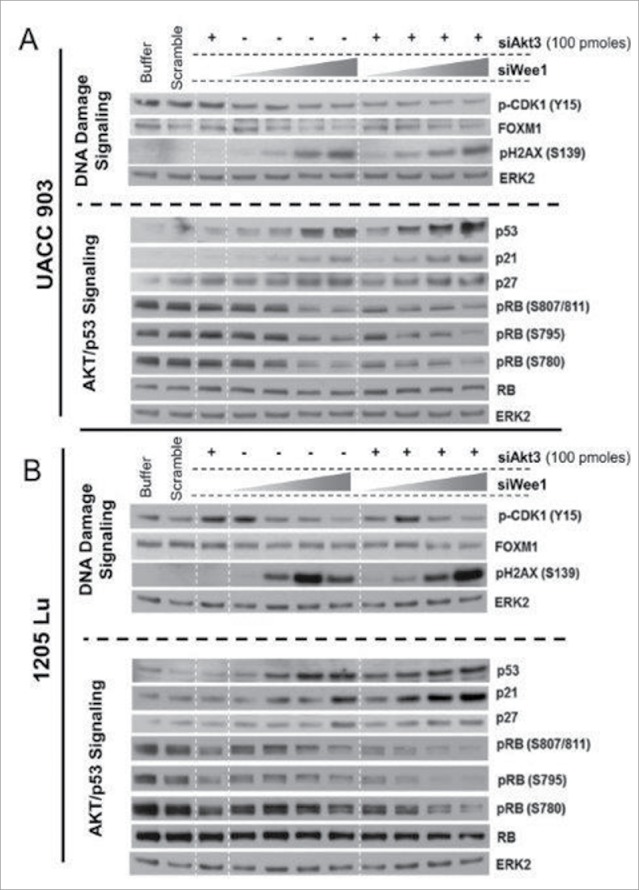
Targeting AKT3 and WEE1 increased p53 while reducing FOXM1 and CDK1 signaling. (A) and (B). Knockdown of WEE1 kinase led to a dose-dependent decrease in the phosphorylation of its substrate CDK1. Dose-dependent increase in the phosphorylation of H2AX was observed. Consequently, this resulted in increased p53, p21 as well as p27 levels, which are known to be inhibitory to cell proliferation. ERK2 served as a control for equal protein loading.
Discussion
The AKT3 signaling cascade is a major target in melanoma, activated in up to 70% of the patients.2 Loss of functional PTEN phosphatase triggers AKT3 activation, which is the most concurrent event occurring along with BRAF mutation.2 One of the functions of AKT3 is to phosphorylate mutant V600EBRAF protein to decrease its activity to levels that promote rather than inhibit melanocytic cell growth.2 Moreover, over-activated AKT signaling also positively regulates cell cycle while negatively regulating cell death cascades.42-44 In melanoma, over-activated AKT abrogates an efficient DNA damage response allowing cells to continuously divide without being eliminated by apoptosis.45 Furthermore, activation of AKT signaling is also implicated in drug resistance to mutant BRAF targeted therapies.16,17 Hence, targeting AKT3 has a significant chemotherapeutic potential to suppress melanoma tumor growth, as well as to reverse melanoma drug resistance.17,35
Unfortunately, targeting AKT3 alone in melanoma patients remains ineffective, requiring the identification of targets with which it can synergize for better therapeutic efficacy.2,46 This study identified WEE1 as a potential target to inhibit in combination with AKT3. Cotargeting AKT3 and WEE1 kinases synergistically reduced cellular proliferation and induced apoptosis. Based on the results of RNA sequencing, RPPA array and Western blotting experiments, a potential signaling map that mediate synergistic tumor inhibition is illustrated in Fig. 5. Accordingly, WEE1 is an important regulator of cell cycle progression and controls the G2/M checkpoint by catalyzing the inhibitory phosphorylation of CDK1 kinase at Tyr15 (Fig. 5#1).22 WEE1 mediated regulation of the mitotic cell cycle ensures that the cell has completed DNA replication and is ready for mitosis. WEE1 inhibition leads to DNA damage and mitotic catastrophe due to premature progression into the cell cycle (Fig. 5#2).47 Indeed, in this study, DNA damage induced by genetic targeting of WEE1 was observed as an increase in phospho H2AX, and resulted in a concomitant increase in p53-p21 signaling. Upregulation of p21 is known to halt S-phase progression by blocking the activity of CDK2.48 As a consequence inhibition of CDK2 leads to decreased phosphorylation of RB1 causing inhibition of E2F family of transcription factors required for the cell cycle progression.48 In this study we also observed a dose-dependent reduction of phosphorylated RB1 levels following the knockdown of WEE1. Taken together, targeting WEE1 induces cell cycle arrest through induction of DNA damage and transcriptional activity of p53.
Figure 5.
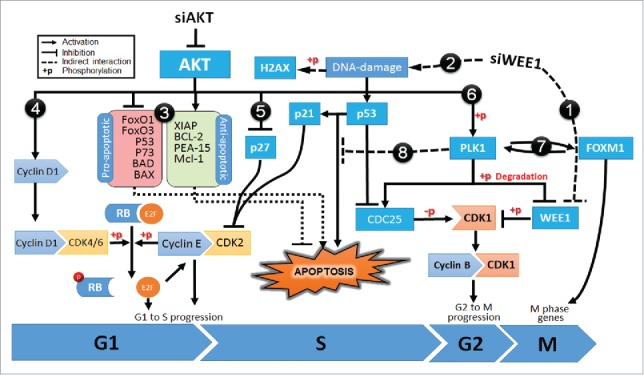
Diagram showing the mechanism of synergism for co-targeting AKT and WEE1 signaling pathways. Inhibition of siWEE1 (1) suppresses inhibitory phosphorylation of CDK1 leading to early-G2/M progression. This leads DNA damage (2) and activates p53 signaling. p53 inhibits cell cycle progression by induction of p21, allowing DNA damage repair. If the DNA damage is not repairable, p53 induces apoptosis. However, in many cancer cells, apoptotic cascades are suppressed by oncogenic alterations. Over-activated AKT inhibits pro-apoptotic factors while inducing antiapoptotic factors (3). AKT signaling also enhances cell cycle progression by CyclinD1 mediated phosphorylation of RB (4) and inhibition of p27 (5). Furthermore, AKT phosphorylates and induces Polo-like kinase 1 (PLK1) (6), which in turn inhibits pro-apoptotic functions of p53 and its family members, p63 and p73 (7). In addition, PLK1 also induces FOXM1 activity and M-phase progression (8). Proteins that were validated by Western blotting are shown in bold.
In steady-state conditions, DNA damage activates repair mechanisms. However, persistent DNA damage triggers apoptosis to eliminate damaged cells.49 In cancer cells, besides promoting cell cycle, over activated AKT bypasses apoptotic signals through multiple pathways (Fig. 5#3 to #5).50 Moreover, activated AKT also disrupts the DNA damage response by modulating p53 activity (Fig. 5#6). AKT not only induces ubiquitin-mediated degradation of p53 but also interferes with its function through stimulating the catalytic activity of PLK1.51-54 As a proto-oncogene frequently overexpressed in tumor cells, PLK1, inhibits the pro-apoptotic functions of p53 via physical interaction and phosphorylation (Fig. 5#7).55,56 In fact, studies have shown that the loss of PLK1 activity can induce pro-apoptotic pathways and inhibit tumor growth.55 PLK1 also promotes the activity of FOXM1, a proto-oncogene transcription factor that regulates G2/M phase progression as well as maintenance of chromosomal segregation and genomic stability (Fig. 5#8).56,57 Inhibition of AKT significantly hinders PLK1 activity and delay metaphase to anaphase transition by reducing FOXM1 activity.54 Indeed, genes that are deregulated by cotargeting of AKT with WEE1 were significantly enriched in the FOXM1 and PLK1 transcription factor networks as well as M/G1 transition and prometaphase genes.
Taken together, cotargeting of AKT and WEE1 kinases could modulate cell cycle and DNA damage responses through a complicated network that involves PLK1 and FOXM1 proteins. Since WEE1 has been identified as downstream of BRAF in the MAPK signaling cascade, this combinatorial approach might also be useful for overcoming acquired resistance to mutant BRAF inhibitors.23 Targeting PI3K-AKT and MAPK signaling is proposed as a strategy to reduce resistance to mutant BRAF inhibitors.9,58 Currently efficacy of WEE1 inhibitor, MK-1775, is being evaluated in clinical trials for the treatment of advanced solid tumors (NCT01748825, NCT02095132 and NCT01827384). Based on the results in this report, further studies to investigate the therapeutic potential of combining MK-1775 together with AKT or PI3K inhibitors are warranted for the treatment of melanoma to make AKT3 targeting more effective in the clinic.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Grant support
National Institutes of Health R01 CA-136667, RO1 CA-1138634 (to Gavin P. Robertson). The Foreman Foundation for Melanoma, The Geltrude Foundation, The Penn State Chocolate Tour Cancer Research Fund.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29. doi: 10.3322/caac.21254. PMID:25559415 [DOI] [PubMed] [Google Scholar]
- 2.Cheung M, Sharma A, Madhunapantula SV, Robertson GP. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res. 2008;68:3429-39. doi: 10.1158/0008-5472.CAN-07-5867. PMID:18451171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs. 2010;19:1355-66. doi: 10.1517/13543784.2010.520701. PMID:20846000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sullivan RJ, Flaherty KT. BRAF in Melanoma: Pathogenesis, diagnosis, inhibition, and resistance. J Skin Cancer. 2011;2011:423239. doi: 10.1155/2011/423239. PMID:22175026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et al. . Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-19. doi: 10.1056/NEJMoa1002011. PMID:20818844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. . Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-16. doi: 10.1056/NEJMoa1103782. PMID:21639808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E, et al. . Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-65. doi: 10.1016/S0140-6736(12)60868-X. PMID:22735384 [DOI] [PubMed] [Google Scholar]
- 8.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, et al. . Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107-14. doi: 10.1056/NEJMoa1203421. PMID:22663011 [DOI] [PubMed] [Google Scholar]
- 9.Jeon SH, Kim SH, Kim Y, Kim YS, Lim Y, Lee YH, Shin SY. The tricyclic antidepressant imipramine induces autophagic cell death in U-87MG glioma cells. Biochem Biophys Res Commun. 2011;413:311-7. doi: 10.1016/j.bbrc.2011.08.093. PMID:21889492 [DOI] [PubMed] [Google Scholar]
- 10.Masuda S, Izpisua Belmonte JC. Trametinib for patients with advanced melanoma. Lancet Oncol. 2012;13:e409;author reply e-10. doi: 10.1016/S1470-2045(12)70419-9. PMID:23026823 [DOI] [PubMed] [Google Scholar]
- 11.Sullivan R, LoRusso P, Boerner S, Dummer R. Achievements and challenges of molecular targeted therapy in melanoma. Am Soc Clin Oncol Educ Book. 2015;35:177-86. doi: 10.14694/EdBook_AM.2015.35.177 [DOI] [PubMed] [Google Scholar]
- 12.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. . Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973-7. doi: 10.1038/nature09626. PMID:21107323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perna D, Karreth FA, Rust AG, Perez-Mancera PA, Rashid M, Iorio F, Alifrangis C, Arends MJ, Bosenberg MW, Bollag G, et al. . BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc Natl Acad Sci U S A. 2015;112:E536-45. doi: 10.1073/pnas.1418163112. PMID:25624498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delmas A, Cherier J, Pohorecka M, Medale-Giamarchi C, Meyer N, Casanova A, Sordet O, Lamant L, Savina A, Pradines A, et al. . The c-Jun/RHOB/AKT pathway confers resistance of BRAF-mutant melanoma cells to MAPK inhibitors. Oncotarget. 2015;6:15250-64. doi: 10.18632/oncotarget.3888. PMID:26098773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson AR, et al. . PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750-60. doi: 10.1158/0008-5472.CAN-10-2954. PMID:21317224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. . Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683-95. doi: 10.1016/j.ccr.2010.11.023. PMID:21156289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atefi M, von Euw E, Attar N, Ng C, Chu C, Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin-Anduix B, et al. . Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PloS One. 2011;6:e28973. doi: 10.1371/journal.pone.0028973. PMID:22194965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lassen A, Atefi M, Robert L, Wong DJ, Cerniglia M, Comin-Anduix B, Ribas A. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol Cancer. 2014;13:83. doi: 10.1186/1476-4598-13-83. PMID:24735930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, et al. . First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29:4688-95. doi: 10.1200/JCO.2011.35.5263. PMID:22025163 [DOI] [PubMed] [Google Scholar]
- 20.Matulonis UA, Wulf GM, Barry W, Birrer MJ, Westin SN, Quy P, Bell-McGuinn KM, Lasonde B, Whalen C, Aghajanian C, Solit DB, Mills GB, Cantley L, and Winer EP, et al. . Phase I study of oral BKM120 and oral olaparib for high-grade serous ovarian cancer (HGSC) or triple-negative breast cancer (TNBC). J of Clin Oncol. 2014;32(15_suppl):2510-2510. doi: 10.1200/jco.2014.32.15_suppl.2510. [DOI] [Google Scholar]
- 21.Algazi AP, Muthukumar AH, O'Brien K, Lencioni A, Tsai KK, Kadafour M, Chapman PB, Daudet A, et al. . Phase II trial of trametinib in combination with the AKT inhibitor GSK 2141795 in BRAF wild-type melanoma. J of Clin Oncol. 2015;33(15_suppl):9068-9068. doi: 10.1200/jco.2015.33.15_suppl.9068. [DOI] [Google Scholar]
- 22.Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14:1878-91. PMID:7743995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma A, Madhunapantula SV, Gowda R, Berg A, Neves RI, Robertson GP. Identification of aurora kinase B and Wee1-like protein kinase as downstream targets of (V600E)B-RAF in melanoma. Am J Pathol. 2013;182:1151-62. doi: 10.1016/j.ajpath.2012.12.019. PMID:23416158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma A, Tran MA, Liang S, Sharma AK, Amin S, Smith CD, Dong C, Robertson GP. Targeting mitogen-activated protein kinase/extracellular signal-regulated kinase kinase in the mutant (V600E) B-Raf signaling cascade effectively inhibits melanoma lung metastases. Cancer Res. 2006;66:8200-9. doi: 10.1158/0008-5472.CAN-06-0809. PMID:16912199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65:2412-21. doi: 10.1158/0008-5472.CAN-04-2423. PMID:15781657 [DOI] [PubMed] [Google Scholar]
- 26.Madhunapantula SV, Sharma A, Gowda R, Robertson GP. Identification of glycogen synthase kinase 3alpha as a therapeutic target in melanoma. Pigment Cell Melanoma Res. 2013;26:886-99. doi: 10.1111/pcmr.12156. PMID:24034838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trapnell C, Pachter L, Salzberg SL. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105-11. doi: 10.1093/bioinformatics/btp120. PMID:19289445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gowda R, Kardos G, Sharma A, Singh S, Robertson GP. Nanoparticle based celecoxib and plumbagin for the synergistic treatment of melanoma. Mol Cancer Ther. 2017;16(3):440-52. doi: 10.1158/1535-7163.MCT-16-0285. PMID:28003325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gowda R, Madhunapantula SV, Kuzu OF, Sharma A, Robertson GP. Targeting multiple key signaling pathways in melanoma using leelamine. Mol Cancer Ther. 2014;13:1679-89. doi: 10.1158/1535-7163.MCT-13-0867. PMID:24688050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gowda R, Madhunapantula SV, Sharma A, Kuzu OF, Robertson GP. Nanolipolee-007, a novel nanoparticle-based drug containing leelamine for the treatment of melanoma. Mol Cancer Ther. 2014;13:2328-40. doi: 10.1158/1535-7163.MCT-14-0357. PMID:25082958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gowda R, Sharma A, Robertson GP. Synergistic inhibitory effects of Celecoxib and Plumbagin on melanoma tumor growth. Cancer Lett. 2017;385:243-50. doi: 10.1016/j.canlet.2016.10.016. PMID:27769779 [DOI] [PubMed] [Google Scholar]
- 32.Kuzu OF, Gowda R, Sharma A, Robertson GP. Leelamine mediates cancer cell death through inhibition of intracellular cholesterol transport. Mol Cancer Ther. 2014;13:1690-703. doi: 10.1158/1535-7163.MCT-13-0868. PMID:24688051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran MA, Gowda R, Sharma A, Park EJ, Adair J, Kester M, Smith NB, Robertson GP. Targeting V600EB-Raf and Akt3 using nanoliposomal-small interfering RNA inhibits cutaneous melanocytic lesion development. Cancer Res. 2008;68:7638-49. doi: 10.1158/0008-5472.CAN-07-6614. PMID:18794153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27-55. doi: 10.1016/0065-2571(84)90007-4. PMID:6382953 [DOI] [PubMed] [Google Scholar]
- 35.Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, Kester M, Sandirasegarane L, Robertson GP. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002-10. doi: 10.1158/0008-5472.CAN-04-1399. PMID:15466193 [DOI] [PubMed] [Google Scholar]
- 36.Madhunapantula SV, Sharma A, Robertson GP. PRAS40 deregulates apoptosis in malignant melanoma. Cancer Res. 2007;67:3626-36. doi: 10.1158/0008-5472.CAN-06-4234. PMID:17440074 [DOI] [PubMed] [Google Scholar]
- 37.Winter GE, Rix U, Carlson SM, Gleixner KV, Grebien F, Gridling M, Müller AC, Breitwieser FP, Bilban M, Colinge J, et al. . Systems-pharmacology dissection of a drug synergy in imatinib-resistant CML. Nat Chem biol. 2012;8:905-12. doi: 10.1038/nchembio.1085. PMID:23023260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dominguez-Kelly R, Martin Y, Koundrioukoff S, Tanenbaum ME, Smits VA, Medema RH, Debatisse M, Freire R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194:567-79. doi: 10.1083/jcb.201101047. PMID:21859861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu MG, Deshpande A, Enos M, Mao D, Hinds EA, Hu GF, Chang R, Guo Z, Dose M, Mao C, et al. . A requirement for cyclin-dependent kinase 6 in thymocyte development and tumorigenesis. Cancer Res. 2009;69:810-8. doi: 10.1158/0008-5472.CAN-08-2473. PMID:19155308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kreahling JM, Foroutan P, Reed D, Martinez G, Razabdouski T, Bui MM, Raghavan M, Letson D, Gillies RJ, Altiok S. Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PloS One. 2013;8:e57523. doi: 10.1371/journal.pone.0057523. PMID:23520471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: A function for each cell compartment? Trends Cell Biol. 2003;13:65-70. doi: 10.1016/S0962-8924(02)00043-0. PMID:12559756 [DOI] [PubMed] [Google Scholar]
- 42.Xu N, Hegarat N, Black EJ, Scott MT, Hochegger H, Gillespie DA. Akt/PKB suppresses DNA damage processing and checkpoint activation in late G2. J Cell Biol. 2010;190:297-305. doi: 10.1083/jcb.201003004. PMID:20679434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell. 2007;129:1261-74. doi: 10.1016/j.cell.2007.06.009. PMID:17604717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khwaja A. Akt is more than just a Bad kinase. Nature. 1999;401:33-4. doi: 10.1038/43354. PMID:10485701 [DOI] [PubMed] [Google Scholar]
- 45.Kandel ES, Skeen J, Majewski N, Di Cristofano A, Pandolfi PP, Feliciano CS, Gartel A, Hay N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol Cell Biol. 2002;22:7831-41. doi: 10.1128/MCB.22.22.7831-7841.2002. PMID:12391152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nissan MH, Solit DB. The “SWOT” of BRAF inhibition in melanoma: RAF inhibitors, MEK inhibitors or both? Curr Oncol Rep. 2011;13:479-87. doi: 10.1007/s11912-011-0198-4. PMID:21997758 [DOI] [PubMed] [Google Scholar]
- 47.Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, Van Tilborg AA, Zwinderman AH, Geerts D, Kaspers GJ, et al. . In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell. 2010;18:244-57. doi: 10.1016/j.ccr.2010.08.011. PMID:20832752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brugarolas J, Moberg K, Boyd SD, Taya Y, Jacks T, Lees JA. Inhibition of cyclin-dependent kinase 2 by p21 is necessary for retinoblastoma protein-mediated G1 arrest after gamma-irradiation. Proc Natl Acad Sci U S A. 1999;96:1002-7. doi: 10.1073/pnas.96.3.1002. PMID:9927683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Norbury CJ, Zhivotovsky B. DNA damage-induced apoptosis. Oncogene. 2004;23:2797-808. doi: 10.1038/sj.onc.1207532. PMID:15077143 [DOI] [PubMed] [Google Scholar]
- 50.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489-501. doi: 10.1038/nrc839. PMID:12094235 [DOI] [PubMed] [Google Scholar]
- 51.King FW, Skeen J, Hay N, Shtivelman E. Inhibition of Chk1 by activated PKB/Akt. Cell Cycle. 2004;3:634-7. doi: 10.4161/cc.3.5.894. PMID:15107605 [DOI] [PubMed] [Google Scholar]
- 52.Tonic I, Yu WN, Park Y, Chen CC, Hay N. Akt activation emulates Chk1 inhibition and Bcl2 overexpression and abrogates G2 cell cycle checkpoint by inhibiting BRCA1 foci. J Biol Chem. 2010;285:23790-8. doi: 10.1074/jbc.M110.104372. PMID:20495005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: A double-edged sword in cell proliferation and genome stability. J Oncol. 2012;2012:951724. doi: 10.1155/2012/951724. PMID:22481935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kasahara K, Goto H, Izawa I, Kiyono T, Watanabe N, Elowe S, Nigg EA, Inagaki M. PI 3-kinase-dependent phosphorylation of Plk1-Ser99 promotes association with 14-3-3 gamma and is required for metaphase-anaphase transition. Nat Commun. 2013;4:1882. doi: 10.1038/ncomms2879. PMID:23695676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu X, Erikson RL. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci U S A. 2003;100:5789-94. doi: 10.1073/pnas.1031523100. PMID:12732729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007;7:847-59. doi: 10.1038/nrc2223. PMID:17943136 [DOI] [PubMed] [Google Scholar]
- 57.Wierstra I, Alves J. FOXM1, a typical proliferation-associated transcription factor. Biol Chem. 2007;388:1257-74. doi: 10.1515/BC.2007.159. PMID:18020943 [DOI] [PubMed] [Google Scholar]
- 58.Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: A new class of treatment for metastatic melanoma. Clin Cancer Res. 2012;18:9-14. doi: 10.1158/1078-0432.CCR-11-2197. PMID:22083257 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.