
Figure 3.
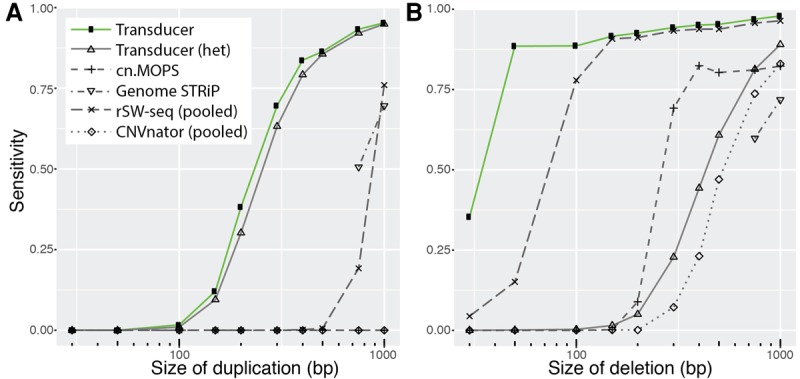
Detecting simulated duplications (A) and deletions (B) present in freshwater, but not marine, individuals. We simulated data analogous to our main data set of 10 marine and 11 freshwater stickleback genomes sequenced to a median coverage of 1.7×, except we randomly placed deletions and duplications that ranged from 30 to 1000 bp in the genomes of all freshwater individuals. We recorded the performance of existing methods (at 0.2 false positives) either based on the annotations of individual genomes (cn.MOPS and Genome STRiP) or after we pooled all marine samples into a pseudo-individual and all freshwater samples into a pseudo-individual (rSW-seq and CNVnator). We also tested the transducer's ability to detect heterozygous duplications and deletions. Heterozygous deletions are especially difficult to detect because, with 36-bp reads, regions with SNP divergence may exhibit reduced mapping efficiency, which results in read coverage similar to that of a heterozygous deletion.