

Abstract
Uncoupling protein-1 (UCP1) facilitates thermogenesis in brown and beige adipocytes and can promote energy expenditure by decreasing mitochondrial respiratory efficiency. Defects in UCP1 and brown adipose tissue thermogenesis subject animals to chronic cold stress and elicit compensatory responses to generate heat. How UCP1 regulates white adipose tissue (WAT) lipid biology and tissue crosstalk is not completely understood. Here, we probed the effect of UCP1 deficiency on FA metabolism in inguinal and epididymal WAT and investigated how these metabolic perturbations influence hepatic lipid homeostasis. We report that at standard housing temperature (21°C), loss of UCP1 induces inguinal WAT de novo lipogenesis through transcriptional activation of the lipogenic gene program and elevated GLUT4. Inguinal adipocyte hyperplasia and depot expansion accompany the increase in lipid synthesis. We also found that UCP1 deficiency elevates adipose stearoyl-CoA desaturase gene expression, and increased inguinal WAT lipolysis supports the transport of adipose-derived palmitoleate (16:1n7) to the liver and hepatic triglyceride accumulation. The observed WAT and liver phenotypes were resolved by housing animals at thermoneutral housing (30°C). These data illustrate depot-specific responses to impaired BAT thermogenesis and communication between WAT and liver in UCP1−/− mice.
Keywords: thermogenesis, fatty acid/metabolism, fatty acid/desaturases, lipid, hepatic, β-oxidation
Obesity is a global epidemic that stems from genetic, dietary, and environmental factors. Recent efforts to thwart rising obesity rates focus on increasing energy expenditure through activation of thermogenesis in fat cells (1). Uncoupling protein-1 (UCP1) facilitates nonshivering thermogenesis in adipocytes by uncoupling the proton motive force generated by the electron transport chain from ATP synthesis to generate heat. Previously available drugs (e.g., dinitrophenol and fenfluramine-phentermine) that have leveraged mitochondrial uncoupling caused severe side effects because they were systemic and nonspecific (2, 3), but thermogenic fat provides a unique avenue for mitochondrial uncoupling and fat reduction because expression of UCP1 is restricted to specific adipocytes (4). In contrast to white adipocytes, which function in energy storage and hormone secretion and contain unilocular lipid droplets, brown and beige fat cells contain multilocular lipid droplets and function in fat oxidation for thermogenesis due to their high mitochondrial density and UCP1 content. Despite a morphology and function that is shared with brown fat cells, beige adipocytes are primarily located within subcutaneous white fat depots in rodents and arise from the same developmental lineage as white adipocytes. Seminal studies revealed that some humans have active brown fat, supporting the relevance of thermogenic adipose tissue in humans (5–9).
The discovery of active thermogenic fat in humans has elicited great interest in UCP1 regulation and function. UCP1−/− mice have aided in investigating the role of UCP1 in energy homeostasis and the efficacy of UCP1 modulation as a weight loss target. UCP1−/− mice fare surprisingly well in terms of thermoregulation and energy expenditure. Mice lacking UCP1 are sensitive to acute cold exposure, but this sensitivity depends on the genetic background, and gradual cold acclimation can ameliorate their cold intolerance (10, 11). Furthermore, loss of UCP1 surprisingly protects mice against high-fat diet-induced obesity at standard housing temperatures (20–26°C) (12). Their protection may stem from compensatory mechanisms of thermogenesis in skeletal muscle and adipose tissue (13–17). A follow-up study reported that, when housed at thermoneutrality (30°C), UCP1−/− mice actually become obese with high-fat diet feeding (18). Together, these studies illustrate that the phenotype of UCP1−/− mice depends greatly on the temperature in which they are housed and that chronic cold stress, and subsequently increased circulating norepinephrine (13), elicits a profound global response in these mice.
Much is known regarding the effects of UCP1 deletion on whole body energy expenditure and thermoregulation, yet how UCP1 deficiency affects tissue-specific FA metabolism and interorgan communication is not completely understood. Lipid metabolism and trafficking are critical regulators of energy homeostasis, and tissue-specific control of FA metabolism is essential for maintaining whole body metabolic health. The pathway of de novo lipogenesis (DNL) includes FA synthesis, desaturation, and elongation (19). Acetyl-CoA carboxylase (ACC) and FAS catalyze the synthesis of palmitate from nonlipid precursors. Stearoyl-CoA desaturase (SCD) converts saturated FAs, 16:0 and 18:0, to monounsaturated FAs, 16:1n7 and 18:1n9, and is a critical regulator of DNL, adipocyte differentiation, and adiposity (20). DNL-derived FAs are used for the synthesis of complex lipids, such as triglycerides (TGs) and phospholipids (PLs); furthermore, recent work demonstrates that specific FAs are lipokines and elicit unique signaling properties (21–23). For example, hepatic-derived oleate (18:1n9) regulates FA synthesis and oxidation, and palmitoleate (16:1n7) has been proposed to influence insulin sensitivity and hepatic inflammation (24–26). Highly regulated at the level of transcription, DNL is induced by the transcription factors SREBP-1c and CHREBP-α and -β. Although hepatic DNL is considered deleterious (27, 28), adipose DNL is proposed to be beneficial and to improve glucose sensitivity (23, 29, 30). Transcriptional studies suggest lipid metabolic change in white adipose tissue (WAT) of UCP1−/− (14, 31, 32), but the consequences of these changes and the differential response between inguinal and epididymal WAT has not been fully investigated. Further, we hypothesized that the response to impaired BAT thermogenesis would involve extensive tissue crosstalk. Thus, we aimed to determine how loss of UCP1 regulates adipose tissue biology in a depot-specific manner and influences hepatic lipid metabolism.
In this study, we report a splicing mutation in Ucp1 that causes loss of UCP1 protein. Using this mouse model, we demonstrated that loss of UCP1 dramatically alters lipid homeostasis in white adipose tissue at the level of gene transcription and substrate flux, inducing FA synthesis and promoting inguinal (i.e., subcutaneous) adipocyte hyperplasia and depot expansion. Further, stearoyl-CoA desaturase-1, -2 and -3 and the FA products, palmitoleate and oleate, are elevated in adipose tissue of UCP1−/− mice. UCP1 deficiency also alters lipid trafficking, promoting inguinal WAT lipolysis and causing adipose-derived palmitoleate to accumulate in the liver.
MATERIALS AND METHODS
Animals
Stearoyl-CoA desaturase-3 flox (SCD3F/F) mice were created by cloning a construct in which Scd3 exon 3 was flanked by LoxP sites. Mice were backcrossed at least six generations with C57BL/6J. SCD3F/F mice were crossed with eIIa cre+/− mice to generate global deletion of SCD3. After germline deletion was achieved, cre was bred out of SCD3−/− mice. SCD3F/F and SCD3−/− mice were bred independently for several years. UCP1 SNP and exon deletion were detected using Sanger sequencing. After mutation discovery, mice were genotyped for the Ucp1 SNP using UCP1 exon 5 PCR amplification followed by MspI restriction digest. All UCP1 SNP mice (called UCP1−/−) used in this study are in the SCD3F/F background and have two functional copies of Scd3. Animals were bred by crossing UCP1+/− with UCP1 +/−, and littermate controls were used for all experiments. UCP1−/− mice were also acquired from Jackson Laboratory (called UCP1(J)−/− mice) and used to confirm phenotypes of UCP1 SNP mice. Unless specified otherwise, all animals used in this study were UCP1 SNP mice. Eleven- to fourteen-week-old male mice were used.
For room temperature experiments (21°C), mice were housed in the animal care facility at the University of Wisconsin-Madison Department of Biochemistry. Mice were maintained on a 12 h light-dark cycle (6AM to 6PM) and had free access to food and water until the day of euthanasia. Unless specified otherwise, mice were fed a standard chow diet (Purina 5008) after weaning and until sacrifice. For high-sucrose, very low-fat diet feeding studies, animals were fed Harlan Teklad TD.03045 (2.5% kcal from fat (corn oil), 76.7% kcal from carbohydrate) for a period of 10 days. At 12 weeks of age, mice were fasted 4 h and euthanized by isoflurane overdose beginning at 10AM. Blood was collected via cardiac bleed and tissues were collected and frozen. Thermoneutrality housing experiments (30°C) were 4 weeks in length and were completed at the University of Wisconsin-Madison Biotron. Animals were fed a standard chow diet (Purina 5008) when housed at thermoneutrality. All in vivo experimental animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin-Madison.
Cold tolerance tests
Twelve-week-old male chow-fed mice were placed at 4°C. Food was withdrawn upon cold exposure, but mice maintained access to water. Body temperature was measured with a rectal thermometer every hour. Animals were euthanized if body temperature dropped to 26°C, or after 6 h of cold exposure (whichever occurred first).
In vivo lipogenesis rates
Rates of FA synthesis were measured as previously described (24). Briefly, mice were fasted 2 h then injected with 50 µCi 3H2O intraperitoneally. Two h after injection, animals were euthanized and tissues were collected and frozen. Tissues were saponifed with 3.3 M potassium hydroxide in ethanol, and lipids were extracted using a modified Folch method. FFAs and cholesterol were separated using TLC (60:40:3 heptane:isopropyl ether:acetic acid, v/v/v). 3H counts (cpm) of scraped bands were measured with PerkinElmer TriCarb 2910 TR scintillation counter.
Ex vivo lipolysis rates
Epididymal and inguinal fat explants (75–125 mg) were incubated in KRH buffer [125 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 2.6 mM MgSO4, 5 mM HEPES (pH 7.2)] with 2% FA-free BSA for 2 h at 37°C. Supernatant was collected and free glycerol content was quantified using a free glycerol determination kit (Sigma-Aldrich). Glycerol release from each sample was normalized to starting tissue mass.
Tissue and plasma lipid analyses
Lipid analyses for adipose tissue, plasma, and liver were conducted as previously described (24). Lipids were extracted using a modified Folch method. Samples were spiked with triheptadecanoic TG, phosphatidylcholine, FFA, diacylglycerol, and cholesteryl ester to control for sample loss (Nu-Chek Prep, Inc.). Lipid extracts were separated using TLC and a heptane:isopropyl ether:acetic acid (60:40:3, v/v/v) chamber solvent. Lipid class bands were scraped from the plate and methylated with boron trifluoride in 14% methanol (v/v). Samples were spiked with heptapentanoic acid before methylation to control for methylation efficiency. FA methyl esters were suspended in hexane and analyzed with gas-liquid chromatography (Agilent 6890). Liver TG content was measured using a colorimetric biochemical assay (Wako Chemicals USA).
Real-time quantitative PCR
Total RNA was isolated using RNeasy Lipid Tissue Mini Kit (Qiagen). cDNA was synthesized from RNA using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative reverse-transcriptase PCR (qPCR) was performed using Power SYBR Green Master Mix and an ABI7500 instrument. Relative mRNA abundance was calculated as relative cycle threshold (CT) value and normalized to a housekeeping gene, Arbp or 18S. To detect Scd3 gene expression, the following primer sequences were used: Forward 5′-CATTGGAGCCGGAGTCCATC-3′ and reverse 5′-GCCATGGTGTTGGCAATGAT-3′. Other primer pairs are available upon request.
Immunoblotting
Liver and adipose tissue homogenates were prepared using RIPA buffer (Cell Signaling Technology) supplemented with 1 mM PMSF and phosphatase inhibitor cocktail (0.5 mM imidazole, 0.25 mM sodium fluoride, 0.3 mM sodium molybdate, 0.25 mM sodium orthovanadate, and 1.0 mM sodium tartrate). Protein concentrations were measured using a modified Lowry assay. A total of 25–40 µg protein was separated by SDS-PAGE gel (10% acrylamide) and transferred onto PVDF membrane. Membranes were blocked in 5% nonfat dry milk (w/v) for 1 h then treated with primary antibody overnight at 4°C. Antibodies from Santa Cruz Biotechnology included SCD1 (sc-14720, 1:650) and SCD2 (sc-34208, 1:200). Antibodies from Cell Signaling Technology included FAS (#3180, 1:1000), ACC (#3676, 1:1000), pACC (#11818, 1:1000), β-actin (#4970, 1:1000), GLUT4 (#2213, 1:1000), HSL (#4107, 1:000), pHSL Ser563 (#4139, 1:1000), pHSL Ser565 (#4137, 1:1000), pHSL Ser660 (#4126, 1:1000), ATGL (#2138, 1:1000), vinculin (#4650, 1:1000), and tubulin (#3873, 1:3000). Antibodies from Abcam included OXPHOS cocktail (ab110412, 1:1000), and VDAC (ab18988, 1:8000).
Histology
Fresh liver tissue was frozen in OCT compound and sectioned and stained with Oil Red O. Liver images were acquired using a Nikon Eclipse Ti Intensilight microscope. Fresh WAT was fixed in formalin then paraffin-embedded for sectioning and staining with hematoxylin and eosin. WAT sections were imaged using Leica DM4000B microscope with Retiga 4000R digital camera. Adipocytes were measured for n = 3 animals per group and three images per animal using Adiposoft (33).
Statistics
Statistical analyses were performed using Student’s t-test (2-sided, unpaired). All data are reported as mean ± SEM. Statistical significance of WT to UCP1−/− mice as denoted by *P < 0.05.
RESULTS
A point mutation in Ucp1 was discovered in SCD3−/− mice and yielded a UCP1−/− mouse
We crossed C57Bl/6J SCD3flox/flox mice with the eIIa-cre+/− mouse to generate mice with global deletion of SCD3. Once germline deletion of SCD3 was established, the eIIa cre gene was bred out to eliminate any potential off-target effects of cre recombinase. After several years of breeding the SCD3−/− and SCD3flox/flox lines independently, we backcrossed the SCD3−/− and SCD3flox/flox lines in order to generate littermate controls. Phenotypic analyses revealed that the SCD3−/− mouse line harbored a second mutation. We identified a 39-nucleotide deletion in the transcript of Ucp1 that occurred at the beginning of exon 5 (Fig. 1A). qPCR analysis of exon 5 of Ucp1 mRNA confirmed deletion of the cDNA region in all mutant animals (Fig. 1A and supplemental Fig. S1B). We sequenced the Ucp1 gene and identified that genomic Ucp1 of mutant mice contains a c.4858C>A SNP at nucleotide 38 of exon 5 of Ucp1 (Fig. 1B). Genomic (g)DNA of this region amplified in mutant mice, but was modestly decreased, signifying imperfections in the primer annealing and confirming the SNP (supplemental Fig. S1C). After mutation identification, the Scd3 and Ucp1 mutations were separated; all UCP1+ or UCP1− animals used in this study contain two functional copies of Scd3.
Fig. 1.

Ucp1 SNP creates an alternative splice site and yields loss of UCP1 protein and thermogenic function. A 39-nucleotide deletion in the Ucp1 transcript (A) and c.4858C>An SNP in the Ucp1 gene (B) were detected using Sanger sequencing. C: An SNP in exon 5 creates an alternative splice site. Abundance of Ucp1 transcript regions in BAT (D) and inguinal WAT (E) in WT and UCP1−/− mice (n = 3-6/group). F: Immunoblot analysis of WT and Ucp1mt/mt (herein called UCP1−/−) demonstrated that Ucp1 SNP causes loss of UCP1 protein. G: Body temperature measurements of UCP1+/− and UCP1−/− mice after exposure to 4°C (n = 6-9/group). Data are expressed as mean ± SEM. *P < 0.05 vs. WT.
The nature of the mutation and the consequent gDNA:cDNA discordance classify this as a splicing mutation, in which an alternative 3′ splice site (AG) is created and the Ucp1 intronic region between exon 4 and 5 is extended (Fig. 1C). Although mutant mice contain defects in exon 5, UTRs and other exons of the Ucp1 transcript were not disrupted (Fig. 1D). The splicing defect was also present in inguinal WAT; all regions of Ucp1 transcript were dramatically elevated except exon 5 (Fig. 1E). The transcriptional activation of Ucp1 in inguinal fat is likely a compensatory attempt. The cDNA deletion did not disrupt the reading frame; however, it caused deletion of 13 amino acids in a proposed helical region that likely caused protein instability (supplemental Fig. S1D) (34). Consequently, UCP1 protein was not present in BAT or inguinal WAT, likely due to decreased protein stability (Fig. 1F, supplemental Fig. S1E). Further underscoring the severity of the mutation on UCP1 function, the deleted cDNA region encodes for D209 and D210, which are required for 80% and 20% of proton transport activity, respectively (35). We identified the epitope and confirmed that the antibody targeted a region of UCP1 encoded by exon 3, an intact region of the Ucp1 transcript in UCP1−/− mice (Fig. 1D, supplemental Fig. S1F). Herein, our UCP1 SNP mice will be called UCP1−/− mice. To further verify that this SNP yields a UCP1−/− mouse comparable to canonical UCP1−/− mice used by other investigators, we subjected UCP1−/− mice to cold and confirmed that these UCP1−/− mice are sensitive to acute cold exposure (Fig. 1G). Consistent with initial reports, the time for body temperature to drop 10°C was variable but 50% of mutant animals dropped in 4–5 h (14, 36).
UCP1−/− mice exhibit altered white adipose tissue distribution and increased inguinal DNL
Mice lacking UCP1 had significantly more inguinal fat mass (a depot of subcutaneous WAT) and modestly less epididymal fat than WT mice at standard housing temperature (Fig. 2A). Body weights were not significantly elevated in mice lacking UCP1 compared with WT mice (supplemental Fig. S2A). Similar results were observed in female mice (data not shown). This predisposition toward inguinal fat storage was also observed in UCP1−/− mice obtained from Jackson Laboratory [which will hereinafter be called UCP1(J)−/− mice] (supplemental Fig. S2B). We investigated alterations in FA metabolism in both depots that could increase inguinal fat mass but spare epididymal fat. We observed significant increases in genes encoding FA synthesis transcription factors Srebp-1c and Chrebp and enzymes Acl, Acc, and Fas in inguinal WAT (Fig. 2B). Notably, Chrepb-β, a transcriptional driver of WAT lipogenesis, was elevated 13-fold. ACC and FAS were elevated at the protein level, but we did not detect a change in the phosphorylation status of ACC in inguinal fat (Fig. 2C). Expression of some lipogenic genes was significantly elevated in epididymal WAT but not to the same degree as in inguinal WAT (Fig. 2B). To follow up on alterations in transcript and protein abundance, we measured the in vivo rates of DNL by assaying incorporation of water-derived tritium into FAs (Fig. 2D). Inguinal fat and epididymal fat lipogenesis was elevated 20-fold and 2-fold, respectively. The strong differential lipogenic response between depots suggested that other differences, such as substrate availability, compounded the transcriptional changes and contributed to lipogenic rates. As such, we measured abundance of glucose transporters and determined that Glut4 gene expression is elevated 4-fold in inguinal WAT but not elevated in epididymal WAT (Fig. 2E). Immunoblotting illustrated a robust increase in protein abundance of the glucose transporter (Fig. 2F).
Fig. 2.

Ucp1 deletion causes inguinal WAT expansion and DNL. Twelve-week old chow-fed male mice were fasted 4 h prior to euthanasia. A: Weights of epididymal and inguinal WAT of UCP1−/− mice (n = 8–12/group). B: Expression of genes involved in FA synthesis in WAT (n = 6/group). C: Immunoblotting of lipogenic enzymes in inguinal WAT. D: In vivo rates of DNL in white adipose tissue depots. Twelve-week-old old male mice were fasted 2 h and 50 mCi of 3H2O was injected intraperitoneally. Mice were euthanized 2 h after injection (n = 3–7/group). E: Gene expression of glucose transporters in WAT depots (n = 6/group). F: Immunoblotting of GLUT4 in inguinal WAT of WT and UCP1−/− mice. G: WAT sections from WT and UCP1−/− were paraffin-embedded, sectioned, stained with H&E, and imaged with 40× magnification. Scale bar represents 50 µm. H: Average adipocyte size in WAT depots (n = 3/group) as measured by Adiposoft (33). I: Total PL content per milligram WAT as measured by TLC-GC (n = 6/group). Data are expressed as mean ± SEM. *P < 0.05 vs. WT.
To determine the nature of inguinal depot expansion, we assessed adipose tissue morphometrics with H&E staining (Fig. 2G). Inguinal adipocyte size was not elevated in UCP1−/− mice, indicating that the increase in depot size stems from adipocyte hyperplasia and not hypertrophy (Fig. 2I). H&E staining of WAT also illustrated that inguinal fat of UCP1−/− mice contained adipocytes with multilocular lipid droplets (Fig. 2G) (32, 36). Consistent with the function of PLs as membrane lipids encapsulating adipocytes and the lipid droplets within, we observed a significant increase in total PL content/mg inguinal WAT, pointing to one specific fate of the de novo synthesized FAs (Fig. 2I).
Loss of UCP1 increases adipose FA desaturation
Because FA desaturation is a critical component of DNL, we next examined the FA composition of white adipose tissue of UCP1−/− mice to further understand the aberrations in adipose lipid metabolism (supplemental Table S1, S2). Inguinal WAT TGs contained increased MUFAs, specifically 16:1n7 and 16:1n10, and decreased saturated FAs, 16:0 and 18:0. We depicted these alterations as relative Δ9 and Δ6 desaturation indices (Fig. 3A, B). Changes in other FAs were variable across adipose tissue depots (supplemental Tables S1, S2), and changes in FA desaturation indices in the PL fraction were weaker than in the TG fraction (supplemental Fig. S3A). FA composition of plasma FFA reflected that of inguinal WAT and contained increased MUFA (Fig. 3C, supplemental Table S3). Concomitantly, we found increased Δ9 and Δ6 desaturase gene expression and protein levels in inguinal fat; notably, Scd3 was dramatically upregulated over 100-fold in inguinal WAT (Fig. 3D, E). We confirmed that Scd3 transcript levels were very high in inguinal WAT of UCP1(J)−/− mice and that the induction of Scd3 was not an artifact of the SCD3F/F;UCP1−/− model (supplemental Fig. S3B). Similar to reports that cold upregulates FA elongation (37, 38), we also observed significant increases in transcript levels of elongase enzymes in WAT of UCP1−/− mice and a 4-fold increase in 20:0/18:0 (supplemental Fig. S3C, S3D).
Fig. 3.

UCP1 deletion increases adipose SCD and D6D gene expression and products. Indices of desaturation in the TG fraction of inguinal WAT (A) and epididymal WAT (B) and in the plasma FFA fraction (C) were determined using TLC-GC (n = 6/group). D: Transcript levels of Δ9 desaturase isoforms (Scd1, 2, and 3) and Δ6 desaturase in WAT as measured by qPCR (n = 6/group). E: Immunoblotting of SCD1 and SCD2 in inguinal WAT. Data are expressed as mean ± SEM. *P < 0.05 vs. WT.
Loss of UCP1 promotes mitochondrial biogenesis and beiging in inguinal WAT
Inguinal fat exhibited a marked induction of FA synthesis, but the inguinal depot of UCP1−/− mice also resembled fat of cold-exposed, oxidative adipose tissue. We next probed a potential futile cycle and assessed mitochondrial biogenesis and beiging in WAT. Transcriptional regulators, Pgc1α, Pparα, and Prdm16, and FA oxidation genes, Cpt1β and Lcad, were markedly upregulated in inguinal WAT but not in epididymal WAT (Fig. 4A). Further, expression of mitochondrial OxPhos genes was significantly elevated in inguinal WAT but not epididymal WAT (Fig. 4B). Consistently, mitochondrial DNA content was increased in inguinal WAT but not epididymal WAT (Fig. 4C). VDAC, the mitochondrial marker voltage-dependent anion channel, and OxPhos subunits were elevated in inguinal WAT (Fig. 4D). The increase in FA oxidation and mitochondrial OxPhos genes is consistent with previous reports that inguinal WAT of UCP1−/− mice exhibits increased O2 consumption (14). We next probed expression of genes implicated in beiging; we detected a dramatic induction of Cidea and Dio2 in inguinal WAT but, surprisingly, did not identify increased transcript levels of the beige cell markers Tmem26, Cd137, and Tbx1 in inguinal fat of UCP1−/− mice (Fig. 4E) (39). After acute cold challenge, employed to better reveal beiging capacity, mitochondrial OxPhos and FA oxidation gene expression as well as mitochondrial OxPhos and VDAC abundance were robustly elevated in UCP1−/− mice compared with heterozygote controls, but beige cell markers remained unchanged (supplemental Fig. S3A, S3B).
Fig. 4.

UCP1 deficiency causes mitochondrial biogenesis in inguinal fat but not epididymal fat. WT and UCP1−/− mice were housed at room temperature. A: Expression of genes encoding transcriptional regulators and FA oxidation enzymes in WAT depots (n = 6/group). B: Gene expression of nuclear-encoded mitochondrial genes (n = 6/group). C: Mitochondrial (mt)DNA content in WAT depots as measured by qPCR. DNA was isolated from WAT and Nd1 abundance was normalized to H19. (n = 5–6/group) D: Immunoblotting of OxPhos proteins and VDAC in inguinal WAT of room temperature-housed WT and UCP1−/− mice. E: Expression of genes associated with beige adipocytes (n = 5–6/group). Data are expressed as mean ± SEM. *P < 0.05 vs. WT.
UCP1 deficiency promotes inguinal WAT lipolysis
Because we observed robust increases in FA oxidation genes, we sought to define other elements of lipid catabolism and examined WAT lipolysis. By measuring glycerol release from fat explants, we observed a 1.75-fold increase in inguinal lipolysis (Fig. 5A). Consistent with increased WAT lipolysis, we found increased total circulating nonesterified FAs (Fig. 5B). Of note, circulating free palmitoleate was elevated over 3.5-fold in plasma of UCP1−/− mice (supplemental Table S3). We detected a significant increase in expression of Atgl transcript and protein levels in inguinal fat of UCP1−/− mice (Fig. 5C, D). Surprisingly, HSL phosphorylation, known to be induced by cold exposure and β-adrenergic signaling, was not increased (Fig. 5D).
Fig. 5.

Loss of UCP1 increases lipolysis in inguinal WAT. A: Rates of lipolysis in 4 h fasted mice as measured by free glycerol release from fat explants (n = 3–7/group). B: Levels of circulating NEFA, as measured by TLC-GC (n = 4–5/group). C: Expression of Atgl in WAT (n = 6/group). D: Immunoblotting and densitometry of inguinal WAT lipolytic proteins, ATGL and HSL, and phospho-HSL. Data are expressed as mean ± SEM. *P < 0.05.
Deletion of UCP1 causes hepatic lipid accumulation
In light of the changes in adipose tissue lipolysis and known crosstalk among metabolic organs, we hypothesized that hepatic lipid metabolism would also be affected by loss of UCP1. We examined the livers of WT and UCP1−/− mice and saw that KO animals exhibited a two-fold increase in total hepatic TG content compared with WT mice under chow feeding (Fig. 6A, supplemental Table S4). We confirmed that this liver phenotype was present in the canonical UCP1(J)−/− mice (supplemental Fig. S5). UCP1−/− mice also had increased liver TG levels after 10-day high-sucrose feeding, a dietary regimen known to highlight changes in lipid accumulation, and Oil Red O staining illustrated the increased hepatic lipid deposition in UCP1−/− mice (Fig. 6B, C). Before declaring lipolysis the mechanism underlying the observed hepatic TG accumulation, we considered more conventional nodes of liver lipid homeostasis, and we sought to identify transcriptional or posttranscriptional changes that could drive increased hepatic FA synthesis. Transcript levels encoding lipogenic transcription factors, Srebp1, Chrebp, and Lxr, and lipogenic and FA oxidation enzymes, Acc, Fas, Cpt1a, and Lcad, were not increased in liver (Fig. 6D). We next measured protein levels of ACC, pSer79 ACC, and FAS but did not identify alterations that would facilitate increased hepatic DNL (Fig. 6E). Furthermore, in the high-sucrose-fed mice, lipid accumulation was paired with a decrease in lipogenic genes and an increase in FA oxidation genes, further discrediting DNL and FA oxidation as players in the liver phenotype of UCP1−/− mice (Fig. 6F). When measuring in vivo rates of DNL, we saw that livers of chow-fed UCP1−/− mice accumulated more de novo synthesized FAs than WT mice (Fig. 6G). However, considering the observed increase in inguinal lipolysis, the in vivo nature of the DNL experiment, and the lack of evidence for hepatic DNL, the de novo synthesized FAs are likely imported products and not liver-derived.
Fig. 6.

UCP1−/− mice exhibit increased hepatic lipid accumulation. Hepatic TG content of 12-week-old chow-fed (A) and 10-day high-sucrose-fed (B) WT and UCP1−/− mice as measured by a biochemical enzymatic assay (n = 4–8/group). C: Livers from 10-day HSVLF-fed WT and UCP1−/− were frozen in OCT, sectioned, stained with Oil Red O, and imaged with 10× magnification. Gene expression (D) and protein levels (E) of key components of FA synthesis and oxidation from livers of chow-fed animals (n = 4–6/group). F: Expression of FA synthesis and oxidation genes from 10-day high-sucrose-fed animals (n = 4–7). G: In vivo rates of de novo synthesized FAs accumulating in liver. Twelve-week-old animals were fasted 2 h and euthanized 2 h after intraperitoneal injection of 50 mCi of 3H2O (n = 3–7/group). Data are expressed as mean ± SEM. *P < 0.05 vs. WT, ‡ P < 0.10 vs. WT.
To further corroborate the proposed adipose-liver axis, we examined the hepatic FA composition of WT and UCP1−/− mice (supplemental Table S4). We observed that UCP1−/− mice exhibited increased indices of desaturation in the hepatic TG, cholesteryl ester, and FFA lipid fraction (Fig. 7A). Notably, nonesterified palmitoleate was seven times higher in UCP1−/− mice than in WT mice (supplemental Table S4). However, SCD1 transcript and protein levels were significantly reduced, suggesting that hepatic FAs are not desaturated within the liver (Fig. 7B, 7C). Scd2 and D6d gene expression was not altered (Fig. 7B), and Scd3 transcript levels were in trace amounts in livers of both WT and UCP1−/− mice. Together, these experiments suggest that increased hepatic lipid accumulation in UCP1−/− mice likely stems from inguinal fat lipolysis and not from intracellular FA homeostasis.
Fig. 7.

Livers of UCP1−/− mice contain increased palmitoleate despite reduced hepatic SCD1. A: Hepatic desaturation indices from livers of chow-fed WT and UCP1−/− mice (n = 6/group). B: Transcript levels of hepatic FA desaturase genes (n = 6/group). C: Immunoblotting and densitometry of hepatic SCD1. Data are expressed as mean ± SEM. *P < 0.05 vs. WT.
Chronic cold stress underlies increased adipose FA desaturation and hepatic lipid accumulation
To determine whether alterations in adipose and hepatic lipid metabolism in UCP1−/− mice stem from cold stress, we housed adult WT and UCP1−/− mice at thermoneutrality for 4 weeks. Thermoneutral housing rescued expression of Δ9 and Δ6 desaturase genes and most of the changes in FA composition in inguinal WAT and plasma FFA (Fig. 8A, B, supplemental Tables S5, S6). Induction of lipogenic and glucose uptake gene expression in inguinal fat was reduced after housing at 30°C, and the inguinal depot size was restored to WT levels (Fig. 8C, D). Weight gain during thermoneutral housing was not different between WT and UCP1−/− mice (supplemental Fig. S6A). Expression of FA oxidation genes was quite variable in UCP1−/− mice (supplemental Fig. S6B). Indeed, H&E staining demonstrated that inguinal fat of some UCP1−/− mice still contained adipocytes with multilocular lipid droplets after thermoneutral housing, although they were less abundant than at room temperature (supplemental Fig. S6C).
Fig. 8.

Thermoneutral housing reduces inguinal fat remodeling and liver lipid accumulation. Male chow-fed animals were housed at thermoneutrality (TNH, 30°C) for 4 weeks. A: Transcript levels of Δ9 desaturase isoforms (Scd1, 2, and 3) and Δ6 desaturase (D6d) in inguinal WAT (n = 5–7/group). B: Indices of desaturation in the TG fraction of inguinal WAT and plasma (n = 5–7/group). C: Inguinal WAT lipogenic gene expression in WT and UCP1−/− mice (n = 5–7/group). D: White fat depot weights after thermoneutral housing. E: Atgl gene expression from inguinal fat (n = 5–7/group). F: Concentration of circulating nonesterified FAs in WT and UCP1−/− mice (n = 3–4/group). G: Liver TG levels as measured by a colorimetric biochemical assay (n = 5–7/group). Data are expressed as mean ± SEM. *P < 0.05 vs. WT.
We next examined how thermoneutral housing influences the observed hepatic lipid phenotype. UCP1−/− mice did not exhibit increased Atgl gene expression, total circulating FFAs, or hepatic TG content after 30°C housing, suggesting that impaired BAT thermogenesis caused the hepatic TG accumulation by eliciting the liberation of FA from WAT (Fig. 8E–G). Surprisingly, after 4 weeks of thermoneutral housing, livers of UCP1−/− mice contained less TG than those of WT mice. Collectively, by housing mice at 30°C, we confirmed that the observed adipose and liver phenotypes were driven by chronic cold stress and were not UCP1-autonomous effects.
DISCUSSION
Therapies to treat obesity are highly desired; increasing thermogenic fat may hold promise in this arena. Surprisingly, UCP1−/− mice are modestly cold intolerant but protected against high-fat diet-induced obesity (12, 36). Later studies reported that UCP1 deletion and impaired BAT thermogenesis causes a peripheral, UCP1-independent response to combat the chronic cold (13–17). It is not completely understood how UCP1 deficiency differentially perturbs lipid metabolism in various white fat depots or impacts tissue crosstalk. Here, we demonstrate that loss of UCP1 dramatically alters lipid homeostasis in inguinal white adipose tissue and liver and that these changes are driven by chronic cold stress (Fig. 9).
Fig. 9.
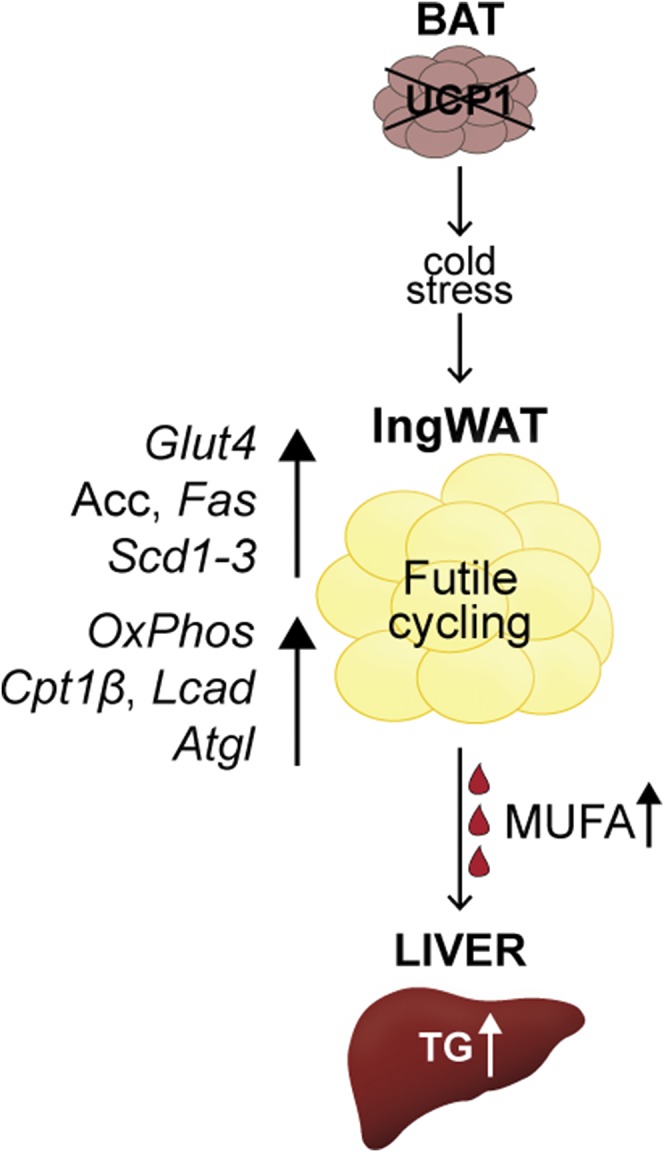
Summary of observed UCP1−/− mouse phenotypes. UCP1 deficiency elicits a global response. Inguinal WAT of UCP1−/− mice exhibit a futile cycle, in which lipid anabolism and catabolism are simultaneously upregulated. Transcriptional changes drive increased inguinal glucose uptake (Glut4), FA synthesis (Acc and Fas), and FA desaturation (Scd1–3). In addition, inguinal WAT mitochondrial content (OxPhos), FA oxidation (Cpt1β, Lcad), and lipolysis (Atgl) are elevated. Enhanced FA synthesis and desaturation paired with elevated lipolysis support elevated circulating MUFAs and hepatic TG accumulation. Alleviation of chronic cold stress, as accomplished by housing animals at thermoneutrality (30°C), restored adipose and hepatic lipid derangements to levels of WT mice.
First, we detected an unintentional splicing mutation in Ucp1 in a mouse line that causes loss of UCP1. This finding should underscore the importance of using littermate controls in experiments. The use of endogenous alternative Ucp1 splice sites in this same intron junction was recently identified in mice lacking IL33 (40). Together, these studies reveal that this intron junction is highly amenable to alternative splicing.
Next, we reported that UCP1 deficiency alters WAT distribution and supports subcutaneous fat deposition over gonadal fat deposition. Notably, inguinal fat pad growth occurs through hyperplasia, not hypertrophy. This is consistent with reports of GLUT4 overexpression in adipose tissue and the increased GLUT4 detected here (30). The introduction of “beige” adipocytes to the inguinal fat pad likely contributes to the increase in cell number. The increase in inguinal fat does not significantly increase body weight as the fat expansion is restricted to one fat depot and adiposity has minor contributions to body weight in chow-fed mice.
Supporting inguinal depot expansion and membrane biogenesis, UCP1−/− mice had increased inguinal WAT DNL. Epididymal WAT from UCP1−/− mice also exhibited increased lipogenic gene expression but to a lesser degree than in inguinal WAT and did not have elevated Glut4 gene expression. Due to differences in substrate flux and weaker activation of the lipogenic gene program, DNL was only elevated 2-fold in epididymal WAT of UCP1−/− mice compared with 20-fold in the inguinal depot. These trends were resolved with thermoneutral housing, demonstrating that lipogenesis was upregulated by chronic cold stress. Indeed, the cold-dependency of this phenotype in UCP1−/− mice is consistent with previous studies evaluating lipid response to long-term cold exposure (41, 42). GLUT4 and Chrebp-β, a transcription factor isoform known to be activated by glucose-mediated stimulation of CHREBP-α (43), were highly upregulated and further studies will shed light on the requirement of this transcription factor in cold-induced lipogenesis. Interestingly, adipose-specific loss of FAS was reported to cause beiging, suggesting that DNL is not required for cold adaptation (44, 45). We surmise, however, that FA production supports thermogenesis by creating substrates for thermogenesis and by providing the building blocks for membrane biogenesis. Further, our studies show that in UCP1−/− mice, increased DNL supports inguinal adipocyte hyperplasia and depot expansion, perhaps because in the context of UCP1 deficiency, increased cold-induced DNL is not met by increased mitochondrial uncoupling and substrate utilization. It is possible that the greater induction in DNL in inguinal fat compared with epididymal fat is partially because inguinal fat is more susceptible to β-adrenergic signaling. Although the quantitative contribution of DNL to obesity in humans is controversial (46, 47), several reports indicate that adipose DNL can impact human metabolic health (48). Specifically, WAT DNL and GLUT4 in humans correlate with CHREBP-β and inversely correlate with insulin resistance and liver steatosis (49).
Consistent with increased ChREBP and DNL, we observed a robust upregulation of Δ9 and Δ6 desaturase genes in inguinal WAT. Thermoneutral housing eliminates this upregulation, demonstrating that chronic cold stress underlies the FA composition changes. All adipose tissue SCD isoforms were upregulated at the level of transcription, and palmitoleate was particularly high in circulation. The function of increasing MUFA or decreasing saturated FA in beiging is unclear but intriguing. Increasing desaturation should increase membrane fluidity, and cold-induced upregulation of Δ9 desaturases has been observed in fish upon cold exposure (50). However, we primarily see upregulation of MUFA in TGs, and less so in PLs, the lipid class to which fluidity is most relevant. Alternatively, SCD may be upregulated to support preadipocyte proliferation or differentiation, as SCD1 and SCD2 are important for these cellular processes (51, 52). Lipidomic analyses describing how the MUFA partition to different lipid species or different organelles may provide clues into the relationship between FA desaturation and cold. Additionally, the dramatic induction of Scd3 is particularly intriguing. The physiological functions of SCD3 are poorly understood, but our group previously established that SCD3 produces primarily palmitoleate and, via expression in sebaceous glands like skin, harderian and preputial gland, supports the unique lipogenic demands of mice (53–55). Further studies are warranted to identify the role of Scd3 in the cold response, and characterization of mice lacking both UCP1 and SCD3 will improve our understanding of SCD3 in the WAT and liver phenotypes presented in this study.
Next, we used this FA signature to track FA trafficking from WAT to the liver and reveal an adipose-liver axis. We reported that UCP1−/− mice have increased hepatic lipid content compared with WT mice at standard housing conditions. Increased inguinal WAT lipolysis likely drives this lipid accumulation, and alleviation of chronic cold stress, as accomplished by thermoneutral housing, ablates the increase in Atgl gene expression and liver lipid excess. We postulated that WAT and BAT thermogenesis is the intended recipient of the liberated FAs, but without UCP1, the excess lipid is deposited in the liver. Interestingly, ectopic expression of UCP1 in white adipose tissue was reported to increase lipolysis in a cell-autonomous fashion (56). In this study, we presented lipolysis regulated peripherally and with cold-induced ATGL induction. Palmitoleate has been reported to increase Atgl gene expression through PPARα in white adipocytes (57). As described above, UCP1 deficiency increases SCD expression and 16:1n7 in adipose tissue, and these FA composition changes may help drive the transcriptional upregulation of the lipolytic gene. Cold-induced transcriptional activation of Pgc1α and Pparα likely also contribute to this upregulation.
We also used UCP1−/− mice to determine which components of inguinal WAT beiging require uncoupling itself. We reported that mice lacking UCP1 exhibit nearly all beiging elements, including the presence of multilocular lipid droplets and upregulation of mitochondrial OxPhos and FA oxidation genes in inguinal WAT. However, we did not detect increases in beige cell markers, Tmem26, Cd137, or Tbx1, at room temperature or after acute cold exposure, showing that uncoupling activity is likely required to upregulate these gene markers. It should be noted that age plays a role in beiging and the perceived phenotype of UCP1−/− mice. Most studies characterizing UCP1−/− mice have used young animals (3–4 months old), but Kontani et al. (58) reported late-onset obesity in high-fat diet-fed UCP1−/− mice at standard housing temperatures, which may be a consequence of decreased beiging capacity with age (59–61). Further, whereas numerous studies demonstrate thermogenic fat in humans, the relative abundance and thermogenic capacity of brown vs. beige adipocytes differs between humans and rodents and must be considered when translating UCP1−/− phenotypes to human therapeutics (1, 62).
Collectively, we demonstrated that UCP1 deficiency promotes inguinal WAT lipogenesis and adipocyte hyperplasia and provokes extensive tissue crosstalk. FA composition and trafficking assays revealed significant transport of adipose-derived MUFA to the liver. Our studies further elucidate the nuanced phenotype of UCP1−/− mice and illustrate the tissue crosstalk and perturbations in systemic lipid homeostasis that result from impaired BAT thermogenesis.
Supplementary Material
Acknowledgments
The authors thank Maggie Burhans and Sabrina Dumas for critical review of the manuscript. Histology was conducted at the University of Wisconsin (UW) School of Veterinary Medicine Histology Laboratory and the UW Carbone Cancer Center Experimental Pathology Laboratory. Sequencing was performed by the UW Biotechnology Center.
Footnotes
Abbreviations:
- ATGL
- adipose triglyceride lipase
- BAT
- brown adipose tissue
- DNL
- de novo lipogenesis
- HSVLF
- high-sucrose very low-fat
- PL
- phospholipid
- qPCR
- quantitative reverse-transcriptase PCR
- SCD
- stearoyl-CoA desaturase
- TG
- triglyceride
- UCP1
- uncoupling protein-1
- VDAC
- voltage-dependent anion channel
- WAT
- white adipose tissue
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (NIH) Grant R01 DK062388, American Diabetes Association 7-13-BS-118, and US Department of Agriculture Hatch W2005 (to J.M.N.). L.M.B. was supported by the National Institutes of Health National Research Service Award T32 GM07215. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
The online version of this article (available at http://www.jlr.org) contains a supplement.
REFERENCES
- 1.Sidossis L., and Kajimura S.. 2015. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. J. Clin. Invest. 125: 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grundlingh J., Dargan P. I., El-Zanfaly M., and Wood D. M.. 2011. 2,4-dinitrophenol (DNP): a weight loss agent with significant acute toxicity and risk of death. J. Med. Toxicol. 7: 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Connolly H. M., Crary J. L., McGoon M. D., Hensrud D. D., Edwards B. S., Edwards W. D., and Schaff H. V.. 1997. Valvular heart disease associated with fenfluramine-phentermine. N. Engl. J. Med. 337: 581–588. [DOI] [PubMed] [Google Scholar]
- 4.Ost M., Keipert S., and Klaus S.. 2017. Targeted mitochondrial uncoupling beyond UCP1 - The fine line between death and metabolic health. Biochimie. 134: 77–85. [DOI] [PubMed] [Google Scholar]
- 5.Cypess A. M., Lehman S., Williams G., Tal I., Rodman D., Goldfine A. B., Kuo F. C., Palmer E. L., Tseng Y. H., Doria A., et al. . 2009. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 360: 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saito M., Okamatsu-Ogura Y., Matsushita M., Watanabe K., Yoneshiro T., Nio-Kobayashi J., Iwanaga T., Miyagawa M., Kameya T., Nakada K., et al. . 2009. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 58: 1526–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Marken Lichtenbelt W. D., Vanhommerig J. W., Smulders N. M., Drossaerts J. M., Kemerink G. J., Bouvy N. D., Schrauwen P., and Teule G. J.. 2009. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 360: 1500–1508. [DOI] [PubMed] [Google Scholar]
- 8.Virtanen K. A., Lidell M. E., Orava J., Heglind M., Westergren R., Niemi T., Taittonen M., Laine J., Savisto N. J., Enerback S., et al. . 2009. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 360: 1518–1525. [DOI] [PubMed] [Google Scholar]
- 9.Nedergaard J., Bengtsson T., and Cannon B.. 2007. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 293: E444–E452. [DOI] [PubMed] [Google Scholar]
- 10.Golozoubova V., Hohtola E., Matthias A., Jacobsson A., Cannon B., and Nedergaard J.. 2001. Only UCP1 can mediate adaptive nonshivering thermogenesis in the cold. FASEB J. 15: 2048–2050. [DOI] [PubMed] [Google Scholar]
- 11.Hofmann W. E., Liu X., Bearden C. M., Harper M. E., and Kozak L. P.. 2001. Effects of genetic background on thermoregulation and fatty acid-induced uncoupling of mitochondria in UCP1-deficient mice. J. Biol. Chem. 276: 12460–12465. [DOI] [PubMed] [Google Scholar]
- 12.Liu X., Rossmeisl M., McClaine J., Riachi M., Harper M. E., and Kozak L. P.. 2003. Paradoxical resistance to diet-induced obesity in UCP1-deficient mice. J. Clin. Invest. 111: 399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rowland L. A., Bal N. C., Kozak L. P., and Periasamy M.. 2015. Uncoupling protein 1 and sarcolipin are required to maintain optimal thermogenesis, and loss of both systems compromises survival of mice under cold stress. J. Biol. Chem. 290: 12282–12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ukropec J., Anunciado R. P., Ravussin Y., Hulver M. W., and Kozak L. P.. 2006. UCP1-independent thermogenesis in white adipose tissue of cold-acclimated Ucp1−/− mice. J. Biol. Chem. 281: 31894–31908. [DOI] [PubMed] [Google Scholar]
- 15.Anunciado-Koza R., Ukropec J., Koza R. A., and Kozak L. P.. 2008. Inactivation of UCP1 and the glycerol phosphate cycle synergistically increases energy expenditure to resist diet-induced obesity. J. Biol. Chem. 283: 27688–27697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kazak L., Chouchani E. T., Jedrychowski M. P., Erickson B. K., Shinoda K., Cohen P., Vetrivelan R., Lu G. Z., Laznik-Bogoslavski D., Hasenfuss S. C., et al. . 2015. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell. 163: 643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kazak L., Chouchani E. T., Lu G. Z., Jedrychowski M. P., Bare C. J., Mina A. I., Kumari M., Zhang S., Vuckovic I., Laznik-Bogoslavski D., Dzeja P., Banks A. S., Rosen E. D., and Spiegelman B. M.. 2017. Genetic depletion of adipocyte creatine metabolism inhibits diet-induced thermogenesis and drives obesity. Cell Metab 26: 660–671 e663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feldmann H. M., Golozoubova V., Cannon B., and Nedergaard J.. 2009. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9: 203–209. [DOI] [PubMed] [Google Scholar]
- 19.Strable M. S., and Ntambi J. M.. 2010. Genetic control of de novo lipogenesis: role in diet-induced obesity. Crit. Rev. Biochem. Mol. Biol. 45: 199–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paton C. M., and Ntambi J. M.. 2009. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol. Endocrinol. Metab. 297: E28–E37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakravarthy M. V., Lodhi I. J., Yin L., Malapaka R. R., Xu H. E., Turk J., and Semenkovich C. F.. 2009. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell. 138: 476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu S., Brown J. D., Stanya K. J., Homan E., Leidl M., Inouye K., Bhargava P., Gangl M. R., Dai L., Hatano B., et al. . 2013. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature. 502: 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yore M. M., Syed I., Moraes-Vieira P. M., Zhang T., Herman M. A., Homan E. A., Patel R. T., Lee J., Chen S., Peroni O. D., et al. . 2014. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell. 159: 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burhans M. S., Flowers M. T., Harrington K. R., Bond L. M., Guo C. A., Anderson R. M., and Ntambi J. M.. 2015. Hepatic oleate regulates adipose tissue lipogenesis and fatty acid oxidation. J. Lipid Res. 56: 304–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao H., Gerhold K., Mayers J. R., Wiest M. M., Watkins S. M., and Hotamisligil G. S.. 2008. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 134: 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo X., Li H., Xu H., Halim V., Zhang W., Wang H., Ong K. T., Woo S. L., Walzem R. L., Mashek D. G., et al. . 2012. Palmitoleate induces hepatic steatosis but suppresses liver inflammatory response in mice. PLoS One. 7: e39286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hudgins L. C., Parker T. S., Levine D. M., and Hellerstein M. K.. 2011. A dual sugar challenge test for lipogenic sensitivity to dietary fructose. J. Clin. Endocrinol. Metab. 96: 861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Postic C., and Girard J.. 2008. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J. Clin. Invest. 118: 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abel E. D., Peroni O., Kim J. K., Kim Y. B., Boss O., Hadro E., Minnemann T., Shulman G. I., and Kahn B. B.. 2001. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 409: 729–733. [DOI] [PubMed] [Google Scholar]
- 30.Shepherd P. R., Gnudi L., Tozzo E., Yang H., Leach F., and Kahn B. B.. 1993. Adipose cell hyperplasia and enhanced glucose disposal in transgenic mice overexpressing GLUT4 selectively in adipose tissue. J. Biol. Chem. 268: 22243–22246. [PubMed] [Google Scholar]
- 31.Keipert S., Kutschke M., Lamp D., Brachthauser L., Neff F., Meyer C. W., Oelkrug R., Kharitonenkov A., and Jastroch M.. 2015. Genetic disruption of uncoupling protein 1 in mice renders brown adipose tissue a significant source of FGF21 secretion. Mol. Metab. 4: 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keipert S., Kutschke M., Ost M., Schwarzmayr T., van Schothorst E. M., Lamp D., Brachthauser L., Hamp I., Mazibuko S. E., Hartwig S., Lehr S., Graf E., Plettenburg O., Neff F., Tschop M. H., and Jastroch M.. 2017. Long-term cold adaptation does not require FGF21 or UCP1. Cell Metab 26: 437–446 e435. [DOI] [PubMed] [Google Scholar]
- 33.Galarraga M., Campion J., Munoz-Barrutia A., Boque N., Moreno H., Martinez J. A., Milagro F., and Ortiz-de-Solorzano C.. 2012. Adiposoft: automated software for the analysis of white adipose tissue cellularity in histological sections. J. Lipid Res. 53: 2791–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klingenberg M., Echtay K. S., Bienengraeber M., Winkler E., and Huang S. G.. 1999. Structure-function relationship in UCP1. Int. J. Obes. Relat. Metab. Disord. 23(Suppl 6): S24–S29. [DOI] [PubMed] [Google Scholar]
- 35.Echtay K. S., Winkler E., Bienengraeber M., and Klingenberg M.. 2000. Site-directed mutagenesis identifies residues in uncoupling protein (UCP1) involved in three different functions. Biochemistry. 39: 3311–3317. [DOI] [PubMed] [Google Scholar]
- 36.Enerbäck S., Jacobsson A., Simpson E. M., Guerra C., Yamashita H., Harper M. E., and Kozak L. P.. 1997. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature. 387: 90–94. [DOI] [PubMed] [Google Scholar]
- 37.Tan C. Y., Virtue S., Bidault G., Dale M., Hagen R., Griffin J. L., and Vidal-Puig A.. 2015. Brown Adipose Tissue Thermogenic Capacity Is Regulated by Elovl6. Cell Reports. 13: 2039–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Westerberg R., Mansson J. E., Golozoubova V., Shabalina I. G., Backlund E. C., Tvrdik P., Retterstol K., Capecchi M. R., and Jacobsson A.. 2006. ELOVL3 is an important component for early onset of lipid recruitment in brown adipose tissue. J. Biol. Chem. 281: 4958–4968. [DOI] [PubMed] [Google Scholar]
- 39.Wu J., Bostrom P., Sparks L. M., Ye L., Choi J. H., Giang A. H., Khandekar M., Virtanen K. A., Nuutila P., Schaart G., et al. . 2012. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 150: 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Odegaard J. I., Lee M. W., Sogawa Y., Bertholet A. M., Locksley R. M., Weinberg D. E., Kirichok Y., Deo R. C., and Chawla A.. 2016. Perinatal Licensing of Thermogenesis by IL-33 and ST2. Cell. 166: 841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flachs P., Adamcova K., Zouhar P., Marques C., Janovska P., Viegas I., Jones J. G., Bardova K., Svobodova M., Hansikova J., et al. . 2017. Induction of lipogenesis in white fat during cold exposure in mice: link to lean phenotype. Int. J. Obes. (Lond.). 41: 372–380. [DOI] [PubMed] [Google Scholar]
- 42.Mottillo E. P., Balasubramanian P., Lee Y. H., Weng C., Kershaw E. E., and Granneman J. G.. 2014. Coupling of lipolysis and de novo lipogenesis in brown, beige, and white adipose tissues during chronic beta3-adrenergic receptor activation. J. Lipid Res. 55: 2276–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herman M. A., Peroni O. D., Villoria J., Schon M. R., Abumrad N. A., Bluher M., Klein S., and Kahn B. B.. 2012. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 484: 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guilherme A., Pedersen D. J., Henchey E., Henriques F. S., Danai L. V., Shen Y., Yenilmez B., Jung D., Kim J. K., Lodhi I. J., et al. . 2017. Adipocyte lipid synthesis coupled to neuronal control of thermogenic programming. Mol. Metab. 6: 781–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lodhi I. J., Yin L., Jensen-Urstad A. P., Funai K., Coleman T., Baird J. H., El Ramahi M. K., Razani B., Song H., Fu-Hsu F., et al. . 2012. Inhibiting adipose tissue lipogenesis reprograms thermogenesis and PPARgamma activation to decrease diet-induced obesity. Cell Metab. 16: 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donnelly K. L., Smith C. I., Schwarzenberg S. J., Jessurun J., Boldt M. D., and Parks E. J.. 2005. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115: 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hellerstein M. K. 2001. No common energy currency: de novo lipogenesis as the road less traveled. Am. J. Clin. Nutr. 74: 707–708. [DOI] [PubMed] [Google Scholar]
- 48.Solinas G., Boren J., and Dulloo A. G.. 2015. De novo lipogenesis in metabolic homeostasis: More friend than foe? Mol. Metab. 4: 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eissing L., Scherer T., Todter K., Knippschild U., Greve J. W., Buurman W. A., Pinnschmidt H. O., Rensen S. S., Wolf A. M., Bartelt A., et al. . 2013. De novo lipogenesis in human fat and liver is linked to ChREBP-beta and metabolic health. Nat. Commun. 4: 1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tiku P. E., Gracey A. Y., Macartney A. I., Beynon R. J., and Cossins A. R.. 1996. Cold-induced expression of delta 9-desaturase in carp by transcriptional and posttranslational mechanisms. Science. 271: 815–818. [DOI] [PubMed] [Google Scholar]
- 51.Christianson J. L., Nicoloro S., Straubhaar J., and Czech M. P.. 2008. Stearoyl-CoA desaturase 2 is required for peroxisome proliferator-activated receptor gamma expression and adipogenesis in cultured 3T3–L1 cells. J. Biol. Chem. 283: 2906–2916. [DOI] [PubMed] [Google Scholar]
- 52.Igal R. A. 2016. Stearoyl CoA desaturase-1: New insights into a central regulator of cancer metabolism. Biochim. Biophys. Acta. 1861: 1865–1880. [DOI] [PubMed] [Google Scholar]
- 53.Miyazaki M., Bruggink S. M., and Ntambi J. M.. 2006. Identification of mouse palmitoyl-coenzyme A Delta9-desaturase. J. Lipid Res. 47: 700–704. [DOI] [PubMed] [Google Scholar]
- 54.Miyazaki M., Gomez F. E., and Ntambi J. M.. 2002. Lack of stearoyl-CoA desaturase-1 function induces a palmitoyl-CoA Delta6 desaturase and represses the stearoyl-CoA desaturase-3 gene in the preputial glands of the mouse. J. Lipid Res. 43: 2146–2154. [DOI] [PubMed] [Google Scholar]
- 55.Zheng Y., Prouty S. M., Harmon A., Sundberg J. P., Stenn K. S., and Parimoo S.. 2001. Scd3–a novel gene of the stearoyl-CoA desaturase family with restricted expression in skin. Genomics. 71: 182–191. [DOI] [PubMed] [Google Scholar]
- 56.Flachs P., Novotny J., Baumruk F., Bardova K., Bourova L., Miksik I., Sponarova J., Svoboda P., and Kopecky J.. 2002. Impaired noradrenaline-induced lipolysis in white fat of aP2-Ucp1 transgenic mice is associated with changes in G-protein levels. Biochem. J. 364: 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bolsoni-Lopes A., Festuccia W. T., Farias T. S., Chimin P., Torres-Leal F. L., Derogis P. B., de Andrade P. B., Miyamoto S., Lima F. B., Curi R., et al. . 2013. Palmitoleic acid (n-7) increases white adipocyte lipolysis and lipase content in a PPARalpha-dependent manner. Am. J. Physiol. Endocrinol. Metab. 305: E1093–E1102. [DOI] [PubMed] [Google Scholar]
- 58.Kontani Y., Wang Y., Kimura K., Inokuma K. I., Saito M., Suzuki-Miura T., Wang Z., Sato Y., Mori N., and Yamashita H.. 2005. UCP1 deficiency increases susceptibility to diet-induced obesity with age. Aging Cell. 4: 147–155. [DOI] [PubMed] [Google Scholar]
- 59.Berry D. C., Jiang Y., Arpke R. W., Close E. L., Uchida A., Reading D., Berglund E. D., Kyba M., and Graff J. M.. 2017. Cellular aging contributes to failure of cold-induced beige adipocyte formation in old mice and humans. Cell Metab. 25: 166–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rogers N. H., Landa A., Park S., and Smith R. G.. 2012. Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose tissue. Aging Cell. 11: 1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoneshiro T., Aita S., Matsushita M., Okamatsu-Ogura Y., Kameya T., Kawai Y., Miyagawa M., Tsujisaki M., and Saito M.. 2011. Age-related decrease in cold-activated brown adipose tissue and accumulation of body fat in healthy humans. Obesity (Silver Spring). 19: 1755–1760. [DOI] [PubMed] [Google Scholar]
- 62.Bartelt A., and Heeren J.. 2014. Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 10: 24–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.