

Abstract
The preparation and characterization of a representative group of novel non-heme metal nitrosyl complexes that have been synthesized over the last decade are discussed here. Their structures are examined and classified based on metal type, the number of metal centers present, and the type of ligand that is coordinated with the metal. The ligands can be phosphorus, nitrogen, or sulfur based (with a few exceptions) and can vary depending on the presence of chelation, intermolecular forces, or the presence of other ligands. Structural and bonding characteristics are summarized and examples of reactivity regarding nitrosyl ligands are given. Some of the relevant physical chemical properties of these complexes, including IR, EPR, NMR, UV–vis, cyclic voltammetry, and X-ray crystallography are examined.
Keywords: Dinitrosyl iron complex, Iron sulfur cluster, Metal nitrosyl, Nitric oxide
1 Introduction
Metal nitrosyl complexes are classified as having at least one nitric oxide group attached to a transition metal atom. The synthesis and study of these molecules began in the mid-1960s and have increased exponentially since the 1990s because of the discovery that nitric oxide can be used within the body for smooth muscle relaxation, tumor regulation, and long-term memory formation [1–3].
There are many different types of metal nitrosyl complexes. Homoleptic nitrosyl complexes containing only a metal atom and nitric oxide groups are very rare [4]. The majority of metal nitrosyl complexes contain two nitric oxide groups and are known as dinitrosyl complexes. However, mononitrosyl complexes are numerous and sometimes represent the products of nitric oxide transfer reactions of their precursor, dinitrosyl complexes. Examples of trinitrosyl complexes are very rare, but do exist. The majority of known nitrosyl complexes have simple structures consisting of a single metal core, but some have several metal atoms in a cluster arrangement. Nearly all metal nitrosyl complexes also have one or more organic ligand molecules of varying size, shape, and donor/acceptor properties. The type of ligand present and the coordination of the ligand to the metal center ultimately define the overall characteristics of the complex.
It has been reported that these complexes exhibit properties that make them useful for pharmaceutical and biological applications; the most useful of these is the storage and transport of nitric oxide [5, 6]. Many recently synthesized complexes demonstrate the ability to deliver NO and a few are currently being used to treat common ailments such as high blood pressure. It is predicted that these nitrosyl complexes can have more complex biological implications, including aiding in long-term memory formation, fighting infection, and treating cancer.
Although this subject has been frequently and comprehensively reviewed [7–11], this review describes a selection of representative metal nitrosyl complexes synthesized during the last decade or so. The representative compounds are chosen in order to give a wide array of structures, including different metal center type, different ligand type and structure, and better synthetic procedures. The synthetic methods, structural characteristics, and spectroscopic properties of those selected complexes are described in detail, while structures similar to those mentioned here are cited but not necessarily discussed. Organometallic metal nitrosyl complexes are minimally discussed with a few examples relating to the reactivity of nitric oxide at transition-metal centers. Work on heme nitrosyls is not discussed, as this subject is the focus of another chapter.
2 Dinitrosyl Complexes Containing a Single Metal Center
Dinitrosyl complexes with single transition metal center are commonly represented with a general formula of M(NO)2(L)2 and are known to possess tetrahedral geometry. Recently, these dinitrosyl units have been detected within living tissues under a wide range of conditions, including inflammatory responses [12]. The [M(NO)2] unit has also been shown to bind to proteins containing cysteine residues and can be formed when a protein with a coordinated iron atom reacts with gaseous nitric oxide [13–15]. These dinitrosyl iron complexes have a nickname of “g = 2.03 species” and are also referred to as non-heme iron nitrosyls in biological systems. Thus, nowadays, dinitrosyl metal complexes are often synthesized with ligands containing sulfur, nitrogen, phosphorus, or oxygen atoms with an ultimate goal of mimicking biological non-heme iron nitrosyls.
2.1 Dicarbonyldinitrosyl Iron
The most common starting material used to synthesize dinitrosyl metal complexes is dicarbonyldinitrosyl iron (DDI). This useful starting material has been synthesized through several different procedures starting in the early 1930s. Some of the different methods included a simple acidification of nitrate ion and Fe(CO)5, or pyrolysis of Hg [Fe(CO)3NO]2, the second of which is only suitable for small amounts of material [16]. Another method involves reacting Fe3(CO)12 or Fe2(CO)9 with nitric oxide [17].
One of the more common procedures for making DDI involves placing a stoichiometric ratio of iron pentacarbonyl and nitrosyl chloride in a stainless steel bomb. This container is sealed and allowed to sit at room temperature for approximately 24 h before cooling to −196°C. Carbon monoxide, the ubiquitous by-product of these reactions, is then pumped off. The contents of the container are distilled under reduced vacuum pressure into a trap cooled to a temperature of −78°C. Analysis by vapor phase chromatography shows that the red liquid collected in the trap is approximately 65% dicarbonyldinitrosyl iron and 35% iron pentacarbonyl [18]. It was found that when the ratio of iron pentacarbonyl to nitrosyl chloride exceeded 1:2, no appreciable amount of dicarbonyldinitrosyl iron is produced.
The most recent method for synthesizing and collecting dicarbonyldinitrosyl iron involves a reaction between iron pentacarbonyl and sodium nitrite and is a slightly modified version of a previously published procedure [19]. The reactions, shown in Eqs. (1) and (2), illustrate the two-step process that occurs. A three-necked round bottom flask is equipped with a condenser column, a nitrogen inlet, and a pressure equalized dropping funnel. The flask is filled with 50 ml of degassed water, 6.0 g sodium nitrite, and 10.0 g sodium hydroxide. Once all of the reactants are dissolved, 5.0 ml of iron pentacarbonyl is injected into the flask. The reaction mixture is refluxed for 3 h with stirring under a gentle nitrogen flow. After the reaction is complete, the temperature is reduced to 30–40°C and a stream of nitrogen is passed through the system, into two traps containing approximately 20 g of calcium chloride, and another two traps cooled to −78°C using an acetone/dry ice bath. A 75% solution of glacial acetic acid and water is added dropwise from the dropping funnel. Brown fumes of DDI begin to form and are collected in the cold trap. A reaction of this scale can be expected to afford up to 60% yield. It is noted that any reaction involving ironpentacarbonyl as a reactant may have trace amounts of the material as a contaminant after the reaction has completed.
| (1) |
| (2) |
2.2 Phosphorus Based Ligands
One of the first DNIC structures synthesized with phosphine ligands was reported in the early 1960s. Since then, many new structures have been synthesized that range from simple derivatives of triphenyl phosphine [20, 21] to more complicated structures that make use of different metal centers [22–24]. We choose to use the following examples to illustrate the structures, spectroscopic properties, and mechanisms of reactions.
Fe(NO)2(CO)(PR3)
Several different nitrosyl complexes were synthesized using phosphine-type ligands PR3, where R = PPh3, OCH3, P(n-Bu)3, PMe2Ph, PEt2Ph. These compounds are synthesized by reacting a 1:1 ratio of Fe(NO)2(CO)2 with the appropriate phosphine ligand at room temperature for approximately 12–15 h [25, 26]. These carbonyl substitution reactions are slow, requiring a longer reaction time for the replacement of the first carbonyl and requiring heating at 85°C for 1–2 days to replace the second carbonyl [27], and is believed to proceed via a conventional associative mechanism. The FT-IR data of these complexes are listed in Table 1. The spectrum of the starting material, Fe(NO)2(CO)2, exhibits two nitrosyl stretches and two carbonyl stretches. Upon replacement of a single carbonyl moiety by a phosphite or phosphine group, the remaining CO absorbs in the lower frequency range 1995–2018 cm−1. The two nitrosyl IR stretches in these complexes also shift to lower wavenumbers. The shifting can be explained by the fact that phosphorus donor increases the electron density at the iron center, which in turn enhances the back-bonding from the filled d-orbitals on the metal to the vacant anti-bonding orbitals of the carbonyl and the nitrosyls, with concomitant weakening of the C=O bond as well as the N≡O bond. This results in shift of both carbonyl and the nitrosyl stretches towards lower wavenumbers, in the regions of ~1995–2018 and 1700–1770 cm−1, respectively. The cyclic voltammogram data for these complexes are listed in Table 2. The family of Fe(NO)2(PR3)(CO) compounds shows a quasi-reversible reduction potential E° from −1.96 V to −2.15 V with large peak-to-peak separations. A correlation between the pKa values and reduction potentials has been observed. This observation can be qualitatively rationalized in that with increasing electron density being donated to the iron center, it is rendered less prone to reduction. A comparison can be made between the decreasing average IR stretching frequencies for the nitrosyls in Table 1 and the increasing pKa values in Table 2.
Table 1.
A list of IR stretching frequencies for the Fe(NO)2(PR3)(CO) family and Fe(NO)2(PR3) (TCNE) family
| Complex | CN stretch (cm−1) | CO stretch (cm−1) | NO stretch(es) (cm−1) | Average NO stretch (cm−1) |
|---|---|---|---|---|
| Fe(NO)2(CO)2 | – | 2090, 2040 | 1817, 1766 | |
| Fe(NO)2(PPh3)(CO) | – | 2007 | 1766, 1718 | |
| Fe(NO)2(PPh3) (η2-TCNE), 1 | 2224 | – | 1834, 1790 | |
| Fe(NO)2[P(OMe3)](CO) | – | 2018 | 1770, 1722 | 1770, 1722 |
| Fe(NO)2[P(OMe)3] (η2-TCNE), 2 | 2230 (2233) | – | 1843, 1790 (1843, 1797) | 1843, 1797 |
| Fe(NO)2[P(n-Bu)3](CO) | – | (1995)a | (1752, 1704) | 1752, 1704 |
| Fe(NO)2[P(n-Bu)3] (η2-TCNE), 3 | 2229 (2230) | – | 1828, 1778 (1824, 1785) | 1824, 1785 |
| Fe(NO)2[PMe2Ph](CO) | – | (2004) | (1754, 1708) | 1754, 1708 |
| Fe(NO)2[PMe2Ph] (η2-TCNE), 4 | 2219 (2226) | 1839, 1792 (1830, 1786) | 1830, 1786 | |
| Fe(NO)2[PEt2Ph](CO) | (2004) | (1755, 1706) | 1755, 1706 | |
| Fe(NO)2[PEt2Ph] (η2-TCNE), 5 | 2225 (2231) | – | 1812, 1755 (1827, 1790) | 1827, 1790 |
Values shown in parentheses were measured in CH2Cl2 solution
Table 2.
Electrochemical potentials for the family of Fe(NO)2(PR3)(TCNE) species vs FeCp2+/FeCp2 at scan rate of 100 mV/s and the pKa values for the phosphines and phosphite
| Complexes | Epc (V) | Epa(P) (V) | E° (ΔE) (V) | pKa (Phosphine) |
|---|---|---|---|---|
| Fe(NO)2[P(OMe)3](η2-TCNE), 2 | −0.990 | −0.398 | 2.6 | |
| Fe(NO)2[PEt2Ph](η2-TCNE), 5 | −1.120 | −0.604 | 6.25 | |
| Fe(NO)2[PMe2Ph](η2-TCNE), 4 | −1.123 | −0.607 | 6.5 | |
| Fe(NO)2[P(n-Bu)3](η2-TCNE), 3 | −1.146 | −0.652 | 8.43 | |
| Fe(NO)2[P(OMe3)](CO), | −1.96 (125 mV) | 2.6 | ||
| Fe(NO)2[PEt2Ph](CO) | −2.108 (131 mV) | 6.25 | ||
| Fe(NO)2[PMe2Ph](CO) | −2.101 (268 mV) | 6.5 | ||
| Fe(NO)2[P(n-Bu)3](CO) | −2.150 (130 mV) | 8.43 |
Fe(NO)2[PR3](η2-TCNE)
The substitution of the CO group by TCNE is further accomplished by treating a 1:1 molar ratio of Fe(NO)2(CO)(PR3) with TCNE, which afforded complexes of Fe(NO)2[PR3](η2-TCNE) (where PR3 = PPh3, 1, P(OCH3)3, 2, P(n-Bu)3, 3, PMe2Ph, 4, and PEt2Ph, 5) [20]. These reactions typically occur over 1–2 h at room temperature with yields over 80%. These complexes are soluble in polar solvents such as CH2Cl2, THF, (CH3)2CO, CH3CN, and MeOH. However, decomposition occurs after a few hours. They are relatively stable when stored in the solid state under a nitrogen atmosphere and at low temperatures.
The rapidity of such a substitution reaction is due to the participation of free radicals, as evidenced by the observation of the intermediates of TCNE− and Fe(NO)2(PPh3)L+ (where L may be CO or a coordinated solvent molecule) radicals using EPR upon mixing of the starting materials. We proposed that the reaction proceeds via an electron transfer autocatalysis mechanism, through a 17-electron paramagnetic intermediate as shown in Scheme 1.
Scheme 1.
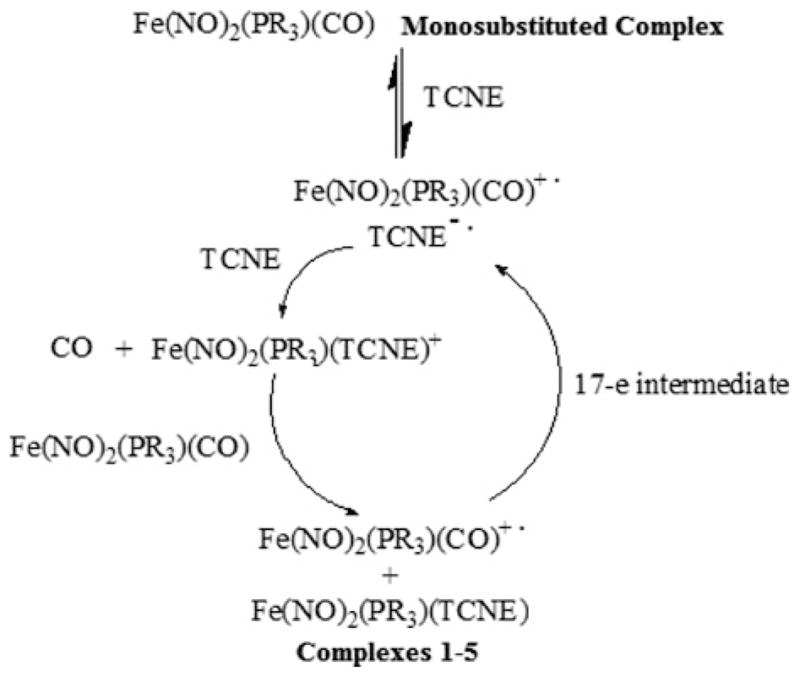
Mechanism of the reactions of Fe(NO)2(CO)(PPR3) with TCNE showing the electron transfer autocatalytic pathway through a 17-electron intermediate
The FT-IR data for 1–5 and other related complexes are listed in Table 1. The IR spectrum shows that the carbonyl stretching band of the CO group disappears during replacement of the carbonyl group by TCNE. Only one broad cyano stretching frequency at 2225 cm−1 in both solid state and in solution is visible in the IR spectra. This gives evidence that the TCNE moiety is in fact π-bonded to the iron. The cyano stretching frequency is shifted to a lower wavenumber in comparison with the corresponding stretching frequencies for free TCNE, which can be interpreted in terms of the efficient back-donation from the filled metal d-orbitals on iron into the vacant π*-orbital of TCNE. This back-donation results in a weakening of the C≡N bond. X-ray crystal structures for compounds 1 and 2 were obtained, and a representative ball-and-stick model of structure 2 is shown in Fig. 1. From the crystal data, the dihedral angle between the plane containing C(1)=C(2), that is perpendicular to the Fe–C(1)–C(2) plane and the plane containing C(1)–C(12)–N(12), and C(11)–N(11) is 15.6°. This loss of planarity presumably results from the back-donation of electron density from the metal to the alkene π* manifold.
Fig. 1.

A ball-and-stick representation of the X-ray crystal structure of compound 2
The 13C-NMR spectrum of 2 at room temperature showed pairs of cyano peaks at 113.1 ppm (JC-P = 4.0 Hz) and 113.3 ppm (JC-P = 5.6 Hz), in addition to the expected methoxy carbon at 55.3 ppm (JC-P = 6.5 Hz). A very weak peak, due to relatively long relaxation time, at 29.0 ppm was also observed and was assigned to the ethylene carbons. Because of coordination to the iron center, the ethylene carbons are significantly shielded compared to those of free TCNE (112.6 ppm). This phenomenon was also observed in the 13C NMR spectrum of 1. The crystallographic data support these observations, especially the lengthening of the C(1)–C (2) distance in the π-bonded TCNE moiety relative to free TCNE. These chemical shifts are indicative of significant sp3-character at the olefinic carbons of the tetracyanoethylene moiety.
A shift of the two nitrosyl peaks to higher frequencies (1750 cm−1 and 1850 cm−1) is also observed. This is explained by the electron-withdrawing effect of TCNE, which reduces back-bonding, which in turn strengthens the nitrosyl bond. The magnitude of this high frequency shift completely compensates for the bond weakening observed on initial incorporation of the phosphine or phosphite moiety. The average N–O distance in 2 (1.158 Å) is similar to the corresponding values found in 1 (1.169 Å) and Fe(NO)2(CO)2 (1.171 Å) [28]. This confirms that the electron-withdrawing effect of the TCNE moiety is sufficient to counteract the corresponding electron-donating strength of the phosphorus ligand, as compared to the Fe(NO)2(CO)2 reference.
Upon replacement of the carbonyl by TCNE, the reduction becomes irreversible. Table 2 lists the reduction potentials, Epc, of complexes 2–5 at a scan rate of 100 mV/s. This reduction has no corresponding oxidation peak, even at a scan rate of 1 V/s, which indicates that the reduction is chemically irreversible at room temperature. Thus, the radical anion, [Fe(NO)2PR3(η2-TCNE)]−, decomposes rapidly to yield a decomposition product. The complexes 2–5 (Epc = −0.990 V ~ −1.146 V) are harder to reduce than the free TCNE ligand (E1/2 = −0.207 V) but are easier to reduce than the corresponding carbonyl compound, Fe(NO)2(PR3)(CO) (E° = −1.96 ~ −2.15 V) [20]. The coordination of TCNE leads to a shift in the reduction potentials to a more negative value compared to free TCNE and to a more positive value in comparison with Fe(NO)2(PR3)(CO). This indicates that the back-bonding from the iron center to the TCNE ligand is stronger than the characteristic covalent bonding arising from σ-donation by the TCNE ligand. This back-donation to the TCNE ligand renders the iron atom partially positive, and thereby easier to reduce. The reduction is presumed to occur at the iron center rather than on the TCNE ligand since the basicity of the phosphorus moiety has an impact on the electrochemical behavior of these compounds.
It is interesting to note that rotation along Fe-TCNE π-bond is restricted on the NMR time-scale. Variable-temperature 13C NMR spectra were recorded as shown in Fig. 2. The peaks coalescence at about 70°C and is consistent with an activation energy barrier of 18.1 kcal/mol. This is rather high compared to the published barriers for the rotation of coordinated olefins, which are typically between 12 and 15 kcal/mol. Clearly, the increased barrier to TCNE rotation is a reflection of the enhanced back-donation from metal d-orbitals into π* of the alkene.
Fig. 2.

Variable-temperature 13C-NMR spectra of structure 1 in CD3CN. Reprinted from Journal of Crganometallic Chemistry Volume 550, A. Horsken, G. Zheng, M. Stradiotto, C. T.C. McCrory, L. Li “Iron Dinitrosyl Complexes of TCNE: A Synthetic, X-Ray Crystallographic High Field NMR and electrochemical Study.” p 1–9, 1998, with permission from Elsevier
One crystallographic feature worthy of noting is that the two nitrosyl groups in 2 are nearly linear with angles of 175.1(7)° and 172.6(6)°, while for 1, angles of 178.0 (5)° and 165.8(5)° were observed. The Fe(NO)2 unit is in an “attracto” conformation in 2 with O–Fe–O and N–Fe–N angles of 116.2° and 120.9°, respectively. “Attracto” conformations generally favor first row transition-metal dinitrosyls containing ligands that are good π-acceptors. The reverse conformation (O–M–O > N–M–N) is called “repulso” and is more common for the second-row and third-row transition metals [29]. For example, the N–M–N and O–M–O bond angles reported for M(NO)2(PPh3)2 complex are 139.2° and 142.7° when M = Ru [30, 31] and 139.1° and 140.6° when M = Os [32]. This is also true for the cationic species, [M(NO)2(PPh3)2]+, where the reported N–M–N and O–M–O bond angles are 157.5° and 173.7° for Rh [33] and 154.2° and 167.5° for Ir [21], respectively. Five coordinated dinitrosyls also fit this correlation, indicating the conformation is an electronic rather than steric effect [34].
(NO)2FeP~PFe(NO)2
Despite the well-established history of dinuclear bis (phosphine) complexes in the field of inorganic chemistry, bis(dinitrosyliron) derivatives of this class are still rare and their properties remain essentially unexplored with the exception of a few select structures [35, 36]. Depending on the reaction conditions employed, either linear diiron constructs connected by one bis(phosphine) linker, (NO)2FeP~PFe(NO)2, or macrocyclic species spanned by two bridging ligands, [(NO)2Fe]2(P~P)2, can be obtained [35].
Several different compounds including a mononuclear complex, (NO)2FeP–X–P (X = CH2, 6), and several linear diiron species, (NO)2FeP–X–PFe(NO)2 (X = CH2, 7, C≡C, 8, (CH2)6, 9, and p-C6H4, 10), could be easily synthesized from Fe(NO)2(CO)2 via addition of the desired ligand (Scheme 2). From those compounds, the cyclic structures 11 and 12 can be synthesized. All of these species are air sensitive and will completely decompose after several hours. Decomposition occurs at a slower rate and is minimized when the compounds are stored in a proper degassed solvent or as a pure solid under a nitrogen atmosphere at ambient temperature.
Scheme 2.

Generalized synthetic pathway to compounds 6–12
The conversion of Fe(NO)2(CO)2 into compounds 6–10 and subsequently into 11 or 12 is readily monitored by the use of infrared spectroscopy; selected FT-IR data are listed in Table 3. The decrease in stretching frequencies observed for the carbonyl and the two nitrosyl ligands in 6–10, relative to Fe(NO)2(CO)2, is characteristic of phosphine substituted dinitrosyl iron complexes. In turn, the nitrosyl stretching frequencies observed for both 11 and 12 appear at even lower wavenumbers. The macrocyclic DPPM-supported complex, 11, exhibits four distinct IR absorptions (1733, 1721, 1687, and 1668 cm−1) in the solid state and in solution, possibly arising from the interaction of the Fe(NO)2 centers, as has been observed in other cyclic systems [37, 38]. This phenomenon appears to depend on ring size, as the related ten-membered ring compound, 12, displays only two nitrosyl stretching signals (1723 and 1679 cm−1) in both the solid and liquid states. Based on the observed IR frequencies, the nitrosyl groups are best described as linear donating NO+ fragments (vide infra). The formation of 6–12 was also followed by the use of NMR spectroscopy, and each of the dinuclear compounds 7–12 exhibits a single 31P NMR resonance in the range of 33–57 ppm, consistent with a disubstituted bis(phosphine) complex.
Table 3.
Nitrosyl and carbonyl IR stretching frequencies for species 6–12 (KBr pellet)
| Compound | νCO (cm−1) | νNO (cm−1) |
|---|---|---|
| [Fe(DPPM)(NO)2(CO)], 6 | 2014, 1994, 2005a | 1763, 1720 (s), 1700, 1761,a 1718a |
| [Fe2(μ-DPPM)(NO)4(CO)2], 7 | 2005, 2004a | 1760, 1719 (s), 1702, 1764,a 1718a |
| [Fe2(μ-DPPA)(NO)4(CO)2], 8 | 2020, 2005 | 1767, 1716 |
| [Fe2(μ-DPPH)(NO)4(CO)2], 9 | 1999 | 1755, 1701 |
| [Fe2(μ-DPPB)(NO)4(CO)2], 10 | 2009, 1999 | 1760, 1707 |
| [Fe2(μ-DPPM)2(NO)4], 11 | – | 1733, 1721, 1687, 1668 |
| [Fe2(μ-DPPA)2(NO)4], 12 | – | 1723, 1679 |
Measured in THF solution
X-ray crystallographic studies of compounds 7, 8, 11, and 12 were conducted and the thermal ellipsoid plots of the refined molecular structures of the DPPM compounds 7 and 11 appear in Figs. 3 and 4, respectively. The iron centers in all four of the crystallographically characterized compounds possess distorted tetrahedral geometries, a structural feature that is common to dinitrosyliron complexes. The iron–iron distances in both the linear (7, ~5.2 Å; 8, ~7.6 Å) and macrocyclic (11, ~4.4 Å; 12, ~7.0 Å) compounds are all significantly longer than the related distances found in other structurally characterized species that are described as possessing a metal–metal bond. The crystallographically determined structures of the linear species, 7 and 8, can be compared with that of [Fe(NO)2Cl]2(μ-DPPE) (DPPE = 1,2-bis(diphenylphosphino)ethane), where DPPE stands as a single bridge joining the two metal centers [35].
Fig. 3.

An X-ray crystallographic representation of species 7 with thermal ellipsoids drawn at 50%. Hydrogen atoms are eliminated for clarity
Fig. 4.

An X-ray crystallographic representation of species 11 with thermal ellipsoids drawn at 50%. Hydrogen atoms are eliminated for clarity
Despite the range in N–Fe–N angles (115.6° to 126.5°), all eight of the Fe(NO)2 units in compounds 7, 8, 11, and 12 exhibit “attracto” conformations where the N–Fe–N angle is greater than the > O–Fe–O angle. The observation of contracted Fe–N distances (~1.64 to ~1.73 Å) and lengthened N–O bonds (~1.16 to ~1.20 Å) in these complexes indicates significant iron-nitrosyl multiple bond character, arising due to appreciable back-donation from the iron fragment into the π*-orbital on the nitrosyl ligand. By comparison, Ray et al. reported crystallographic data for a series of trigonal bipyramidal, iron nitrosyl complexes, in which the Fe–N(O) and N–O distances are in the range of ~1.73–1.75 Å and ~1.12–1.15 Å, respectively [39]. These X-ray structural data and the IR results suggest that the NO units in 7, 8, 11, and 12 function as three-electron donors. Compounds 8 and 12 represent the first examples of crystallographically characterized species containing the Fe(NO)2(μ-DPPA).
2.3 Nitrogen-Based Ligands
Dinitrosyl iron complexes in biological systems have been investigated intensively by EPR techniques and usually exhibit a characteristic isotropic g-factor of 2.03 [21, 31–43]. Three types of EPR-active “g = 2.03” complexes have been identified in mammalian ferritins, which have been attributed to iron-nitrosyl complexes with imidazole groups of histidine, thiol groups of cysteine, and carboxylate groups of aspartate and glutamate [44]. While there are plenty of examples of in situ characterization by IR or EPR spectroscopy, the isolation and structural determination of these compounds are both extremely tedious and difficult. It was not until 1994 that the first “g = 2.03” species, Fe(NO)2(1-MeIm)2, 13, was isolated [45]. Since then, other structures utilizing ligands pertaining to the imidazole family [46, 47] or containing metals other than iron [48] have been reported. In this section, the physical and chemical properties of several well-studied structures are thoroughly examined. General synthesis steps and some spectroscopic data are given for other representative compounds in order to demonstrate the array of structures that have been synthesized so far.
Fe(NO)2(1-MeIm)2
Single crystals of structure 13 suitable for X-ray analysis were obtained from diethyl ether by mixing a 1:1 ratio of DDI with 1-methylimidazole. The X-ray crystal structure is shown in Fig. 5. The complex is pseudo-tetrahedral with a d10 iron center. The nitrosyl groups are linear with Fe–N–O angles of 167.5° and 170.1° and are displaced at the N(NO)–Fe–N(Im) angles of 111.3° and 107.8° away from the imidazole ligands. The Fe–N(NO) bond distances are 1.648 and 1.650 Å. The Fe–N–O groups are bent symmetrically, with a O–Fe–O angle of 107.3° as compared to the N(NO)–Fe–N(NO) angle of 116.6°. This is considered an “attracto” conformation because the N–M–N bond angle is less than 130° and the two oxygen atoms bend towards each other. The Fe–N(Im) bond distances are 2.048 and 2.044 Å. The horizontal plane through each 1-methylimidazole ligand is skewed 106.7° away from the other [45].
Fig. 5.

A ball-and-stick representation of the X-ray crystal structure of complex 13
The reaction of Fe(NO)2(CO)2 with L-histidine and a series of substituted imidazoles, such as 1-methylimidazole, 4-methyl imidazole (4-MeIm), imidazole (Im), benzimidazole (BenzIm), and 5,6-dimetheylbenzimidazole (Me2BenzIm) were also investigated by EPR spectroscopy in order to establish the validity of using this ligand as the biomimetic model. Figure 6 shows a typical reaction monitored by EPR spectroscopy. The starting material, Fe(NO)2(CO)2, underwent an auto-oxidation in the solution and produced a broad singlet with g = 2.0275 and ΔHpp = 18.5 G corresponding to [Fe(NO)2(CO)2]+. Upon the addition of one of the ligands, a new set of signals, corresponds to the 17e species, overlapping with the broad singlet was observed after approximately 15 min. A list of the EPR parameters measured at 240 K in solution and some of the IR stretching frequencies of the isolated product are shown in Table 4. The g-values for the imidazole and substituted imidazole radicals fall in the range of 2.0151–2.0338. Computer simulations gave rise to two sets of equivalent nitrogens (14N, I = 1) and aN in the range of 2.28–3.90 G. The hyperfine structure is due to coupling of two equivalent 14N nuclei from the nitrosyls and two equivalent 14N nuclei from the 1-MeIm, yielding a structure of Fe(NO)2(I-MeIm)2+. For benzimidazole and substituted benzimidazole, the g-values are slightly higher (2.0341–2.0352) while the aN values are smaller (1.88 G–2.12 G). For L-histidine, both g-value (2.0222) and hyperfine couplings (2.66 G and 3.01 G) fall in the range of substituted imidazoles.
Fig. 6.

The evolution of Fe(NO)2(Me-Im)2+ upon addition of Me-Im to a solution of Fe(CO)2(NO)2. Reprinted with permission from N. Reginato, C.T.C. McCrory. D. Pervitsky and L. Li. 1999. J. Am. Chem. Soc. 121, pp. 10217–10218. Copyright 1999 American Chemical Society
Table 4.
A list of EPR parameters measured at 240 K and IR stretching frequencies of the isolated product
| Compound | g-value | aN (G) | νNO (cm−1) |
|---|---|---|---|
| Fe(NO)2(1-MeIm)2+ | 2.0151 | aN1 = 3.60, aN2 = 3.90 | 1673, 1616 |
| Fe(NO)2(4-MeIm)2+ | 2.0338 | aN1 = 2.33, aN2 = 2.64 | 1677, 1620 |
| Fe(NO)2(Im)2+ | 2.0337 | aN1 = 2.28, aN2 = 2.44 | 1680, 1622 |
| Fe(NO)2(BenzIm)2+ | 2.0341 | aN1 = 1.97, aN2 = 2.12 | 1682, 1625 |
| Fe(NO)2(5,6-dimethylBenzIm)2+ | 2.0352 | aN1 = 1.88, aN2 = 2.04 | 1683, 1625 |
| Fe(NO)2(L-Histidine)2+ | 2.0222 | aN1 = 2.66, aN2 = 3.01 | – |
FT-IR investigations showed that upon addition of the imidazole ligands to Fe(NO)2(CO)2, the νNO’s were lowered by approximately 140 wavenumbers and no carbonyl stretching frequencies were observed. This indicates that two Im ligands had replaced the two CO ligands. The nitrosyls fall into the region of 1650–1940 cm−1, indicating they are linear. Table 4 lists the NO stretching frequencies observed for these complexes. Addition of 1-MeIm shifted the IR stretching frequencies of the nitrosyl groups from 1810 and 1767 cm−1 [νNO for Fe(NO)2(CO)2] to 1673 and 1616 cm−1, suggesting that 1-MeIm acts as a strong σ-donor. To explain the trend in the IR stretching frequencies, EHMO calculations were made. The results show that the LUMO of the Fe(NO)2 unit has more contribution to the overall molecular orbitals. Thus, effectively, imidazole units act as electron donors to the LUMO of Fe(NO)2 fragment.
Fe(NO)2(N,N′-chelate)
Despite the fact that coordination of N,N′-chelating ligands to transition metals is very common, only a limited number of examples on isolated iron-nitrosyl with nitrogen donor ligands involving N,N′-chelation could be found. Earlier work includes: a reaction of [Fe(NO)2Cl]2 with 1,4,-diaza-1,3-butadiene, which yielded a DNIC with N,N′-chelation [49], a salt [Fe(bipy)3][Fe(NO)2Cl2]2, which was isolated by using large excess of bipy vs. [Fe(NO)2Cl]2 (10:0.75) [50], and [(spartiene)Fe(NO)2] was prepared by reacting spartiene with Fe(CO)2(NO)2 [51].
Recently, we reported the synthesis, structures, and spectroscopic and electrochemical properties of three new dinitrosyl iron complexes with bidentate [N,N] chelating ligands; 2,2′-bipyridine, 14, 2,2′,2′′-terpyridine, 15, and 1,10-phenathroline, 16 [52]. Complexes 14–16 were prepared by mixing 1:1 ratio of Fe(NO)2(CO)2 and the proper chelating ligands, bipy, terpy, and phen in THF and was stirred 48 h at room temperature under nitrogen atmosphere as shown in Scheme 3 (yields between 82 and 88%). X-ray quality single crystals of 14 and 15 were obtained by slow evaporation of either THF or methanol. Complexes 14–16 are stable in the solid state under nitrogen, and all three complexes are relatively soluble in most polar organic solvents including dichloromethane, methanol, and THF but are insoluble in diethyl ether and hexane.
Scheme 3.

Synthesis scheme for compounds 14–16
Similar to the imidazole-based ligands, upon reacting, the typical carbonyl stretching frequencies from Fe(NO)2(CO)2 disappeared, indicating that both carbonyl groups are replaced by the bidentate ligands 2,2′-bipyridine, 2,2′,2′′-terpyridine, or 1,10-phenanthroline as observed in the crystal structures. In the meantime, the characteristic IR absorptions of nitrosyl groups (νNO, 1807, 1760 cm−1) shifted ~120–146 cm−1. These values are located in the range of NO+, suggesting again that these nitrogen-containing ligands act as strong σ donors rather than π-acceptors. The peaks close to 1619 cm−1, 1621 cm−1, and 1614 cm−1 in complexes 14–16 are assigned to the coordinated bidentate ligands by comparing them with the IR spectra of the free ligands. The characteristic nitrosyl stretching frequencies are similar to the reported values for the mononuclear metal complex Fe(NO)2(sparteine) [44] and for Fe(NO)2(1-MeIm)2 [38]. However, they are clearly lower than the values 1774 cm−1, 1712 cm−1 for the anionic complex [Fe(NO)2(Im-H)2]− [53], and 1796 cm−1, 1726 cm−1 for the tetranuclear iron complex [Fe(NO)2(Im-H)]4 (Im-H = imidazolate). The results show that the NO stretching frequencies are related to the oxidation levels of dinitrosyl iron complexes and the observed νNOs’ in complexes 14–16 make them {Fe(NO)2}10 according to the Enemark-Feltham notation [54].
These complexes are diamagnetic thus, well-resolved 1H-NMR and 13C-NMR spectra were recorded in DMSO. The NMR results indicate that both complexes 14 and 16 possess high symmetry. Four sets of resonances between 7.54 and 8.66 ppm were observed in the 1H-NMR of complex 14, and five distinct peaks in the aromatic carbon region for the 13C-NMR, which corresponds to the coordinated bipy ligand. Complex 16 exhibits four sets of resonances between 7.74 and 8.86 ppm with two of them very close to each other and the 13C-NMR shows six distinct carbons which correspond to the protons and carbons in the coordinated phen ligands, respectively. On the contrary, complex 15 displays seven sets of resonances between 7.07 and 9.21 ppm, which are attributed to the magnetic inequivalence of the three pyridyl rings in the terpy ligand, meaning this compound bears low symmetry. These observations are consistent with the results found in the crystal structures of complexes 14 and 15. These show that the two NOs are symmetrically located on two sides of Fe(bipy) plane in complex 14, while in complex 15, the two NOs are distributed in two sides of plane consisted of Fe and two coordinated pyridyl rings but almost parallel to the uncoordinated pyridyl ring.
As shown in the X-ray crystal structures (Fig. 7), complex 14 crystallizes in triclinic P-1 space group and the asymmetric unit consists of one Fe(NO)2(bipy) molecule with NO groups in the two sides of Fe(bipy) plane, while complex 15 crystallizes in monoclinic P21/n space group. In both cases, the iron center is connected to four nitrogen atoms, which include two from the nitrosyls and two nitrogen atoms from the ligands, with a pseudo-tetrahedral geometry. In complex 15, the uncoordinated pyridyl is perpendicular to the plane consisted of iron atom and two coordinated pyridyl rings, but almost parallel to the Fe(NO)2 plane.
Fig. 7.

Ball-and-stick representations of the X-ray crystal structures of complexes 14 and 15. Reprinted with permission from R. Wang, X. Wang, E. B. Sundberg, P. Nguyen, G. Paola, G. Grant, C. Sheth, Q. Zhao, S. Herron, K. A. Kantardjieff, and L. Li. 2009. Inorg. Chem. 48, pp 9779–9785. Copyright 2009 American Chemical Society
The average Fe–N(NO) and Fe–N(N,N-L) bond lengths are 1.650 Å and 2.046 Å for complex 14 and 1.648 Å and 2.048 Å for 15, respectively. The average Fe–N(NO) bond distance of 1.649 Å is similar to those reported in other mononuclear complexes but is clearly shorter than those found in the tetranuclear [Fe(NO)2(Im-H)]4 (Im-H = imidazolate) and the dinuclear complexes, [(N2C5H7) Fe(NO)2]2 (N2C5H7 = 3,5-dimethylpyrazolyl). The average Fe–N–O bond angle of 167.9° is thought to be close to linear formation, indicating that the nitrosyl moieties exhibit sp hybridized NO+ character in complex 14, while the N(N,N-L)–Fe1–N(N,N-L) bond angle of 78.4° shows that the Fe(N4) possesses severe distorted tetrahedral environment.
Complexes 14 and 15 also exhibit “attracto” conformations with the Fe–N–O groups symmetrically bent with the average O(NO)–Fe–O(NO) and N(NO)–Fe–N(NO) angles of 104.1° and 114.6°, respectively. In both complexes 14 and 15, Fe–N(NO) bond distances are all clearly shorter than Fe–N(N,N-L) distances, indicating the NO groups are much better π-acceptors than either 2,2′-bipyridine or 2,2′,2′′-terpyridine.
Both complexes 14 and 15 reveal a layering effect on the crystal packing diagrams, in which different layers interact by the π–π stacking and H-bonding effects (Fig. 8). Both molecules of different layers are staggered with the iron nitrosyls in the opposite sides. The uncoordinated pyridyl groups are clearly oriented away from each other in 15. Interestingly, in both complexes, the aromatic rings almost completely overlap, which is expected because the space hindrance is compensated by the H-bonding interactions of nitrogen and oxygen atoms of nitrosyls with the hydrogen atoms from the adjacent bipyridine or pyridyl ligands. The distances between the planes are 3.5880 Å and 3.4254 Å for 14 and 15, respectively. This indicates that there are quite strong π–π stacking interactions between the two bipyridine ligands and the two coordinated pyridyl rings of terpyridine ligands. However, almost no interactions are observed between the adjacent uncoordinated pyridyls.
Fig. 8.

Ball-and-stick representations of the molecular packing in the crystal structures of 14 and 15. Reprinted with permission from R. Wang, X. Wang, E. B. Sundberg, P. Nguyen, G. Paola, G. Grant, C. Sheth, Q. Zhao, S. Herron, K. A. Kantardjieff, and L. Li. 2009. Inorg. Chem. 48, pp 9779–9785. Copyright 2009 American Chemical Society
The electronic absorption spectra of complexes 14–16 are recorded and the data are shown in Table 5 together with cyclic voltammetry (CV) results. The relative low-energy absorption bands at 374–471 nm can be assigned mainly to the MLCT (dFe-π*NO, dFe-π*N,N-L), while the absorptions at higher energy are attributed to combined contributions from LMCT (π*NO-dFe, π*N,N-L-dFe), πNO (πN,N-L)→ dFe, πNO→π*NO, and ligand-localized transitions of the coordinated [N,N] ligands, πN,N-L→π*N,N-L.
Table 5.
List of electrochemical redox potentials and the UV–vis absorptions of complexes 14–16
| Complex | E° 1/2, expt (V) (vs. Fc+/Fc) | Absorptions | ||
|---|---|---|---|---|
| Fe(NO)2(N2C10H8), 14 | −0.48 | −2.07 | 221(s), 245(m), 294(m), 390(w), 471(w) | |
| Fe(NO)2(N3C15H11), 15 | −1.09 | −1.85a | −2.07 | 211(s), 272(m), 304(m), 374(w) 470(w) |
| Fe(NO)2(N2C12H8), 16 | −0.50 | −1.80a | −2.05 | 212(s), 224(s), 271(m), 389(w), 470(w) |
Irreversible redox of a solvated species
As shown in Table 5, complex 14 exhibits two quasi-reversible one-electron reductions at −0.48 V and −2.07 V [versus E1/2° (Cp2Fe/Cp2Fe+)], while complexes 15 and 16 show two quasi-reversible one-electron reductions at −1.09 V, −2.07 V and −0.50 V, −2.05 V, and one irreversible reduction at −1.85 V and −1.80 V, respectively. The first reductions of all complexes are assigned to the [LFe(NO)2]/[LFe(NO)2]− couple, while the reductions close to −2.07 V are believed to be the usual ligand-based reductions. The half-wave potential of the first reduction peak for complex 15 is clearly more negative than the corresponding values for complexes 14 and 16, showing that complex 15 is more difficult to reduce. It is consistent with the greater electron donor effect of the uncoordinated pyridyl ring in complex 15. The results indicate that the electronic property of the ligands has important influence on the electrochemical properties of the relevant complexes.
[(TPA)Fe(NO)2][BF4] and [(PDI)Fe(NO)2][BF4]
Several new structures have been synthesized that result in 5 and 6 coordinate compounds [55]. These structures make use of a single ligand, which contains three or four nitrogen atoms that all coordinate with the metal center. The structures of these ligands are shown in Fig. 9. The first compound was synthesized by 1:1 ratio of tris(2-methylpyridine)amine (TPA) ligand and [Fe(CO)2(NO)2][BF4] in THF at 0°C. This reaction led to a product containing two IR vNO stretching frequencies at 1720 and 1619 cm−1. This cationic six-coordinate [(NO)2Fe(TPA)][BF4] molecule was characterized using several different methods. In a similar fashion as above, a five-coordinate DNIC was also be synthesized by 1:1 ratio of Fe(CO)2(NO)2 with 2,6-bis[1-(2,6-diisopropylphenylimino)ethyl]pyridine (PDI). This reaction generated a cationic five-coordinate [(NO)2Fe(PDI)][BF4] molecule. The IR stretching frequencies (νNO 1794 s, 1721 s cm−1) imply the formation of the five-coordinate DNIC, coordinated by the tridentate PDI ligand. It was noticed that the IR spectra of the six-coordinate, five-coordinate, and four-coordinate DNICs ([(NO)2FeL2] (L=thiolate, imidazolate)) all had the same peak patterns but differed in the position of νNO and the separation of qNO in the NO stretching frequencies (qNO, ~101 cm−1 for six-coordinate vs. ~73 cm−1 for five-coordinate vs. ~60–45 cm−1 for four-coordinate DNICs). Interestingly, several other 4 and 5 coordinate nitrosyl complexes have also been synthesized using cobalt instead of iron [56].
Fig. 9.

Structures for TPA and PDI ligands
While the majority of metal dinitrosyl complexes have ligands containing phosphorus, nitrogen, or sulfur atoms, coordination of other ligands such as oxygen or arsenic has become more common [44, 57]. Recent work is also focusing on compounds that are harder to synthesize, like those involving rare or radioactive metals [58, 59], or the isolation of unstable products with metals that usually do not form nitrosyls such as copper [60] while other molecules are synthesized for their specific abilities, such as hydrogen bonding [61].
3 Nitrosyl Complexes Containing Two or More Metal Centers
3.1 Complexes Containing “M2S2” Core
Sulfur-containing ligands have been known to coordinate with the [M(NO)2] unit for some time. These complexes were discovered as being bound to the cysteine residues of proteins within body tissues. It was also found that proteins containing [Fe–S] clusters, such as rubredoxin and ferredoxin, can react with nitric oxide to form protein-bound DNICs and Roussin’s Red Ester types of complexes [62]. The dinitrosyl iron complexes and RREs were found to be interchangeable when binding to proteins. These molecules were tested for their effects on tumor cell growth and were found to be efficient NO donors that lead to eventual cell death [63].
RREs and RBEs are a class of compounds frequently used as a starting material in the synthesis of novel metal nitrosyl complexes (Fig. 10). Many structures resembling Roussin’s Red and Black Esters have been prepared [64–70]. RREs may be synthesized through the alkylation of Roussin’s Red Salt (RRS) with an alkyl halide or treatment of Fe2(μ-I)2(NO)4 with an organic thiol compound in the presence of a proton acceptor [71]. Many substituted complexes are also generated by reacting DDI with the proper ligand under an inert atmosphere [72, 73] or by reacting a protein containing a biological Rieske center with nitric oxide [74]. A wide variety of structures mimicking Rieske centers have also been reported [73, 75–80]. Ford and others studied the photochemical NO release of RREs and found that the photochemical response and the light harvesting ability improved when the R groups were changed [70, 81, 82]. Here, several structures are discussed in depth to illustrate the chemical and physical characteristics. General synthesis steps for several other compounds are also given in order to display the wide variety of structures discovered in the past decade.
Fig. 10.

Roussin’s Red and Black Salt Structures
[Fe2(μ-RS)2(NO)4]
The Roussin’s Red Salt Esters [Fe2(μ-RS)2(NO)4] (R = n-Pr, 17, t-Bu, 18, 6-methyl-2-pyridyl, 19, and 4,6-dimethyl-2-pyrimidyl, 20), were prepared by mixing Fe(NO)2(CO)2 with equal molar of the corresponding ligands, 1-propanethiol, 2-methyl-2-propanethiol, 4,6-dimethyl-2-mercaptopyrimidine, or 6-methyl-2-mercaptopyridine in CH2Cl2 in the presence of potassium carbonate and was stirred 72 h at ambient temperature under nitrogen atmosphere (Scheme 4) [72]. The reaction solution was filtered to remove undissolved potassium carbonate, and methanol was slowly added to the filtrate, then the mixed solution was kept at −35°C overnight to crystallize. Black crystals suitable for X-ray crystallography were collected by filtration and washed with methanol. The solids were dried under vacuum for several hours and yields ranging from 49% to 69% were obtained [72].
Scheme 4.

A general synthesis method for complexes 17–24
Another method of preparing 18 was also reported, in which Fe(NO)2(CO)2 and sodium 2-methyl-2-propanethiolate were dissolved in methanol and stirred 48 h at room temperature under nitrogen atmosphere. The solution turned from green to dark brown. The reaction solution was filtered and recrystallized from a mixture of CH2Cl2 and methanol.
These complexes are soluble in common organic solvents such as CH2Cl2, THF, and acetone. The infrared spectra of 17–20 were studied in both KBr pellets and in THF solution, as shown in Fig. 11. The characteristic carbonyl stretching frequencies disappeared, indicating that both carbonyl groups were replaced by the sulfur-containing ligands, while the typical IR absorptions of nitrosyl groups (νNO) shift from 1807, 1760 cm−1 to 1805–1823, 1770–1793, and 1743–1759 cm−1, suggesting that these sulfur-containing ligands only act as weak electron donors.
Fig. 11.

Infrared spectra of the nitrosyl stretching region of complexes 18 and 22
In solution, complexes 17–20 display one weak and two strong NO stretching frequencies. However, in the solid state, only two strong NO stretching frequencies were observed. It was proposed that in solution, the cis- and trans-isomers coexist, while in the solid state, only trans-isomers are present. To explain the vibrational modes, frequency calculations using density functional theory (DFT) were carried out for cis- and trans-isomers using complex 17 as a model. The calculated results for the cis-isomer show four different vibrational modes, which correspond to the two symmetric and two anti-symmetric vibration modes, whereas the trans-isomer results in only two vibrational modes derived from two anti-symmetric vibrational modes. The bands at 1775 and 1748 cm−1 are the result of the overlap of those bands derived from symmetric and anti-symmetric vibration modes of the cis- and trans-isomers, but the band at 1810 cm−1 is only derived from one of symmetric vibration modes of cis-isomer. Hence, these complexes actually show only three vibrational bands for the NO moieties in the experimental solution IR spectra, as shown in the theoretical simulation in Fig. 12.
Fig. 12.

Theoretical IR simulation of NO stretching frequencies
Geometry optimizations using density functional theory (DFT) were also performed on the cis- and trans-isomers of complex 17 and the results showed that the energy difference is only about 3 kcal/mol, as shown in Fig. 13. Such a small energy difference of two spatial isomers could easily be supplied by the solvent in solution, which allows both isomers to coexist. Indeed, experimentally, with the increase of spatial hindrance of the R group from complex 17 to 20, the intensity of the weak absorption band at ~1810 cm−1 derived from the cis-isomer gradually decreases in comparison with two strong ones in both THF and CH2Cl2 solution. In solid state, complexes 17–20 only contain the trans-isomer as shown by the X-ray crystal structures; hence, their IR spectra only display two strong NO stretching frequencies (Fig. 14).
Fig. 13.

Geometry optimizations using DFT showing conversion of the cis- and trans-isomers of complex 17
Fig. 14.

The molecular structure of complex 17 with thermal ellipsoids drawn at the 30% probability displaying the chair shape and the trans-isomer found in the solid state
The redox behavior of complexes 17–20 was studied by cyclic voltammetry (CV) in CH2Cl2. All of the complexes exhibited irreversible oxidations, which is consistent with the fact that these complexes are very unstable in air. As shown in Fig. 15, complexes 17 and 18 exhibit two quasi-reversible one-electron reductions at −1.16, −1.84 V and −1.20, −1.81 V, respectively, but complexes 19 and 20 only show one quasi-reversible one-electron reduction at −0.99 and −0.91 V, respectively. All of these reductions can be attributed to iron–sulfur-based redox processes. The half-wave potentials for the first reduction peak clearly turn to more positive values in the order of complex 18, 17, 19, and 20, showing these complexes are easier to reduce along the sequence. This is consistent with the less electron donor effect of the R group in this order. These results indicate that the electronic properties of the R group of the RREs significantly influence the electrochemical properties of the relevant complexes.
Fig. 15.

Cyclic voltammograms of a 2 mM solution of complexes 17–20 in 0.1 M [NBu4][PF6]/CH2Cl2
The molecular structures of complexes 17–20 were determined by X-ray diffraction analysis, and it showed that all these complexes possess a “chair-shape” structure with the two R groups are almost parallel to each other along opposite direction and form an angle of ~110° with the 2Fe–2S plane as shown in Fig. 14. The Fe(1)–Fe(1a) distance of 2.70 Å suggests that there is fairly strong interaction between the two iron centers. The Fe–Fe distance and the average Fe–S bond length for complex 20 are 2.741 and 2.278 Å, respectively, which are longer than the corresponding values (2.698, 2.708, 2.708 and 2.257, 2.257, 2.270 Å) for complexes 17–19, indicating that complex 20 is essentially more unstable. The observations are consistent with the electrochemical studies where complex 20 possesses the most positive reduction potential amongst the four complexes.
The average Fe–N(NO) bond distance for complex 17–20 is ~1.670 Å, which is slightly shorter than the reported value in the complex Fe(NO)2[(SC6H4-o-NHC(O) CH3)2]− (average value: 1.681 Å), the dinuclear complex [(N2C5H7)Fe(NO)2]2 (N2C5H7 = 3,5-dimethylpyrazolyl) (average value: 1.696 Å), and the tetramer [Fe(NO)2(Im-H)]4 (Im-H = imidazolate) (average value: 1.694 Å), but significantly longer than the found value in the dinuclear complex Fe2(μ-L)2(NO)4 (L = Ph2PCH2PPh2 and Ph2PC≡CPPh2) (average value: 1.644 and 1.656 Å).
[Fe2(μ-RS)2(NO)4]− anionic form
The corresponding family of reduced species, [Fe2(μ-RS)2(NO)4]−, was prepared by the reaction of neutral [Fe2(μ-RS)2(NO)4] with a slight excess of cobaltocene or Li(BHEt3) in THF. The dark brown solution turned to dark green after the reduction. Complex 20 slowly decomposes during the course of the reduction reaction.
IR spectra of the monoanionic complexes [Fe2(μ-RS)2(NO)4]− (21–24) were collected and exhibit the characteristic νNO stretching frequencies at 1673, 1655 (21); 1670, 1650 (22); 1690, 1670 (23) and 1693, 1674 cm−1 (24) in THF. The corresponding νNO bands are shifted by 100 cm−1 to a lower energy in comparison with neutral species due to their negative charge. Roussin’s Red Salt Esters are diamagnetic and EPR-silent. The reduced species, [Fe2(μ-RS)2(NO)4]−, are EPR active and exhibit an isotropic signal at g = 1.998–2.004 without hyperfine splitting in the temperature range from 180 K to 298 K. This is quite different from the typical DNICs, where the g-values are close to 2.03 and the hyperfine structures rising from the coupling between the unpaired electron and the nitrogen of the NO.
In order to interpret the difference, optimizations were first performed on neutral and negatively charged complexes with the spin-unrestricted method. The optimized structures were all obtained at minimum energy as proved by no negative frequencies evident in frequency calculations. The spin density distributions of the singly occupied molecular orbit (SOMO) for the complexes 21–24 were obtained by single point calculations on the four optimized structures of the anionic complexes by including all electrons (Fig. 16). The results show that there are 60–63% of the electron delocalized on two irons, 25.0–25.8% of the electron delocalized on two sulfurs, and only 2–6% of the electron delocalized on four NOs. Since most of the unpaired electron is delocalized over the Fe and S atoms and the most natural abundance of isotopes of these are 56Fe and 32S, whose nuclear spins (I) are zero, the lack of hyperfine splitting in the EPR spectra of these complexes is expected.
Fig. 16.

Spin density distribution of the SOMO for the complexes 21–24 and the calculated composition (in percent) of the SOMO in terms of Fe, S, and NO fragments
In order to understand the differences between the g-values for 21–24 (~2.000) and the typical DNICs (2.03), the distribution of electron density on the SOMO of complex [Fe(NO)2(CO)2]+ was also calculated by using DFT as shown in Fig. 17.
Fig. 17.

Comparison of the distribution of electron density on the SOMO of complex [Fe(NO)2(CO)2]+ and RRE calculated by DFT
The results showed that Fe and NO moiety possessed 54.4% and 41.8% of the electron density of the SOMO, respectively. It should be pointed out that the calculated distribution of electrons on the iron in DNICs (54%) is lower than the values obtained by 57Fe-enriched EPR experiments on other g = 2.03 species [83]. However, it is well known that DFT calculations often produce an over-delocalized distribution. Nonetheless, for complexes 21–24, the amount of electron density on each iron is in the range of 30–32%, and the total percentage of the electrons on the NO moieties is only 2–6%. Therefore, the difference between the EPR g-values for 21–24 (~2.000) and the typical DNICs (2.03), and the lack of hyperfine couplings from the NO group in complexes 21–24, can be well explained by the percentage of the electrons on the metal and NO moiety.
In order to validate the above-mentioned results, additional calculations were carried out to obtain the g-values by using complex 22 as an example. The calculated isotropic g-value is 1.995, which is very close to the experimental data of 1.999. The calculated anisotropic parameters, g⊥ = 2.014 and g|| = 1.958 are in agreement with the experimental data g⊥ = 2.009 and g|| = 1.965, for this complex. These theoretical results explain clearly why hyperfine structures were not observed and corroborate that the electron density of the SOMO is mostly delocalized on the Fe and S atoms. In fact, the single point calculations show that the extra electron goes mainly on the Fe atoms, which further explains why adding electrons causes weakening the bond between two irons. The molecular orbital characters of the SOMOs shown in Fig. 17 are clearly anti-bonding: thus adding electrons into the SOMO actually increases electron–electron repulsion between iron atoms and contributes to the weakening of the Fe–Fe bond. The composition of the SOMO mainly comes from the d orbital of the metal but also has some 3p character, although in a very small amount for all four complexes, while the sulfur contribution is solely from its p orbital.
[PPN]2[Fe2(μ-StBu)2(NO)2]
Several other RRE-like structures have been synthesized. One of these was formed by dissolving a precursor complex containing an iron atom bound to several chelating oxygen atoms in an appropriate amount of CH3CN solvent. To this mixture, 2 molar equivalents of [StBu]− was added [55] and the product was easily isolated and is surprisingly stable. This product was characterized by IR, UV–vis, and other methods. The FT-IR νNO bands of this product [1637 m, 1613 s, 1578 s, 1567 s cm−1 (KBr)] shifted by ~30 cm−1 from those of the precursor molecule, indicating a complete reaction. The UV–vis spectrum of the product displays absorptions 270 and 396 nm (CH3CN). This is in direct contrast to that of the reduced RRE [(NO)2Fe(μ-StBu)]2− which displays an intense transition absorption around 982 nm. It is thought that the conversion of the precursor molecule into the product is due to the capability of the thiolate ligand to bridge two {Fe(NO)2}10 fragments. This is also attributed to the preference of {Fe(NO)2}10 motifs which are connected by the electron-donating thiolate group.
Another branch of nitrosyl complex exists containing sulfur ligands that do not resemble Roussin’s Red or Black Esters. These complexes frequently seek to mimic amino acids such as cysteine [84]. Several different structures have been discussed pertaining to the dinuclear {Fe(NO)2}10–{Fe(NO)2}10 and mononuclear {Fe(NO)2}10 constructs. These complexes rapidly convert between the DNIC and RRE forms [85] or into the RBE forms [86, 87]. Recently, Lippard’s group reported reactions of NO with synthetic [2Fe–2S] and [4Fe–4S], yielded the DNIC, [Fe(NO)2(SR)2]−, (R = Ph, p-tolyl, 4-MeC6H4) [88]. Other work focuses on the effects that thiolate ligands play on the conversion between DNICs and RREs [89] or the means by which nitric oxide is delivered [90]. Reaction pathways of iron nitrosyls in sulfur-rich biological coordination environments are also a well-studied area [91, 92].
[Fe2(μ-SCys)2(NO)4]
It has been proven that DNICs as well as other metal nitrosyl complexes can form upon reacting with biological Rieske centers. In order to determine this, the Rieske protein, toluene/o-xylene monooxygenase component C (ToMOC) from Pseudomonas was exposed to several different components [74]. The Rieske center of this protein was slowly exposed to Na2S2O4. As the reaction took place, three new maxima characteristic of reduced ToMOC began appear at 380, 420, and 520 nm and steadily increased. Once Na2S2O4 was added to the protein, the spectra ceased to change. This lack of further changes indicated a complete reduction of the iron–sulfur cluster. The optical features of ToMOCox and ToMOCred were used as standards to determine whether the various redox states of the Rieske cluster will undergo reaction with NO(g) and NO donors such as DEANO and SNAP under anaerobic conditions. The charge-transfer bands of ToMOCox were bleached which is consistent with cluster disassembly, upon addition of a large excess (~20 equivalents) of DEANO or SNAP under anaerobic conditions. When the reaction was complete, a new optical feature indicative of an iron dinitrosyl species was evident at 367 nm. This is indicative of the ability of DNICs and/or other nitrosyl complexes to form at Rieske centers of certain proteins.
[Fe2(μ-SEt)2(NO)4]2+(cationic form)
A separate body of work has studied the effects of sulfur ligands on the ability of an Fe–S cluster to convert into DNICs [89]. This work began with the synthesis of DNIC cations, which is a reactive species that can undergo further transformations under proper condition. A precursor complex, [Fe(μ-SEt)(NO)2]2 coordinated with the anion of choice, was dissolved in an appropriate amount of THF and stirred at ambient temperature for approximately 20 min. During this time, a reduction reaction occurred to yield complexes of [Fe(μ-SEt)(NO)2]2+ (cation) coordinated with PPN+, Na+-18-crown-6-ether or, Me4N+. These complexes were identified by IR, UV–vis, EPR, and single-crystal X-ray diffraction. The complex coordinated with the PPN ligand exhibits diagnostic IR νNO stretching frequencies at 1673 s, 1655 s cm−1 (THF). In contrast to Roussin’s red esters with {Fe(NO)2}9–{Fe(NO)2}9 coupling, rationalizing the absence of paramagnetism and the EPR signal, the anionic Roussin’s red ester using PPN is best described as a fully delocalized [{Fe(NO)2}9–{Fe(NO)2}10] complex.
[Fe2(SC6H4-o-NHC(O)Ph)2(NO)4]
Another metal nitrosyl complex that falls into this category is a mixed ligand structure. In this case, the DNIC contains sulfur, nitrogen, and oxygen coordinated ligands. Several different complexes were obtained by varying the ligand structure slightly [93]. In order to synthesize this particular compound freshly prepared Fe(CO)2(NO)2 liquid was dissolved in degassed THF solvent. Separately bis(o-benzamidophenyl) disulfide was dissolved in THF solvent. The second solution was added to the first by cannula under a positive N2 pressure at 0°C. The resulting solution was allowed to stir at ambient temperature overnight. The solution was filtered through Celite to remove solid contaminants. An appropriate amount of hexane was then added to the filtrate to precipitate the brown-green solid [Fe(SC6H4-o-NHC(O)Ph)(NO)2]2 (Yield 90%). This product was characterized by 1H NMR, IR, and UV–vis spectra.
[(ON)Ni(S(CH2)2S(CH2)2S)Fe(NO)2]
This interesting compound makes use of two different metals to form the nitrosyl complex [94]. It was prepared by mixing the starting materials: [PPN][Fe(NO)2(SePh)2], NaNO2, and [Ni(S(CH2)2S(CH2)2S)] into a Schlenk flask and CH2Cl2 was added and stirred at 50°C overnight. A dark green solid was isolated by recrystallization from CH2Cl2 and hexane. IR (νNO): 1805 m, 1767 s, 1725 s cm−1 (CH2Cl2); 1798, 1763 s, 1723 s cm−1 (THF).
(μ-depdt)[Fe(NO)2PMe3]2PF6
Here, a rather complicated, mixed-valent molecule is shown with two iron centers and several different ligand groups [95]. This species was synthesized through addition of a cold (−78°C) CH2Cl2 solution of a precursor molecule to a dry ice cooled solution of Fe[PF6]2. This resulted in an immediate color change from red to purple red. The reaction was stirred for 10 min and then warmed to −42°C. Addition of precooled hexane formed purple/red precipitates. X-ray quality crystals were obtained by mixture of CH2Cl2 with pentane/diethyl ether solution. IR (cm−1, in CH2Cl2): νCO 2041 (s), 2005 (s) 1993 (sh), 1874 (w).
[Fe2(C14H12N3S)2(NO)4]
A dinuclear iron nitrosyl complex was also prepared [Fe2(C14H12N3S)2(NO)4], 25, (C14H12N3S = 2-mercapto-1-[2-(4-pyridyl)-ethyl]-benzimidazolyl) by stirring Fe(NO)2(CO)2 with 2-mercapto-1-[2-(4-pyridyl)-ethyl]-benzimidazole in 1:1 ratio in CH3OH (30 ml) at ambient temperature under nitrogen atmosphere for 96 h [66]. The reaction solution was filtered to remove any dissolved impurities. The residue was redissolved in CH2Cl2 and methanol was slowly added to the solution. The solution was then left at −35°C in a glove box overnight to crystallize. Black crystals suitable for X-ray crystallography were collected by filtration, washed with methanol, and dried under vacuum for several hours. Yield: 40%.
The complex shown in Fig. 18 is soluble in common organic solvents such as CH2Cl2, THF, and acetone and is characterized by IR, UV–vis, electrochemistry, and single-crystal X-ray diffraction. IR spectrum displays two strong characteristic NO stretching frequencies (νNO) in solution and in solid state. Cyclic voltammetry shows one irreversible, two quasi-reversible, and two reversible one-electron reductions and irreversible oxidizations. This result is consistent with the fact that complex 25 is very unstable and ready to lose NO in the air. The single-crystal X-ray diffraction of complex 25 shows a “chair-shape” structure by the connections of two iron centers and S–C–N frames of benzimidazole. The dihedral angle of benzimidazole ring and 2Fe–2S plane is 73.6° (Fig. 18).
Fig. 18.

A molecular drawing and X-ray crystal structure of complex 25
3.2 Iron–Sulfur and Iron–Selenium Nitrosyl Clusters
As the only structurally authenticated example iron–sulfur nitrosyl cluster of [Fe4S3] was foremost found in the Roussin’s Black Salt anion [Fe4S3(NO)7]−, several iron–sulfur nitrosyl clusters, such as [Fe4S3(NO)7] n−, (n = 1–3), [Fe4S4(NO)4]0, 1− and [Fe6S6(NO)6]2− were also obtained [96–99]. However, some clusters such as [Fe6S6(NO)6]2− were obtained only by complicated multistep procedures [98]. Subsequently, a new [Fe8S6(NO)8]2− cluster [100] was synthesized with improved one-step synthetic method, in which [Fe4S3(NO)7]− was used as one of the starting materials. Yet, despite many known examples of iron–sulfur nitrosyl clusters, iron–selenium nitrosyl clusters are extremely rare. Prior to our recent work on [(n-Bu)4N]2[Fe6Se6(NO)6], which will be described in more detail below, only one iron–selenium nitrosyl cluster, (Ph4As)[Fe4Se3(NO)7], has been reported, with no characterization other than its structure [101].
[(n-Bu)4N]2[Fe6Se6(NO)6]
Recently, a new complex [(n-Bu)4N]2[Fe6Se6(NO)6], 26, was synthesized by mixing [(n-Bu)4N][Fe(CO)3NO], selenium, and methanol in a vial under a nitrogen atmosphere [102]. The vessel was then sealed and heated at 85°C for 48 h in an autoclave, which was subsequently allowed to cool to room temperature. After the reaction solution was filtered and washed using methanol, black solids were obtained, which was then redissolved in acetonitrile, and diethyl ether was slowly added to the solution. The mixed solution was placed in a glove box at −35°C overnight and yielded black crystals, which were collected by filtration, washed with methanol, and dried under vacuum for several hours. Yield: 52 mg (85%, based on [(n-Bu)4N][Fe(CO)3NO]). This complex is soluble in most polar organic solvents including acetonitrile, dichloromethane, and THF but is insoluble in methanol, ethyl ether, and hexane.
The IR spectrum of 26 displays one strong characteristic NO stretching frequency at 1694 cm−1 (νNO) in solution with the characteristic of NO+. The X-ray crystallographic study shows two parallel “chair-shape” structures, consisting of three iron and three selenium atoms, and are connected by Fe–Se (Fig. 19). The Fe–Se bonds have an average distance of 2.341 Å, and each iron center is bonded to three selenium atoms and a nitrogen atom from nitrosyl ligand with pseudo-tetrahedral center geometry. The average Fe–Fe distance of 2.730 Å suggests that there is a fairly strong interaction between the two iron centers. The Fe–N bond distances range from 1.661 to 1.665 Å with an average of 1.663 Å. The N–O bond lengths range from 1.172 to 1.186 Å with an average of 1.180 Å. The Fe–N–O bond angles range from 174.5° to 178.9° with an average of 176.5°, which is close to linear. These nitrosyl moieties exhibit sp hybridized NO+ character, which indicates a considerable amount of charge transfer between the NO and the metal took place.
Fig. 19.

The X-ray crystal structure of complex 26 with thermal ellipsoids drawn at 50% probability. Reprinted with permission from R. Wang, W. Xu, J. Zhang, and L. Li. Inorg. 2010. Chem. 49, pp 4814–4819. Copyright 2010 American Chemical Society
The electrochemistry of this complex was studied by cyclic voltammetry and showed two cathodic current peaks at Epc = −0.42 and −1.36 V and three anodic peaks at Epa = −0.04, −0.38 and −1.30 V. The peak at Epc = −0.42 V is unusually strong. Detailed electrochemical studies indicate that it consists of a minimum of three processes as shown in Fig. 20. One is the quasi-reversible reduction at E1/2° = −0.41 V and the other two are from an irreversible electrochemical process that occurred at Epc = −0.42 V, in which the compound went through a typical electron transfer and chemical reaction (ECE) mechanism of which its product is easier to reduce than the original one, resulting in an overlap of the reduction potentials and subsequently, a very strong peak (Table 6). The peak at Epa = −0.04 V is the product from such a chemical reaction. The electronic absorption spectra of the complex show bands in the range of 259–562 nm (Fig. 21), which are assigned to the transitions between orbitals delocalized over the Fe–S cluster, the ligand to metal charge transfer (LMCT), π*NO-dFe, and the metal to ligand charge transfer (MLCT), dFe-π*NO.
Fig. 20.

Cyclic voltammograms of a 1 mM solution of compound 26 in 0.1 M (NBu4)(PF6)/CH3CN at scan rate of 100 mV/S. Reprinted with permission from R. Wang, W. Xu, J. Zhang, and L. Li. Inorg. 2010. Chem. 49, pp 4814–4819. Copyright 2010 American Chemical Society
Table 6.
List of redox potentials and IR frequencies for complexes 26–28
| Redox potentials vs. Fc+/Fc (V)
|
IR frequencies
|
||||
|---|---|---|---|---|---|
| Compounds | Epa | E°1/2 | (νNO) (cm−1) | ||
| [(n-Bu)4N]2[Fe6Se6(NO)6], 26 | −0.04 | −0.41 | −1.33 | 1694 | |
| [(n-Bu)4N]2[Fe6S6(NO)6], 27 | 0.07 | −0.33 | −1.32 | 1698 | |
| (Me4N)[Fe4S3(NO)7], 28 | −1.09 | −1.71 | −2.21 | 1799, 1744, 1710 | |
Fig. 21.

The UV–vis spectra of complexes 26 (solid line), 27 (dashed line) and 28 (dotted line). Reprinted with permission from R. Wang, W. Xu, J. Zhang, and L. Li. Inorg. 2010. Chem. 49, pp 4814–4819. Copyright 2010 American Chemical Society
[(n-Bu)4N]2[Fe6S6(NO)6]
A hexanuclear iron–sulfur nitrosyl cluster, [(n-Bu)4N]2[Fe6S6(NO)6], 27, was synthesized using similar solvent-thermal reactions described in 26, at 120°C using sulfur (32 mg, 1 mmol) and the product was obtained in high yield of 92% based on [(n-Bu)4N][Fe(CO)3NO] [102]. Complex 27 is also soluble in most polar organic solvents including acetonitrile, dichloromethane, and THF. However, it is insoluble in methanol, ethyl ether, and hexanes. The IR spectrum of complexes 27 is similar to 26 and displays one strong characteristic NO stretching frequency at 1698 cm−1 (νNO) in solution with the characteristic of NO+.
The X-ray crystal structure of complex 27 was determined and it was found to be consistent with the reported one [99]. The average Fe–Fe distance is 2.644 Å, which is shorter than that of 26 (2.730 Å), which is understandable as the radius of selenium atom is larger than that of sulfur. The Fe–N bond distances for compound 27 range from 1.659 to 1.672 Å with an average of 1.667 Å, which is similar to 26. Accordingly, the N–O bond lengths in compound 27 range from 1.168 to 1.197 Å with an average of 1.182 Å, which is also similar to compound 26. The average Fe–N–O bond angle of 174.4° is similar to the average value of 176.5° found in complex 26, 176.9° in [Fe8S6(NO)8]2−, 177.6° in [Fe4S4(NO)4], 177.5° in [Fe4S4(NO)4]−, and the Fe–N–O bond angles of 177.6° arising from the apical Fe(NO) in [Fe4S3(NO)7]2−.
The redox behavior of compound 27 was also studied by CV and showed two cathodic current peaks at Epc = −0.30 and −1.29 V and three anodic peaks at Epa = 0.08, −0.23, and −1.19 V at scan rate of 100 mV/s (Table 6). It is similar to compound 26 in that the first reduction peak is much stronger than the second one. Various scan rates from 0.1 to 1.0 V/s were recorded. When faster scan rates were applied, the first reduction peak was separated to two reductions, and the faster the scan rate, the clearer the separation between the two reduction peaks. In addition, the peaks correspond to the ECE process did not disappear even at the scan rate of 1 V/s, which indicates that the chemical step is quite fast.
The electronic absorption spectra of complexes 27 show bands in the range of 259–562 nm (Fig. 21), which are assigned to the transitions between orbitals delocalized over the Fe–S cluster, the ligand to metal charge transfer (LMCT), π*NO-dFe, and the metal to ligand charge transfer (MLCT), dFe-π*NO.
(Me4N)[Fe4S3(NO)7]
A tetranuclear cluster, (Me4N)[Fe4S3(NO)7], 28, was also prepared through a solvent-thermal reaction by mixing FeCl2·4H2O, thiourea, (CH3)4NCl, NaNO2 in methanol in a vial under nitrogen atmosphere. The vessel was sealed and heated at 85°C for 48 h [102]. The autoclave was then allowed to cool to room temperature. The solution was filtered and washed using methanol and the solid mixture was dissolved in the acetonitrile and filtered to remove the undissolved white solid. Subsequently, diethyl ether was slowly added to the solution, and the mixed solution was placed in a glove box at −35°C overnight to crystallize. The black crystals were collected by filtration, washed with methanol, and dried under vacuum for several hours. Yield: 88%, based on FeCl2·4H2O. Complex 28 is more or less soluble in all organic solvents and is fairly stable in the solid state and in solution under air. IR spectrum of complex 28 shows three stretching frequencies at 1799, 1744, and 1710 cm−1. The UV–vis spectrum for 28 was also recorded and is shown in Fig. 21.
X-ray crystal structure of complex 28 was determined. The average Fe–Fe distance is 2.705 Å, which is similar to [Fe6S6(NO)6]2− (2.644 Å), [Fe6Se6(NO)6]2− (2.730 Å), but is clearly shorter than the relevant value of 2.764 Å for dianion [Fe4S3(NO)7]2−. This difference was explained by the HOMO of [Fe4S3(NO)7]2−, which contains an unpaired electron, and has anti-bonding character involving all pairs of iron atoms of the Fe4S3 core. This leads to the increase of Fe–Fe bond lengths. When comparing [Fe4S3(NO)7]2− and [Fe4S3(NO)7]− (28), the Fe–N interactions are evidently strengthened in the dianion (average value: 1.646 Å vs. 1.671 Å in the monoanion) owing to more back-donation from dFe to π*NO. On the other hand, the average N–O bond lengths are 1.176 Å for [Fe4S3(NO)7]2− and 1.166 Å for 28 – an opposite trend. These observations are consistent with the results of IR spectra, which display that the absorptions of nitrosyl groups (νNO) appear at higher frequencies for complex 28.
The Fe–N–O bond angles is 178.3°, which is clearly larger than the average Fe–N–O bond angles of 167.9° and 166.6° arising from the three sets of Fe(NO)2 of [Fe4S3(NO)7]2− and complex 27, respectively. When comparing complex 28 with other Roussin’s Black Salt, no differences could be attributed to effects of the counter ion besides the packing effects. These results show that the Fe–N–O bond angles of iron–sulfur (selenium) clusters are irrelevant to their dimension and charge, but relevant to the number of nitrosyls attached to the iron atoms and the localized symmetry of the iron atoms. This also means that the variance of NO+ (linear, sp hybridized) and NO− (bent, sp2 hybridized) may be brought out because of the greater deviations of the Fe–N–O bond angles from 180° in the iron dinitrosyl units for complex 28 and [Fe4S3(NO)7]2−.
Compound 28 has three quasi-reversible reductions with half-wave potentials of −1.09, −1.71, and −2.21 V (Table 6). The electronic absorption spectra of complexes 28 show bands in the range of 259–562 nm (Fig. 21), which are assigned to the transitions between orbitals delocalized over the Fe–S cluster, the ligand to metal charge transfer (LMCT), π*NO-dFe, and the metal to ligand charge transfer (MLCT), dFe-π*NO.
3.3 Multinuclear Metal Nitrosyl Complexes
In addition to the iron–sulfur or iron–selenium nitrosyl clusters, a few multinuclear metal nitrosyl clusters have been synthesized. Here a well-documented structure is examined and its physical and chemical characteristics are explained in thorough detail.
Fe4(NO)8(Im-H)4
One example of a tetra-nuclear DNIC was synthesized when one equivalent of Fe(NO)2(CO)2 was reacted with two equivalents of imidazole, Im, in methylene chloride at room temperature in a glove box under nitrogen atmosphere [103]. After stirring for 24 h, a dark reddish brown precipitate was obtained and collected by filtration with 50% yield. Single crystals suitable for X-ray diffraction were isolated by recrystallization of 29 from acetone at −38°C under nitrogen atmosphere.
The structure shows four iron centers that are linked together through four deprotonated imidazole bridging ligands forming a neutral 16-membered rhombic macrocycle with alternating imidazolates and irons as shown in Fig. 22. Each iron center possesses a pseudo-tetrahedral geometry and is coordinated with four nitrogen atoms, two from the nitrosyl ligands and two from the imidazolate ligands (Im-H). The molecule has dimensions of 8.18 × 8.70 Å (Fe1· · ·Fe1 × Fe2· · ·Fe2). A solvent molecule, acetone, is crystallized inside the cavity. Upon detailed examination of the crystal structure, it is found that the two C–N bond lengths are approximately equal to the mean value of 1.336 Å, while the crystal structure of imidazole free ligand shows two different C–N bond lengths of 1.349 Å and 1.326 Å. This is due to the deprotonation of the bridging ligand, which created a six-electron aromatic ring with expected delocalization. The deprotonation also created a negative charge on the imidazolate ligand, which has to be balanced by one positive charge on each iron center, since the whole molecule is neutral. This is reflected by both the nitrosyl IR stretching frequencies of the complex, the relatively longer Fe–NO bond distances, and shorter N–O bond distances.
Fig. 22.

X-ray crystal structure of complex 29 with anisotropic thermal displacement ellipsoids drawn at 50%. An acetone molecule is crystallized inside of the cavity
The average Fe–N (Im-H) bond distances is 2.005 Å, which is shorter than the values reported for Fe(NO)2(1-MeIm)2 (average value of 2.046 Å). The average N–O bond length is 1.166 Å, which is also shorter than the N–O distance of 1.189 Å in Fe(NO)2(1-MeIm)2. This is because the positive iron atom in complex 29 donates less electron density to the π* orbitals of the nitrosyl ligands than the neutral iron of Fe(NO)2(1-MeIm)2. This is also reflected in the average Fe–N (NO) bonds length (1.694 Å), which is longer than those in Fe(NO)2(1-MeIm)2 due to the less extensive π back-bonding.
The νNO values for Fe(NO)2(CO)(Im), 30, (1731 cm−1 and 1687 cm−1) appear at lower frequencies than those of Fe(CO)2(NO)2, consistent with electron-donating property of the imidazole ligand. Since both carbonyls have been replaced by a donator ligand on each metal, one would expect the nitrosyl stretching frequencies of complex 29 to shift to even lower wavenumbers. However, the nitrosyl stretching frequencies νNO occur at 1796 cm−1 and 1726 cm−1 for 29, which is even higher than that of 30, suggesting that the oxidation of the Fe(NO)2 units took place to balance deprotonation of the bridging ligands. The observed νNO in 29 makes it {Fe(NO)2}9, according to the Enemark-Feltham notation.
EPR studies of complex 29 in the solid-state at room temperature showed a very broad peak at g = 2.023 with a peak-to-peak width of 250 G, indicating that the metal centers are weakly coupled to each other. In THF solution, a signal at g = 2.031 with ΔHpp = 13.2 G was observed at room temperature and it becomes a well-resolved 9-line spectrum at 170 K as shown in Fig. 23. The computer simulation of the low temperature spectrum indicates that the hyperfine structure is a result of the coupling of two equivalent 14N nuclei from the nitrosyls and one 14N from the imidazolate, with hyperfine coupling constants of 2.54 G and 4.50 G, respectively. From this observation, one concludes that the tetrameric molecule has fragmented and most likely solvated to give the seventeen electron species Fe (NO)2(Im-H)(THF) or its protonated analog [Fe(NO)2(Im-H)(THF)]+ as shown in Scheme 5. The presence of the solvated species, [Fe(NO)2(Im-H)(THF)], was further investigated using FT-IR spectroscopy and the results showed that the more polar the solvent, the more solvated products were observed. The characteristic g-value close to 2.03 and the small hyperfine coupling constants (2–3 G) indicate that the unpaired electrons are localized on the Fe center.
Fig. 23.

Comparison of the experimental and simulated EPR spectra of the 17-electron solvated species derived from complex 29
Scheme 5.

Preparation of compounds 29 and 30 and the formation of the 17-electron solvated species in polar solvents
Nitric oxide release from complex 29 was also studied by photolytic and thermo methods. It showed four steps of weight losses at different temperature ranges determined by TGA attributed to stepwise loss of imidazole and NO and a slow decomposition under photolytic conditions. Other tetranuclear compounds that are very similar to 29 have also been isolated [104, 105].
4 Mononitrosyl and Trinitrosyl Complexes
4.1 Mononitrosyl Complexes
Majority of the mononitrosyl complexes are existed as pentacoordinated. Earlier work pioneered by Mingos [106–108] laid out the foundations for these complexes and a few representative work will be discussed in more detail in the following. Nowadays, many mononitrosyl complexes are often synthesized with a purpose of comparing their structures and properties with the products of nitric oxide transfers in order to confirm a successful reaction. For instance, using sterically hindered β-Diketiminate ligand, a new complex [Fe(NO)(Ar-nacnac)], Ar-nacnac = anion of [(2,6-diisopropylphenyl)NC(Me)]2CH, was isolated and studied by Lippard recently [109]. Some mononitrosyl complexes are stabilized by highly coordinated metal centers [110], while others contain metal centers besides iron, such as (OEP)Ru(NO)(p-C6H4F) (where OEP = octaethylporphyrinato dianion) [111], [Mn(PaPy3)(NO)]ClO4 [112], and [NBu4][cis-RuCl4(4-pyha)NO] (4-pyha=4-pyridinehydroxamic acid) [113] giving a wide variety of mononitrosyl complexes. Some of these structures have demonstrated the ability to release nitric oxide under visible light, making them possibly useful candidates for nitric oxide transfer reactions [114–117]. Other structures make use of porphyrins as a bulky ligand in order to stabilize the nitric oxide group, and an axially bound NO on cobalt porphyrins with the formula (T(p/m-X)PP)Co(NO) (p/m-X = p-OCH3, p-CH3, m-CH3, p-H, m-OCH3, p-OCF3, p-CF3, p-CN) [118, 119], and with a ruthenium metal [120, 121]. Photoliability studies on mixed-type structures with both S and N ligands were also examined [122, 123]. X-ray absorption spectroscopic study of nitric oxide binding to iron in active sites was also reported [124]. Here, several different compounds are summarized along with some relevant spectroscopic data.
[Ir(NO)(PPh3)2L1L2] and [IrH(NO)(PPh3)3][ClO4]
The structures of the complexes with formula [Ir(NO)(PPh3)2L1L2], (L1 = I, L2 = Me and L1 = L2 = Cl) have been determined [106, 107] and showed that they form a square–pyramidal geometries with the nitrogen atom of the nitrosyl group occupying the apical position and the other ligands lying in the basal plane. The nitrosyl ligand is coordinated with iridium in a nonlinear fashion, with an iridium–nitrogen–oxygen bond angle between 120 and 123°, which is described formally as an NO− to a transition metal. To the contrary, the structure [IrH(NO)(PPh3)3][ClO4] is described as distorted trigonal bipyramidal with the hydrido and nitrosyl ligands occupying the axial positions and the triphenylphosphine ligands the equatorial positions [108]. The nitrosyl ligand is linear (Ir–N–O bond angle of 175°) and best described as an NO+. A general bonding model for linear and bent transition metal nitrosyl complexes was also reported and it was concluded that nitric oxide, unlike carbon monoxide and dinitrogen, forms linear and bent complexes primarily because it has a low-lying π* orbital [125]. Further investigations on molecular orbital model of pentacoordinate nitrosyls indicate that the σ- or π-donating capability of the basal ligands in a square pyramid affects the degree of bending of the nitrosyl ligand [126].
Mingo also pioneered the use of 15N NMR to differentiate linear and bent nitrosyls in transition metal complexes, including nitro and nitrito ligands in 4-, 5-, and 6-coordinate transition metal complexes [127, 128]. Nitrogen-15 solid-state NMR of [RuCl(NO)2(PPh3)2](BF4) using high-resolution cross-polarization magic-angle spinning (CP/MAS) also showed a large chemical shift anisotropy for the bent as compared with the linear nitrosyl ligand [129].
[TcCl(NO)(DPPE)2]
Several other noteworthy structures include a novel technetium complex with phosphine ligands [130]. To synthesize this particular compound, a sample of (NH4)[TcO4] was evaporated to dryness and added to a fivefold excess amount hydroxylamine hydrochloride in dry methanol. The reaction mixture was heated to the reflux temperature and allowed to react for approximately 45 min. This reaction yielded a red solution. Six equivalents of diphenylphosphinoethane (DPPE) were added to the refluxed solution. This mixture was then heated and refluxed for an additional 45 min. One equivalent of ammonium hexafluorophosphate was then added to the reaction mixture. The solvent was evaporated from the reaction mixture yielding a red oily liquid. This product was dissolved in methylene chloride and then layered with methanol and diethyl ether. After being allowed to sit at room temperature for approximately 24 h, the solution yielded red-brown crystals. Yield: 373 mg (93%). Analytical results IR (KBr): m (Tc=N), 1098 cm−1, m (N=O), 1723 cm−1.
[Ni(NO)(nP3)]X
Several different compounds of nickel have been synthesized that also make use of phosphine ligands. The basic formula of these compounds is [Ni(NO)(nP3)]+ where nP3 is tris(2-diphenylphosphinoethyl)amine and X is the coordinating anion. Four structures are given that utilize four different anions [22].
X = BF4
A solution was made up of np3 ligand dissolved in acetone. A mixture containing [NiBF4]·6H2O dissolved in ethanol was added to the previous solution. The resulting mixture was concentrated by evaporation. Crystals were obtained from this solution. The formula of these crystals was [NiHx (nP3)]BF4 where (x = 0.06–0.26). In order to form the final product, the purified crystals were dissolved in dichloromethane. Nitrogen oxide was bubbled through the solution for approximately 20 min until the color changed to a deep violet purple (this solution is further reacted with later mentioned reagents to form new products). The solution was diluted with ethanol and then concentrated again. This yielded dark black–violet crystals with the formula [Ni(NO)(nP3)]BF4.
X = BPh
Another member of this family of compounds makes use of a different coordinated ligand. This product was formed by dissolving NaBPh4 in ethanol to the earlier mentioned violet solution. The solution was concentrated and recrystallized from a THF-ethanol mixture.
X = I
This alternate product was prepared using the same procedure as the tetrafluoroborate derivative. The starting reagents used were NiI dissolved in dichloromethane. This mixture was added to the solution containing nP3 ligand in acetone and reacted in the same manner.
X = NO3
In order to synthesize this compound, a suspension of Ni(NO3) was dissolved in THF. The solution was stirred and NO was bubbled through it. The solution turned black–violet and was concentrated to yield the solid product.
[(N-N)Ru(NO)Cl3]
A family of complexes has been made that makes use of diimine ligands [131]. The members of this family, fac-[(N-N)Ru(NO)Cl3], (N-N = R′N=CR-CR=NR′), differ in size, shape, and formula of the R groups present on the diimine ligands. The synthetic procedures for the family Ru(II) nitrosyl complexes [(N-N)Ru(NO)Cl3] were obtained through the single step reactions between Ru(NO)Cl3·5H2O dissolved in an appropriate solvent. To this mixture, 1 molar equivalent of the appropriate ligand molecule was added. The products of these reactions were collected and purified. Interestingly, a novel unexpected by-product, which was classified by the rearrangement of one of the NO ligands, was also isolated (very low yield of 5%) from the reaction mixture.
Monoanionic [(NO)Mn(S,S-C6H3-R)2]− and dianionic [(NO)Mn(S,S-C6H3-R)2]2−
An interesting family of new structures based on sulfur ligand is discussed here. These structures make use of manganese as the coordinated metal center instead of the usual iron atom. Several compounds within this family were obtained by changing the substituents on the ring of the benzenedithiolate ligand. Two different complexes were formed here with the ligands bound to manganese in a bidentate and a monodentate manner [132]. These structures were synthesized by dissolving the proper precursor molecule in THF solvent. An NO(g) mixture (10% NO + 90% N2) was then bubbled through the solution. This reaction leads to the formation of the final product, which was isolated and characterized. IR and UV–vis spectra implied the formation of the anionic {Mn(NO)}5 complex [(NO) Mn(S,S-C6H3-R)2]− (R = H or Me). The simple H containing complex was isolated in solid form and characterized by single-crystal X-ray analysis. Several structures have been synthesized that are very similar to these, but make use of different metal centers [133, 134].
[Ru(L)(PPh3)2(NO)](ClO4) (L = pyridine 2,6-dicarboxylic acid)
Another example of a mixed ligand molecule is discussed here. This molecule makes use of a ruthenium atom as its metal center [135]. A precursor complex [Ru(L1) (PPh3)2(Cl)] was reacted with NO that was generated in situ by an acidified NaNO2 solution. The brownish-red color of the initial solution changed to yellow after approximately 1 h of stirring. The resultant yellow compound was isolated as a perchlorate salt. The precursor complex possesses a band near 400 nm in UV–Vis spectrum. This band was not present in the final product spectrum, where a peak near 320 nm was observed. The IR spectrum of the final product provided νNO at ~1890 cm−1 and the presence of perchlorate ion was confirmed by peaks near 1090 and 623 cm−1.
(OEP)Os(NO)(OiPr)
One of the interesting structures using porphyrin as a ligand makes use of osmium as the metal center [136]. To form this product (OEP) Os(NO)-(OEt) was dissolved in 2-propanol. To this solution, HBF4·Et2O was added in the absence of light. The resulting dark red solution was brought to the reflux temperature and allowed to react for 2 h. The solvent was then removed using a vacuum; the resulting residue was redissolved in a solvent mixture made up of CH2Cl2 and pyridine. This mixture was stirred at room temperature for 5 min to completely dissolve the residue. The volatile solvents were removed from the solution again using a vacuum. The red residue was dissolved again in a minimum amount of CH2Cl2 and purified by being quickly chromatographed on a short silica gel column by using an eluent solution of CH2Cl2/THF (5:1). The bright red band was collected and evaporated to dryness, and the alkoxide compound (OEP)Os (NO)(OiPr) was obtained as a red solid (65% yield).
[Fe(NO)(cyclam-ac)](ClO4)Cl·H2O
A molecule similar to that of the afore-mentioned porphyrin structure is an octahedral pentadentate molecule described here [93]. This compound has a mixed chelated structure, having both nitrogen and oxygen coordinating atoms that are part of the same ligand molecule. In order to synthesize this molecule the precursor molecule was dissolved in degassed acetonitrile solvent at ambient temperature with stirring. To this solution (tris (4-bromophenyl)ammoniumyl) hexachloroantimonate was carefully added in a 1:1 molar ratio. The mixture was allowed to stir for 5 min. This yielded a light yellow solution and upon sitting undisturbed, colorless tris(4-bromophenyl)amine precipitated. This solid was filtered off. Following this, a threefold excess of tetra-n-butyl ammonium perchlorate was dissolved in degassed CH3CN and added to the previous yellow solution. A slow flow of Ar gas was passed through the solution in order to slowly evaporate off the solvent. After ~12 h a yellow solid had precipitated and was collected by filtration. The solid was recrystallized in water and yellow crystals were obtained with a 72% yield.
4.2 Trinitrosyl Complexes
Trinitrosyl metal complexes are rarely isolated due to their relative instability. However, a few structures have been reported which make use of large bulky ligands that stabilize the three nitrosyl groups.
[(IMes)Fe(NO)3][BF4]
One form of trinitrosyl iron complex (TNIC) was synthesized by the Darensbourg group [137]. This new compound was only made possible with the use of a bulky carbene ligand that stabilizes the three NO groups on the iron center. This was synthesized by reacting an appropriate amount of Fe (NO)2(CO)2, with 1 molar equivalent of the carbene ligand (abbreviated here as IMes) to form the precursor DNIC molecule (IMes)Fe(CO)(NO)2. This precursor molecule was then reacted with NOBF4 and left to stir overnight. Purification using hexane yielded the final product [(IMes)Fe(NO)3][BF4]. Green crystals of X-ray quality were obtained that are air and moisture stable, as well as being thermally stable in THF with a very slow decomposition rate. This molecule was qualitatively tested using IR spectroscopy and a nitric oxide trapping agent. The results showed that this new structure was able to transfer nitric oxide under these conditions.
5 Organometallic Metal Nitrosyl Complexes
Organometallic metal nitrosyl complexes represent a large portion of the subject and the methods of preparing these complexes have been published in several helpful texts [7, 28, 138, 139]; therefore, it is minimally discussed here. Instead we picked a few recent examples that deal with controlling the reactivity of nitric oxide at transition-metal centers, because of their fundamental significance and possible biological relevance.
Legzdins’ group recently reported an intramolecular insertion of bound NO into an adjacent metal–ligand bond in the presence of an oxygen-atom transfer reagent [140]. Treatment of the dialkyl compounds Cp*M(NO)(CH2CMe3)2 (where Cp* = η5-C5Me5, M = Mo or W) with cumene hydroperoxide induced the insertion of NO into one of their metal–carbon bonds and resulted in the formation of the η2-nitrosoalkane oxo complexes Cp*M(O)(η2-ONCH2CMe3)(CH2CMe3). Further treatment of Cp*M(O)(η2-ONCH2CMe3)(CH2CMe3) with excess O2 yielded the dioxo alkyl complexes Cp*M(O)2(CH2CMe3).
Recently, Bergman et al. reported several examples of double additions of C–N bond-forming reactions of metal nitrosyls, some of which are shown in Fig. 24 [141, 142]. For instance, the dinitrosyl complex [RuCl2(NO)2(THF)], in the presence of an additional neutral chelating ligand, binds alkenes to the nitrosyl nitrogen atoms at room temperature [142]. This was achieved by the reaction of [{(cymene)RuCl2}2] with 10 equiv of norbornadiene in the presence of NO in THF, which yielded a six-coordinate ruthenium dinitro-soalkane adduct [(THF)2RuCl2{(μ-NO)2(C7H8)}] in high yield.
Fig. 24.

Examples of double additions of C–N bond-forming reactions of metal nitrosyls
Using a combination of experimental techniques (XRD, EPR, UV–vis, NIR, and X-ray absorption spectroscopies) and computational studies (DFT, time-dependent DFT, complete active space self-consistent field (CASSCF), and multireference configuration interaction (MRCI) calculations), the ground-state electronic structure of a four-coordinate Co nitrosyl complex: Tp*Co(NO) (Tp* = hydro-tris (3,5-dimethylpyrazolyl)borate) was well described. A detailed understanding of the interaction between the metal center and the nitrosyl ligand was obtained from correlating between experiment and theory [143].
6 Conclusion
In summary, the preparation of some novel metal nitrosyl complexes, which have been synthesized over the last decade, has been summarized and several selected examples have been discussed in detail. From those examples, one can see that dinitrosyl complexes are typically prepared by using Fe(NO)2(CO)2 as the starting material, which is then reacted with appropriate ligands. Final products are formed as a result of simple substitution reactions. These processes can easily be followed through the use of FT-IR spectroscopy, as the remaining carbonyl group(s) can help to differentiate between single or double substituted complexes. Depending on the donating/accepting properties of the ligands, the IR stretching frequencies of the nitrosyl group will shift into different directions, as well as the electrochemical reduction potentials. As shown in the X-ray crystallographic studies, all of these complexes possess tetrahedral geometry at their metal centers with the exception of a very few. NMR is also useful in studying the geometry changes of the ligands and in some cases, a restricted rotation of the ligand has been observed. The 17-electron paramagnetic form of these complexes is EPR active and shows exclusively well-resolved spectra with g-factors close to 2.03. This high g-value is related to the high electron density on the metal center, and the hyperfine couplings are attributed to the nitrogens of the nitrosyl ligands (though the coordinated ligands also contribute). These complexes are closely related to the biological non-heme iron nitrosyl complexes, the so-called g = 2.03 species.
Nitrosyl complexes containing two or more metal centers have also been summarized. The complexes containing Fe2S2 cores are closely related to the RRE structures and are often used as biomimics of NO bound to the cysteine residues of proteins. Formation of DINCs at Rieske centers of certain proteins has been achieved. These complexes are typically synthesized through the reaction of Fe(NO)2(CO)2 with the appropriate thiol or thiolating ligands. A few mixed metal complexes have also been prepared. Characterizations of these complexes are usually done through a combination of X-ray crystallographic methods and FT-IR spectroscopy. The structures in their solid state are almost exclusively trans-isomers, while in solution, cis- and trans-isomers coexist. DFT calculations show that the two isomers can interchange and the activation energy barrier is only ~3 kcal/mol. The electrochemical studies show ligand-based redox behavior with SR group influence of the reduction potentials. These complexes are diamagnetic and EPR silent. The reduced form of the complexes are EPR active and show a single line at g = 1.99 without any hyperfine coupling. The electron distribution calculated by DFT methods shows that the unpaired electron is delocalized between the iron and sulfur atoms with ~60% and 25%, respectively, and very small amount (~2%) on the nitrosyl groups. In contrast, the unpaired electron in DNICs is mostly on the iron atom and there are significantly larger spin densities on the nitrogens of the nitrosyl ligands. These results explain the lack of hyperfine coupling in the reduced RREs and the g-value difference between the RREs (g = 1.99) and the DNICs (g = 2.03).
The method of using simple one-step solvent-thermal reactions to prepare iron–sulfur and iron–selenium nitrosyl clusters is also discussed in detail. The results show that the solvent-thermal reaction is a more effective and simple procedure for the synthesis of polynuclear iron nitrosyl compounds than traditional methods, in which other iron–sulfur nitrosyl clusters are used as starting materials. The structural analysis done by single-crystal X-ray diffraction shows that each iron center is bonded to three selenium (sulfur) atoms and a nitrogen atom from the nitrosyl ligand with pseudo-tetrahedral geometry. The UV–vis spectra are also presented along with the electrochemical study, in which an ECE mechanism is identified. The formation and structural characteristics of multinuclear metal nitrosyl complexes containing four iron and four imidazole or four substituted imidazoles have also been presented.
Mononitrosyl complexes are usually prepared through the treatment of a precursor molecule with source of NO. Many mononitrosyl complexes are often synthesized with the intention of comparing their structures and properties with the products of nitric oxide transfer reactions. Trinitrosyl metal complexes are harder to prepare due to their relative instability and the example presented shows that it is made possible only with the use of a bulky ligand that stabilizes the three NO groups on the iron center. The subject of organometallic metal nitrosyl complexes is minimally discussed here with a few recent examples related to the reactivity of nitric oxide at transition-metal centers.
Many of the aforementioned structures show promising properties including the ability to bind to proteins, transfer nitric oxide, and reversibly convert into Roussin’s Red or Black Esters. Despite this intensive study, further work is needed in order to completely understand the chemical properties of these compounds. Quantitative studies of the NO transfer and delivery have yet to be accomplished on many of these complexes. The full potential of these complexes can be unlocked once these properties are quantified and fully understood.
Acknowledgments
We wish to thank the National Institute of Health (NIH) MBRS Score program (Grant # 5 SC3 GM 092301) for their continued financial support.
References
- 1.Koshland DE. Science. 1992;258:1861–1902. doi: 10.1126/science.1470903. [DOI] [PubMed] [Google Scholar]
- 2.Feldman PL, Griffith OW, Stuehr DJ. Chem Eng News. 1993;71:26–38. [Google Scholar]
- 3.de Mel A, Murad F, Seifalian AM. Chem Rev. 2011;111:5742–5767. doi: 10.1021/cr200008n. [DOI] [PubMed] [Google Scholar]
- 4.Lin Z, Chiou T, Liu K, Hsieh C, Yu JK, Liaw WF. Inorg Chem. 2012;51:10092–10094. doi: 10.1021/ic3018437. [DOI] [PubMed] [Google Scholar]
- 5.Vanin FA, Serezhenkov VA, Mikoyan VD, Genkin MV. Nitric Oxide. 1998;2:224–234. doi: 10.1006/niox.1998.0180. [DOI] [PubMed] [Google Scholar]
- 6.Muller B, Kleschyov AL, Stoclet JC. Br J Pharmacol. 1996;119:1281–1285. doi: 10.1111/j.1476-5381.1996.tb16034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richter-Addo GB, Legzdins P. Chem Rev. 1988;88:991–1010. doi: 10.1021/cr010188k. [DOI] [PubMed] [Google Scholar]
- 8.Saraiva J, Oliveira SS, Cicillini SA, Eloy JO, Marchetti JM. J Drug Deliv. 2011:1–16. doi: 10.1155/2011/936438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reglinski J, Butler AR, Glidewell C. Appl Organomet Chem. 1994;8:25–31. [Google Scholar]
- 10.Mingos DMP, Sherman DJ. Adv Inorg Chem. 1989;34:293–393. [Google Scholar]
- 11.Ford PC, Laverman LE. Coord Chem Rev. 2005;249:391–403. [Google Scholar]
- 12.King SB. Free Radical Biol Med. 2004;37:735–737. doi: 10.1016/j.freeradbiomed.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 13.Lancaster JR, Hibbs JB., Jr Proc Natl Acad Sci USA. 1990;87:1223–1227. doi: 10.1073/pnas.87.3.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang PG, Xian M, Tang X, Wu X, Wen Z, Cai T, Janczuk AJ. Chem Rev. 2002;102:1191–1200. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 15.Vanin AF. FEBS Lett. 1991;289:1–3. doi: 10.1016/0014-5793(91)80894-9. [DOI] [PubMed] [Google Scholar]
- 16.Hieber W, Klingshirn W. Z Anorg Allgem Chem. 1963;323:292. [Google Scholar]
- 17.King B. Organometallic synthesis. 1. Academic; New York: 1965. pp. 167–168. [Google Scholar]
- 18.McBride DW, Stafford SL, Stone FGA. Inorg Chem. 1962;1:386–388. [Google Scholar]
- 19.Klein A, Mering Y, Uthe A, Butsch K, Schaniel D, Mockus N, Woike T. Polyhedron. 2012;29:2553–2559. [Google Scholar]
- 20.Li L. Commun Inorg Chem. 2002;23:335–353. [Google Scholar]
- 21.Mingos DMP, Ibers JA. Inorg Chem. 1970;9:1105–1111. [Google Scholar]
- 22.Divara M, Ghilardi CA, Sacconi L. Inorg Chem. 1976;15:1555–1561. [Google Scholar]
- 23.Darensbourg DJ, Decuir TJ, Stafford NW, Robertson JB, Draper JD, Reibenspies JH, Kathó A, Joó F. Inorg Chem. 1997;36:4218–4226. [Google Scholar]
- 24.Matsukawa S, Kuwata S, Hidai M. Inorg Chem. 2000;39:791–798. doi: 10.1021/ic990805b. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Enright GD, Preston KF. Organometallics. 1994;13:4686–4688. [Google Scholar]
- 26.Horsken A, Zheng G, Stradiotto M, McCrory CTC, Li L. J Organomet Chem. 1998;558:1–9. [Google Scholar]
- 27.Allano VG, Araneo A, Bellon PL, Ciani G, Manassero M. J Organomet Chem. 1974;67:413. [Google Scholar]
- 28.Richter-Addo GB, Legzdins P. Metal nitrosyls. Oxford University Press; New York: 1992. p. 81. [Google Scholar]
- 29.Martin RL, Taylor D. Inorg Chem. 1976;15:2970–2976. [Google Scholar]
- 30.Gaughan AP, Corden BJ, Eisenberg R, Ibers JA. Inorg Chem. 1974;13:786–791. [Google Scholar]
- 31.Bhaduri S, Sheldrick GM. Acta Crystallogr Sect E. 1975;31:897. [Google Scholar]
- 32.Haymore BL, Ibers JA. Inorg Chem. 1975;14:2610–2617. [Google Scholar]
- 33.Kaduk JA, Ibers JA. Inorg Chem. 1975;14:3070–3073. [Google Scholar]
- 34.Laing M, Reimann RH, Singleton E. Inorg Chem. 1970;18:2666–2670. [Google Scholar]
- 35.Guillaume P, Wah HLK, Postel M. Inorg Chem. 1991;30:1828–1831. [Google Scholar]
- 36.Li L, Reginato N, Urschey M, Stradiotto M, Liarakos DJ. Can J Chem. 2003;81:468–475. [Google Scholar]
- 37.Atkinson FL, Blackwell HE, Brown NC, Connelly NG, Crossley JG, Orpen AG, Rieger AL, Rieger PH. J Chem Soc Dalton Trans. 1996:3491–3502. [Google Scholar]
- 38.Bryar TR, Eaton DR. Can J Chem. 1992;70:1917–1926. [Google Scholar]
- 39.Ray M, Bolombek AP, Hendrich MP, Yap GPA, Liable-Sands LM, Rheingold AL, Borovik AS. Inorg Chem. 1999;38:3110–3115. [Google Scholar]
- 40.Vanin AF, Van Faasen E. Radicals for life, the various forms of nitric oxide. Elsevier; Oxford: 2007. pp. 19–74. [Google Scholar]
- 41.Foster MW, Cowan JA. J Am Chem Soc. 1999;121:4093–4100. [Google Scholar]
- 42.Heo J, Campbell SL. Biochemistry. 2004;43:2314–2322. doi: 10.1021/bi035275g. [DOI] [PubMed] [Google Scholar]
- 43.Sanina NA, Aldoshin SM. Russ Chem Bull Int Ed. 2004;53:2428–2448. [Google Scholar]
- 44.Lee M, Arosio P, Cozzi A, Chasteen ND. Biochemistry. 1994;33:3679–3687. doi: 10.1021/bi00178a026. [DOI] [PubMed] [Google Scholar]
- 45.Reginato N, McCrory CTC, Pervitsky D, Li L. J Am Chem Soc. 1999;121:10217–10218. [Google Scholar]
- 46.Tsai FT, Kuo TS, Liaw WF. J Am Chem Soc. 2009;131:3426–3427. doi: 10.1021/ja808743g. [DOI] [PubMed] [Google Scholar]
- 47.Tsai ML, Liaw WF. Inorg Chem. 2006;45:6583–6585. doi: 10.1021/ic0608849. [DOI] [PubMed] [Google Scholar]
- 48.Puiu SC, Warren TH. Organometallics. 2003;22:3974–3976. [Google Scholar]
- 49.Dieck HT, Bruder H, Kuhl E, Junghans D, Hellfeldt K. New J Chem. 1989;13:259–268. [Google Scholar]
- 50.Wah HLK, Postel M, Tomi F, Mordenti L. New J Chem. 1991;15:629–633. [Google Scholar]
- 51.Hung MC, Tsai MC, Lee GH, Liaw WF. Inorg Chem. 2006;45:6041–6047. doi: 10.1021/ic0605120. [DOI] [PubMed] [Google Scholar]
- 52.Wang R, Wang X, Sundberg EB, Nguyen P, Grant GPG, Sheth C, Zhao Q, Herron S, Kantardjieff KA, Li L. Inorg Chem. 2009;48:9779–9785. doi: 10.1021/ic901368t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang HW, Tsou CC, Kuo TS, Liaw WF. Inorg Chem. 2008;47:2196–2204. doi: 10.1021/ic702000y. [DOI] [PubMed] [Google Scholar]
- 54.Enemark JH, Feltham RD. Coord Chem Rev. 1974;13:339–406. [Google Scholar]
- 55.Shih WC, Lu TT, Yang LB, Tsai FT, Chiang MH, Lee JF, Chiang YW, Liaw WF. J Inorg Biochem. 2012;113:3–93. doi: 10.1016/j.jinorgbio.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 56.Crimmin MR, Rosebrugh LE, Tomson NC, Weyhermüller T, Bergman RG, Toste FD, Wieghardt K. J Organomet Chem. 2011;696:3974–3981. [Google Scholar]
- 57.Yeh SW, Lin CW, Li YW, Hsu IJ, Chen CH, Jang LY, Lee JF, Liaw WF. Inorg Chem. 2012;51:4076–4087. doi: 10.1021/ic202332d. [DOI] [PubMed] [Google Scholar]
- 58.Siladke NA, Meihaus KR, Ziller JW, Fang M, Furche F, Long JR, Evans WJ. J Am Chem Soc. 2012;134:1243–1249. doi: 10.1021/ja2096128. [DOI] [PubMed] [Google Scholar]
- 59.Kozhukh J, Lippard SJ. J Am Chem Soc. 2012;134:11120–11123. doi: 10.1021/ja305011g. [DOI] [PubMed] [Google Scholar]
- 60.Wright AM, Wu G, Hayton TW. J Am Chem Soc. 2010;132:14336–14337. doi: 10.1021/ja105930b. [DOI] [PubMed] [Google Scholar]
- 61.Chiou SJ, Wang CC, Chang CM. J Organomet Chem. 2008;693:3582–3586. [Google Scholar]
- 62.Chiang CY, Miller ML, Reibenspies JH, Darensbourg MY. J Am Chem Soc. 2004;126:10867–10874. doi: 10.1021/ja049627y. [DOI] [PubMed] [Google Scholar]
- 63.Lin Z, Lo F, Li C, Chen C, Huang W, Hsu I, Lee J, Horng J, Liaw WF. Inorg Chem. 2011;50:10417–10431. doi: 10.1021/ic201529e. [DOI] [PubMed] [Google Scholar]
- 64.Chang HH, Huang HJ, Ho YL, Wen YD, Huang WN, Chiou SJ. Dalton Trans. 2009:6396–6402. doi: 10.1039/b902478f. [DOI] [PubMed] [Google Scholar]
- 65.Baltusis LM, Karlin KD, Rabinowitz HN, Dewan JC, Lippard SJ. Inorg Chem. 1980;19:2627–2632. [Google Scholar]
- 66.Wang R, Xu W, Zhang J, Li L. J Mol Struc. 2009;923:110–113. doi: 10.1016/j.molstruc.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee CM, Chen CH, Chen HW, Hsu JL, Lee GH, Liaw WF. Inorg Chem. 2005;44:6670–6679. doi: 10.1021/ic050108l. [DOI] [PubMed] [Google Scholar]
- 68.Harrop TC, Song D, Lippard SJ. J Inorg Biochem. 2007;101:1730–1738. doi: 10.1016/j.jinorgbio.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 69.Conradie J, Quarless DA, Jr, Hsu HF, Harrop TC, Lippard SJ, Koch SA, Ghosh A. J Am Chem Soc. 2007;129:10446–10456. doi: 10.1021/ja0719982. [DOI] [PubMed] [Google Scholar]
- 70.Wecksler SR, Hutchinson J, Ford PC. Inorg Chem. 2006;45:1192–1200. doi: 10.1021/ic051723s. [DOI] [PubMed] [Google Scholar]
- 71.Rauchfuss TB, Weatherill TD. Inorg Chem. 1982;21:827–830. [Google Scholar]
- 72.Wang R, Camacho-Fernandez MA, Xu W, Zhang J, Li L. Dalton Trans. 2009:777–786. doi: 10.1039/b810230a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsou CC, Lu TT, Liaw WF. J Am Chem Soc. 2007;129:12626–12627. doi: 10.1021/ja0751375. [DOI] [PubMed] [Google Scholar]
- 74.Tinberg CE, Tonzetich ZJ, Wang H, Do LH, Yoda Y, Cramer SP, Lippard SJ. J Am Chem Soc. 2011;132:18168–18176. doi: 10.1021/ja106290p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsou CC, Lin ZS, Lu TT, Liaw WF. J Am Chem Soc. 2008;130:17154–17160. doi: 10.1021/ja806050x. [DOI] [PubMed] [Google Scholar]
- 76.Tonzetich ZJ, Do LH, Lippard SJ. J Am Chem Soc. 2009;131:7964–7965. doi: 10.1021/ja9030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hsu IJ, Hsieh CH, Ke SC, Chiang KA, Lee JM, Chen JM, Jang LY, Lee GH, Wang Y, Liaw WF. J Am Chem Soc. 2007;129:1151–1159. doi: 10.1021/ja065401e. [DOI] [PubMed] [Google Scholar]
- 78.Tsai ML, Chen CC, Hsu IJ, Ke SC, Hsieh CH, Chiang KA, Lee GH, Wang Y, Liaw WF. Inorg Chem. 2004;43:5159–5167. doi: 10.1021/ic0494915. [DOI] [PubMed] [Google Scholar]
- 79.Tsai FT, Chiou SJ, Tsai MC, Tsai ML, Huang HW, Chiang MH, Liaw WF. Inorg Chem. 2005;44:5872–5881. doi: 10.1021/ic0505044. [DOI] [PubMed] [Google Scholar]
- 80.Lu TT, Chiou SJ, Chen CY, Liaw WF. Inorg Chem. 2006;45:8799–8806. doi: 10.1021/ic061439g. [DOI] [PubMed] [Google Scholar]
- 81.King SB. Free Radical Biol Med. 2004;37:737–738. doi: 10.1016/j.freeradbiomed.2004.02.073. [DOI] [PubMed] [Google Scholar]
- 82.Ford PC. Int J Photoenergy. 2001;3:161–169. [Google Scholar]
- 83.Li L, Morton JR, Preston KF. Magn Reson Chem. 1995;31:s14–s19. [Google Scholar]
- 84.Roncaroli F, Olabe JA. Inorg Chem. 2005;44:4719–4727. doi: 10.1021/ic048156d. [DOI] [PubMed] [Google Scholar]
- 85.Lu TT, Huang WH, Liaw WF. Inorg Chem. 2009;48:9027–9035. doi: 10.1021/ic9012679. [DOI] [PubMed] [Google Scholar]
- 86.Wecksler SR, Mikhailovsky A, Korystov D, Buller F, Kannan R, Tan LS, Ford PC. Inorg Chem. 2007;46:395–402. doi: 10.1021/ic0607336. [DOI] [PubMed] [Google Scholar]
- 87.Chen TN, Lo FC, Tsai ML, Shih KN, Chiang MH, Lee GH, Liaw WF. Inorg Chim Acta. 2006;359:2525–2533. [Google Scholar]
- 88.Harrop TC, Tonzetich ZJ, Reisner E, Lippard SJ. J Am Chem Soc. 2008;130:15602–15610. doi: 10.1021/ja8054996. [DOI] [PubMed] [Google Scholar]
- 89.Lu TT, Tsou CC, Huang HW, Hsu IJ, Chen JM, Kuo TS, Wang Y, Liaw WF. Inorg Chem. 2008;47:6040–6050. doi: 10.1021/ic800360m. [DOI] [PubMed] [Google Scholar]
- 90.Chen YJ, Ku WC, Feng LT, Tsai ML, Hsieh CH, Hsu WH, Liaw WF, Hung CH, Chen YJ. J Am Chem Soc. 2008;130:10929–10938. doi: 10.1021/ja711494m. [DOI] [PubMed] [Google Scholar]
- 91.Harrop TC, Song D, Lippard SJ. J Am Chem Soc. 2006;128:3528–3529. doi: 10.1021/ja060186n. [DOI] [PubMed] [Google Scholar]
- 92.Sellmann D, Blum N, Heinemann FW, Hess BA. Chem Eur J. 2007;7(9):1. doi: 10.1002/1521-3765(20010504)7:9<1874::aid-chem1874>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 93.Serres RG, Grapperhaus CA, Bothe E, Bill E, Weyhermuller T, Neese F, Wieghardt K. J Am Chem Soc. 2004;126:5138–5153. doi: 10.1021/ja030645+. [DOI] [PubMed] [Google Scholar]
- 94.Liaw WF, Chiang CY, Lee GH, Peng SM, Lai CH, Darensbourg MY. Inorg Chem. 2000;39:480–484. doi: 10.1021/ic990631y. [DOI] [PubMed] [Google Scholar]
- 95.Hsieh CH, Erdem OF, Harman SD, Singleton ML, Reijerse E, Lubitz W, Popescu CV, Reibenspies JH, Brothers SM, Hall MB, Darensbourg MY. J Am Chem Soc. 2012;134:13089–13102. doi: 10.1021/ja304866r. [DOI] [PubMed] [Google Scholar]
- 96.D’Addario S, Demartin F, Grossi L, Iapalucci MC, Laschi F, Longoni G, Zanello P. Inorg Chem. 1993;32:1153–1160. doi: 10.1021/ic950482t. [DOI] [PubMed] [Google Scholar]
- 97.Chu CTW, Lo FYK, Dahl LF. J Am Chem Soc. 1982;104:3409–3422. [Google Scholar]
- 98.Scott MJ, Holm RH. Angew Chem Int Ed Engl. 1993;32:564–566. [Google Scholar]
- 99.Geiser U, Williams JM. Acta Crystallogr. 1998;C54:292–293. [Google Scholar]
- 100.Kalyvas H, Coucouvanis D. Inorg Chem. 2006;45:8462–8464. doi: 10.1021/ic060417r. [DOI] [PubMed] [Google Scholar]
- 101.Barnes JC, Glidewell C, Lees A, Howie R. Acta Crystallogr. 1990;C46:2051–2053. [Google Scholar]
- 102.Wang R, Xu W, Zhang J, Li L. Inorg Chem. 2010;49:4814–4819. doi: 10.1021/ic9014509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang X, Sundberg EB, Li L, Kantardjieff KA, Herron SR, Lim M, Ford PC. Chem Commun. 2005:477–479. doi: 10.1039/b412086h. [DOI] [PubMed] [Google Scholar]
- 104.Hess JL, Hsieh CH, Reibenspies JH, Darensbourg MY. Inorg Chem. 2011;50:8541–8552. doi: 10.1021/ic201138f. [DOI] [PubMed] [Google Scholar]
- 105.Hess JL, Hsieh CH, Brothers SM, Hall MB, Darensbourg MY. J Am Chem Soc. 2011;133:20426–20434. doi: 10.1021/ja208384d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mingos DMP, Ibers JA. Inorg Chem. 1971;10:1035–1042. [Google Scholar]
- 107.Mingos DMP, Robinson WT, Ibers JA. Inorg Chem. 1971;10:1043–1048. [Google Scholar]
- 108.Mingos DMP, Ibers JA. Inorg Chem. 1971;10:1479–1486. [Google Scholar]
- 109.Tonzetich ZJ, Heroguel F, Do LH, Lippard SJ. Inorg Chem. 2011;50:1570–1579. doi: 10.1021/ic102300d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lehnert N, Galinato MGI, Paulat F, Richter-Addo GB, Sturhahn W, Xu N, Zhao J. Inorg Chem. 2010;49:4133–4148. doi: 10.1021/ic902181e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fry NL, Mascharak PK. Acc Chem Res. 2011;44:289–298. doi: 10.1021/ar100155t. [DOI] [PubMed] [Google Scholar]
- 112.Richter-Addo GB, Wheeler RA, Hixson CA, Chen L, Khan MA, Ellison MK, Schulz CE, Scheidt WR. J Am Chem Soc. 2001;123:6314–6326. doi: 10.1021/ja010276m. [DOI] [PubMed] [Google Scholar]
- 113.Schweitzer D, Ellison JL, Shoner SC, Lovell S, Kovacs JA. J Am Chem Soc. 1998;120:10966–10977. [Google Scholar]
- 114.Eroy-Reveles AA, Leung Y, Mascharak PK. J Am Chem Soc. 2006;128:7166–7167. doi: 10.1021/ja061852n. [DOI] [PubMed] [Google Scholar]
- 115.Rose MJ, Olmstead MM, Mascharak PK. J Am Chem Soc. 2007;129:5342–5343. doi: 10.1021/ja070247x. [DOI] [PubMed] [Google Scholar]
- 116.Eroy-Reveles AA, Leung Y, Beavers CM, Olmstead MM, Mascharak PK. J Am Chem Soc. 2008;130:4447–4458. doi: 10.1021/ja710265j. [DOI] [PubMed] [Google Scholar]
- 117.Rose MJ, Fry NL, Marlow R, Hinck L, Mascharak PK. J Am Chem Soc. 2008;130:8834–8846. doi: 10.1021/ja801823f. [DOI] [PubMed] [Google Scholar]
- 118.Lee J, Kovalevsky AY, Novozhilova IV, Bagley KA, Coppens P, Richter-Addo GB. J Am Chem Soc. 2004;126:7180–7181. doi: 10.1021/ja0488986. [DOI] [PubMed] [Google Scholar]
- 119.Cheng L, Chen L, Chung HS, Khan MA, Richter-Addo GB, Young VG., Jr Organometallics. 1998;17:3853–3864. [Google Scholar]
- 120.Richter-Addo GB, Hodge SJ, Yi GB, Khan MA, Ma T, Van Caemelbecke E, Guo N, Kadish KM. Inorg Chem. 1996;35:6530–6538. doi: 10.1021/ic960031o. [DOI] [PubMed] [Google Scholar]
- 121.Kadish KM, Adamian VA, Van Caemelbecke E, Tan Z, Tagliatesta P, Bianco P, Boschi T, Yi GB, Khan MA, Richter-Addo GB. Inorg Chem. 1996;35:1343–1348. doi: 10.1021/ic950799t. [DOI] [PubMed] [Google Scholar]
- 122.Rose MJ, Betterley NB, Mascharak PK. J Am Chem Soc. 2009;131:8340–8341. doi: 10.1021/ja9004656. [DOI] [PubMed] [Google Scholar]
- 123.Madhani A, Patra AK, Miller TW, Eroy-Reveles AA, Hobbs AJ, Fukuto JM, Mascharak PK. J Med Chem. 2006;49:7325–7330. doi: 10.1021/jm0604629. [DOI] [PubMed] [Google Scholar]
- 124.Schweitzer D, Ellison JL, Shoner SC, Lovell S, Kovacs JA. J Am Chem Soc. 1998;120:9237–9245. [Google Scholar]
- 125.Mingos DMP. Inorg Chem Corres. 1973;12:1209–1211. [Google Scholar]
- 126.Hoffmann R, Chen MML, Elian M, Rossi AR, Mingos DMP. Inorg Chem. 1974;13:2666–2675. [Google Scholar]
- 127.Bell LK, Mason J, Mingos DMP, Tew DG. Inorg Chem. 1983;22:3497–3502. [Google Scholar]
- 128.Mason J, Mingos DMP, Sherman D, Wardleb RWM. J Chem Soc Chem Commun. 1984:1223–1227. [Google Scholar]
- 129.Mason J, Mingos DMP, Schaefer J, Sherman D, Stejskal EO. J Chem Soc Chem Commun. 1985:444–446. [Google Scholar]
- 130.Nicholson T, Muller P, Davison A, Jones AG. Inorg Chim Acta. 2006;359:1296–1298. [Google Scholar]
- 131.Xu HJ, Lu XY, Cheng Y, Sun JF, Chen XT, Xue ZL. Organometallics. 2009;28:6687–6694. [Google Scholar]
- 132.Lin CH, Chen CG, Tsai ML, Lee GH, Liaw WF. Inorg Chem. 2008;47:11435–11443. doi: 10.1021/ic801553s. [DOI] [PubMed] [Google Scholar]
- 133.Chen HW, Lin CW, Chen CC, Yang LB, Chiang MH, Liaw WF. Inorg Chem. 2005;44:3226–3232. doi: 10.1021/ic049105j. [DOI] [PubMed] [Google Scholar]
- 134.Tennyson AG, Dhar S, Lippard SJ. J Am Chem Soc. 2008;130:15087–15098. doi: 10.1021/ja803992y. [DOI] [PubMed] [Google Scholar]
- 135.Tsai ML, Hsieh CH, Liaw WF. Inorg Chem. 2007;46:5110–5117. doi: 10.1021/ic0702567. [DOI] [PubMed] [Google Scholar]
- 136.Cheng L, Powell DR, Khan MA, Richter-Addo GB. Inorg Chem. 2001;40:125–133. doi: 10.1021/ic0007512. [DOI] [PubMed] [Google Scholar]
- 137.Hsieh CH, Darensbourg MY. J Am Chem Soc. 2010;132:14118–14125. doi: 10.1021/ja104135x. [DOI] [PubMed] [Google Scholar]
- 138.Hayton TW, Legedins P, Sharp WB. Chem Rev. 2002;102:935–991. doi: 10.1021/cr000074t. [DOI] [PubMed] [Google Scholar]
- 139.Bitterwolf TE. Coord Chem Rev. 2006;250:388–413. [Google Scholar]
- 140.Graham PM, Buschhaus MSA, Baillie RA, Semproni SP, Legzdins P. Organometallics. 2010;29:5068–5072. [Google Scholar]
- 141.Schomaker JM, Boyd WC, Stewart IC, Toste FD, Bergman RG. J Am Chem Soc. 2008;130:3777–3779. doi: 10.1021/ja800738d. [DOI] [PubMed] [Google Scholar]
- 142.Crimmin MR, Bergman RG, Toste FD. Angew Chem Int. 2011;50:4484–4487. doi: 10.1002/anie.201100816. [DOI] [PubMed] [Google Scholar]
- 143.Tomson NC, Crimmin MR, Petrenko T, Rosebrugh LE, Sproules S, Boyd WC, Bergman RG, DeBeer S, Toste FD, Wieghardt K. J Am Chem Soc. 2011;133:18785–18801. doi: 10.1021/ja206042k. [DOI] [PubMed] [Google Scholar]