

Abstract
Background
We intended to evaluate diagnostic utility of a targeted gene sequencing by using next generation sequencing (NGS) panel in patients with intractable early-onset epilepsy (EOE) and find the efficient analytical step for increasing the diagnosis rate.
Methods
We assessed 74 patients with EOE whose seizures started before 3 years of age using a customized NGS panel that included 172 genes. Single nucleotide variants (SNVs) and exonic and chromosomal copy number variations (CNVs) were intensively examined with our customized pipeline and crosschecked with commercial or pre-built software. Variants were filtered and prioritized by in-depth clinical review, and finally classified according to the American College of Medical Genetics and Genomics guidelines. Each case was further discussed in a monthly consensus meeting that included the participation of all laboratory personnel, bioinformaticians, geneticists, and clinicians.
Results
The NGS panel identified 28 patients (37.8%) with genetic abnormalities; 25 patients had pathogenic or likely pathogenic SNVs in 17 genes including SXTBP1 (n = 3), CDKL5 (n = 2), KCNQ2 (n = 2), SCN1A (n = 2), SYNGAP1 (n = 2), GNAO1 (n = 2), KCNT1 (n = 2), BRAT1, WWOX, ZEB2, CHD2, PRICKLE2, COL4A1, DNM1, SCN8A, MECP2, SLC9A6 (n = 1). The other 3 patients had pathogenic CNVs (2 duplications and 1 deletion) with varying sizes (from 2.5 Mb to 12 Mb). The overall diagnostic yield was 37.8% after following our step-by-step approach for clinical consensus.
Conclusions
NGS is a useful diagnostic tool with great utility for patients with EOE. Diagnostic yields can be maximized with a standardized and team-based approach.
Electronic supplementary material
The online version of this article (10.1186/s12920-018-0320-7) contains supplementary material, which is available to authorized users.
Keywords: Diagnostic yield, Early-onset epilepsy, Next-generation sequencing
Background
Early-onset epilepsy (EOE) in infancy or early childhood is often a devastating form of epilepsy that is associated with severe cognitive impairment. Etiologies remain unidentified despite comprehensive structural, metabolic, immunologic, infection, and genetic investigations due to their heterogeneity [1]. Recently, several studies demonstrated the usefulness of targeted next-generation sequencing (NGS) for diagnosing EOE using a genetic approach [2–6]. Previous studies have reported increased diagnostic yields with NGS panels compared to those obtained with other genetic tests [2, 7]. NGS strategies have revolutionized epilepsy genomic research by increasing the detection of de novo variants using targeted sequencing and whole-exome sequencing (WES).
Nevertheless, clinical implications of diagnostic NGS panel for EOE are currently limited. Previous studies focused on identifying the efficacy of NGS in large, multicenter patient cohorts. Limited clinical descriptions of patients were provided. Clinical questions regarding the identification of patients who are most likely to show positive genetic results, and strategies to increase the diagnostic yields of NGS panel still remain to be explored.
Here, we used a customized NGS panel to clinically and genetically investigate a highly selected group of patients who developed EOE in infancy or early childhood (≤3 years of age). All analytical steps were performed within our center according to a systematized diagnostic pipeline. Our goal was to optimize the clinical use of NGS panel for EOE by finding the most important analytical step that determines the diagnosis rate and identifying patients who are most likely to show genetic abnormalities.
Methods
Patients and clinical information
A total of 74 unrelated pediatric patients with EOE without a known cause were recruited from the epilepsy clinic in Severance Children’s Hospital from March 2015 to May 2016. As a nationwide referral center for EOE, patient population generally includes severe epilepsy patients with unknown causes. All patients met the following criteria: (1) seizure onset before the age of 3 years; (2) multiple epileptiform discharges with severely disorganized background activity on electroencephalography (EEG); (3) diagnosed with drug-resistant epilepsy and progressive developmental delay or with a known epileptic encephalopathy syndrome; (4) no structural lesion detected with brain magnetic resonance imaging (MRI); (5) no metabolic abnormalities; (6) no abnormalities detected with previous genetic tests; and (7) offspring of asymptomatic Korean parents. For the diagnosis of specific epilepsy syndrome, patients were classified according to the 2010 International League Against Epilepsy classification [8] and previous diagnostic criteria.
All EEGs were reviewed by one of four pediatric epileptologists (S.H.K, H.K, J.S.L, and H.D.K). All EEG recordings were obtained according to the 10–20 international system, using a 21-channel digital EEG system (Xltek, Natus Medical Incorporated, San Carlos, CA, USA). Data were recorded using a sampling rate of 200 Hz with filter settings of 1–70 Hz. Brain MRI was performed according to standard epilepsy protocols, including oblique coronal view on T2-weighted and fluid-attenuated inversion recovery at 1.5−3.0 Tesla. Investigations for metabolic disorders included plasma amino acid analysis, urine organic acid analysis, homocysteine level, acylcarnitine profile (total and free carnitine levels), blood gas analysis, and serum lactate, pyruvate, and ammonia. Previous genetic tests in each patient varied but usually included chromosomal analysis, Sanger sequencing for specific genes such as MECP2, and multiplex ligation-dependent probe amplification (MLPA) using the SALSA MLPA P245 Microdeletion Syndrome kit (MRC Holland, Amsterdam, The Netherlands), which were mainly performed before referral from various other hospital to Severance Children’s Hospital.
For clinical analysis, previous seizure history was reviewed in detail for the age of seizure onset, seizure types, history of neonatal seizures, history of status epilepticus, and febrile seizures. Information regarding admission to the neonatal intensive care unit (NICU), developmental state, and premature deaths were collected. This study was approved by the Institutional Review Board of Severance Hospital (IRB 4–2016-0080). Informed consent was obtained from patients or their parents.
Targeted gene sequencing using NGS panel
For the customized NGS panel, we selected 172 candidate genes implicated in EOE based on an extensive literature review and the Online Mendelian Inheritance in Man (OMIM) database (http://www.ncbi.nlm.nih.gov/omim) (listed in online Additional file 1: Table S1). These genes include genes related to not only EOE, but also epilepsies of inborn errors of metabolism and conditions that are indications or contraindications of the ketogenic diet.
Genomic DNAs were extracted from leukocytes of whole blood samples using the QIAamp Blood DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Intact DNA was quantified and adjusted to a concentration of 5 ng/μL using a Qubit 2.0 Fluorometer (Invitrogen, Waltham, MA, USA) and the Qubit dsDNA HS assay kit (Invitrogen). Precapture libraries were constructed according to the manufacturer’s sample preparation protocol. Each patient’s genomic DNA was fragmented to a median size of 300 base pair (bp). The DNA fragments were end-repaired, phosphorylated, and adenylated on the 3′ ends. The index adaptors were ligated to the repaired ends, DNA fragments were amplified, and fragments of 200−500 bp were isolated. Pooled libraries were sequenced using a MiSeq sequencer (Illumina, San Diego, CA, USA) and the MiSeq Reagent Kit v2 (300 cycles).
NGS data analysis
Data analysis was performed primarily through our custom pipeline. Briefly, raw sequence data were mapped to GRCh37 (hg19) using the Burrows-Wheeler Aligner algorithm, followed by removal of duplicate reads, realignment of insertions and deletions, base quality recalibration, and variant calling using the Genome Analysis Toolkit (GATK). Small nucleotide variants called by GATK were further crosschecked with BaseStudio software (Illumina). Every variant that was suspected to be pathogenic, likely pathogenic, or variant of unknown significance (VUS) was confirmed by visual inspection of the bam file using Integrated Genomics Viewer version 2.3 (IGV; Broad Institute, Cambridge, MA, USA). Quality metrics were calculated for each sample using the FastQC software and TEQC package.
Split-read-based detection of large insertions and deletions was conducted using the Pindel and Manta algorithms, and both results were finally crosschecked. Read-depth-based detection of structural rearrangements was conducted using the ExomeDepth software. Chromosomal copy number variations (CNVs) detected by ExomeDepth were further crosschecked using our custom pipeline; this retrieved base-level depth-of-coverage for each bam file using SAMtools software and normalized the depths against those of other samples in the same batch. We performed off-target analysis of chromosomal copy number alterations using the CopywriteR software.
The following databases were used for variant annotation: OMIM, Human Gene Mutation Database (HGMD), ClinVar, dbSNP, 1000 Genome, Exome Aggregation Consortium (ExAC), Exome Sequencing Project, and Korean Reference Genome Database (KRGDB). The pathogenicity of missense variants was predicted using five in silico prediction algorithms, including Sorting Tolerant from Intolerant (SIFT), Polymorphism Phenotyping v2 (PolyPhen-2), MutationTaster, MutationAssessor, and Functional Analysis through Hidden Markov Models (FATHMM) implemented in dbNSFP version 3.0a. Effects on splicing were predicted using the SPIDEX version 1.0 and dbscSNV version 1.1.
Annotation and interpretation of variants
Identified variants were described according to nomenclature recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen). The interpretation of variants followed the 5-tier classification system recommended by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) [9] using a step-by-step approach (Fig. 1). Briefly, ACMG guideline defined 28 criteria that address evidence such as population data, case-control analyses, functional data, computational predictions, allelic data, segregation studies, and de novo observations. We evaluated all the possible components in every patient by three-step analyses. First, variants were filtered by their frequencies in population control databases, including ExAC (non-TCGA dataset; frequencies were calculated according to ethnic subgroups), ESP6500, 1000 Genomes Project, and KRGDB. To maximize the diagnostic yield, variants with a minor allele frequency (MAF) greater than 5% in any of the population subgroups rather than conventional 1% criteria were classified as absolutely benign, whereas those that were absent from the general population were considered to have moderate evidence as pathogenic. Second, literature and database searches for previous reports and functional studies were performed using the Alamut Visual 2.6 software (Interactive Biosoftware, France) and HGMD professional database. When all in silico analyses showed consistent predictions, the results were regarded as evidence of benign or pathogenic variants.
Fig. 1.
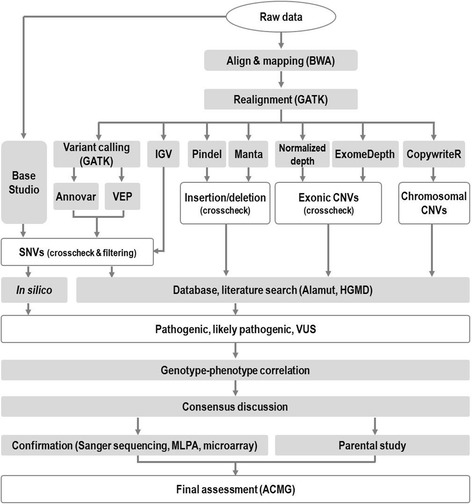
Flow chart of our bioinformatics pipeline and variant classification results with interpretation
Phenotype review and consensus discussion
The last step involved genetic specialists or laboratory physicians presenting a preliminary report to the patient’s attending physicians or pediatric epileptologists, which listed all possible pathogenic variants, likely pathogenic variants, and VUSs. The clinicians performed an in-depth review of each patient’s phenotype and gave an opinion from their point of view. Each case was further reviewed and discussed in a monthly consensus meeting that included the participation of all laboratory personnel, bioinformaticians, geneticists, and clinicians. When pathogenic or likely pathogenic variants were consistent with the patient’s phenotype, final validation using other confirmatory assays and a parental study was planned. VUSs, especially missense variants, were prioritized according to population frequency, ACMG score, and the patient’s clinical phenotype. A parental study was scheduled to detect de novo occurrence for the candidate pathogenic, likely pathogenic, and VUS variants in all available trios.
Confirmation using other methods
For small nucleotide variations, pathogenic and likely pathogenic variants as well as VUSs needing parental study were examined using Sanger sequencing on a 3730 DNA Analyzer with the BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA). Sequencing data were aligned against appropriate reference sequences and analyzed using the Sequencher 5.3 software (Gene Codes Corp., Ann Arbor, MI, USA).
Large exonic deletions and duplications were confirmed using the MLPA kit (MRC Holland). Chromosomal copy number alterations were confirmed using the Infinium CytoSNP 850 K array (Illumina) and BlueFuse Multi software (Illumina).
Statistical analysis
To compare clinical presentations among subgroups according to epilepsy syndrome type or presence of definitive genetic causes revealed by this study, Fisher’s exact test, Chi-square test, and independent t-test were utilized for comparisons of numerical and categorical variables. Bonferroni correction was utilized for multiple testing correction. Statistical analyses were computed using the PASW statistics software (version 18.0, SPSS Inc., Chicago, IL, USA). P-value <0.05 was considered statistically significant.
Results
Demographics and clinical characteristics of patients
Demographics and clinical characteristics of all patients are summarized in online Additional file 1: Table S2. Mean age of seizure onset was 7.5 ± 7.8 months (standard deviation). Seizures started before the age of 1 year in 85.1% of patients (63/74). Global developmental delay was observed in 83.8% of patients (62/74). Among them, 17 (23.0%) patients could not make eye contact. Epileptic spasms (70.3%) were the most common seizure type, followed by generalized (33.8%) and focal (21.6%) seizure types. Two patients had all three seizure types. Many patients had a remarkable perinatal history, which included admission to the NICU in 23.0% of patients (17/74) and a history of neonatal seizures in 15.0% of patients (11/74). Six (8.1%) of the 74 patients had a history of status epilepticus, and two (2.7%) had unexpected premature deaths. The causes of death were pneumonia and sudden infantile death syndrome. The epilepsy syndrome was diagnosed most commonly as infantile spasm (IS) (n = 51), followed by Dravet syndrome (n = 2), malignant migrating focal seizures in infancy (MMFI) (n = 1), and Doose syndrome (n = 1).
NGS run and quality metrics
Quality metrics of NGS runs for all patients are summarized in online Additional file 1: Table S3. On average, more than 8.7 million reads were sequenced per sample, and approximately 8.0 million reads (93.5%) were mapped on references. The average horizontal coverage, which was interpreted as percentage of regions with more than 20× coverage, was 99.8%.
Spectrum and statistics of pathogenic variants
Among the 28 patients with pathogenic or likely pathogenic variants, 25 had single nucleotide variants (SNVs) or nucleotide/exon deletions in 17 epilepsy-associated genes (Table 1), and 3 had deletion or duplication of large genomic segments (Table 2). Among the SNVs, missense variants (n = 14) were the most common type, followed by nonsense (n = 5), frameshift (n = 4), and splice-site (n = 2) variants (Table 1). Two patients (P16 and P17) were compound heterozygous for one SNV and one exon-level deletion/duplication (Table 1, Fig. 2a). The most commonly affected gene was STXBP1.
Table 1.
Results of mutation analysis in 25 patients with pathogenic or likely pathogenic mutations involving single nucleotide or several exons according to ACMG guideline
| Patient | Gene | Inheritance | NM | Nucleotide change | Amino acid change | Zygosity | ACMG classification | ACMG evidence components | Origin of variant |
|---|---|---|---|---|---|---|---|---|---|
| P1 | STXBP1 | AD | NM_003165.3 | c.733C > G | p.His245Asp | Heterozygosity | Pathogenic | PS2, PM2, PM5, PP3, PP4 | de novo |
| P2 | STXBP1 | AD | NM_003165.3 | c.874C > T | p.Arg292Cys | Heterozygosity | Pathogenic | PS1, PS2, PM2, PP4 | de novo |
| P3 | STXBP1 | AD | NM_003165.3 | c.1216C > T | p.Arg406Cys | Heterozygosity | Pathogenic | PS1, PS2, PM2, PM5, PP3, PP4 | de novo |
| P4 | CDKL5 | XD | NM_003159.2 | c.511 T > A | p.Tyr171Asn | Heterozygosity | Pathogenic | PS2, PM2, PM5, PP3, PP4 | de novo |
| P5 | CDKL5 | XD | NM_003159.2 | c.282 + 1G > A | splice site | Heterozygosity | Pathogenic | PVS1, PS2, PM2, PP4, PP5 | de novo |
| P6 | KCNQ2 | AD | NM_172107.2 | c.917C > T | p.Ala306Val | Heterozygosity | Pathogenic | PS2, PM2, PP2, PP3, PP4, PP5 | de novo |
| P7 | KCNQ2 | AD | NM_172107.2 | c.593G > A | p.Arg198Gln | Heterozygosity | Pathogenic | PS2, PM2, PP2, PP3, PP4, PP5 | de novo |
| P8 | SCN1A | AD | NM_001165963.1 | c.5068_5069delinsG | p.Ser1690AlafsTer25 | Heterozygosity | Pathogenic | PVS1, PM2, PP4 | NA |
| P9 | SCN1A | AD | NM_001165963.1 | c.1209dupT | p.Val404CysfsTer46 | Heterozygosity | Pathogenic | PVS1, PM2, PP4 | NA |
| P10 | SYNGAP1 | AD | NM_006772.2 | c.980 T > C | p.Leu327Pro | Heterozygosity | Likely pathogenic | PS2, PM2, PP3, PP4, PP5 | de novo |
| P11 | SYNGAP1 | AD | NM_006772.2 | c.1735C > T | p.Arg579Ter | Heterozygosity | Pathogenic | PVS1, PM2, PP4, PP5 | NA |
| P12 | GNAO1 | AD | NM_020988.2 | c.118G > T | p.Gly40Trp | Heterozygosity | Pathogenic | PS2, PM2, PM5, PP3, PP4 | de novo |
| P13 | GNAO1 | AD | NM_020988.2 | c.155A > C | p.Gln52Pro | Heterozygosity | Likely pathogenic | PS2, PM2, PP3, PP4 | de novo |
| P14 | KCNT1 | AD | NM_020822.2 | c.2800G > A | p.Ala934Thr | Heterozygosity | Likely pathogenic | PS1, PM2, PP4 | NA |
| P15 | KCNT1 | AD | NM_020822.2 | c.1038C > G | p.Phe346Leu | Heterozygosity | Likely pathogenic | PS2, PM2, PP2, PP4 | de novo |
| P16 | BRAT1 | AR | NM_152743.3 | c.1576C > T | p.Gln526Ter | Heterozygosity | Likely pathogenic | PVS1, PM2, PP4 | maternal inheritance |
| P16 | BRAT1 | AR | NM_152743.3 | exon 2–3 deletion | _ | Heterozygosity | Likely pathogenic | PM2, PM3, PP4, PP5 | NA |
| P17 | WWOX | AR | NM_016373.2 | c.1060C > T | p.Gln354Ter | Heterozygosity | Pathogenic | PVS1, PM2, PM3, PP4 | maternal inheritance |
| P17 | WWOX | AR | NM_016373.2 | exon 6–8 duplication | _ | Heterozygosity | Likely pathogenic | PM2, PM3, PP4, PP5 | paternal inheritance |
| P18 | ZEB2 | AD | NM_014795.3 | c.1956C > A | p.Tyr652Ter | Heterozygosity | Pathogenic | PVS1, PS1, PM2, PP4 | NA |
| P19 | CHD2 | AD | NM_001271.3 | c.1269dupA | p.Glu424ArgfsTer3 | Heterozygosity | Pathogenic | PVS1, PM2, PP4 | NA |
| P20 | PRICKLE2 | AD | NM_198859.3 | c.2129_2147del | p.Arg710LeufsTer2 | Heterozygosity | Likely pathogenic | PVS1, PM2 | NA |
| P21 | COL4A1 | AD | NM_001845.4 | c.2897G > A | p.Gly966Glu | Heterozygosity | Likely pathogenic | PS2, PM2, PP2, PP3, PP4 | de novo |
| P22 | DNM1 | AD | NM_004408.2 | c.1195A > G | p.Arg399Gly | Heterozygosity | Likely pathogenic | PS2, PM2, PP2, PP3, PP4 | de novo |
| P23 | SCN8A | AD | NM_014191.3 | c.782G > T | p.Cys261Phe | Heterozygosity | Likely pathogenic | PS2, PM2, PP2, PP3, PP4 | de novo |
| P24 | MECP2 | XD | NM_004992.3 | c.1164_1207del | p.Pro389Ter | Heterozygosity | Pathogenic | PVS1, PM1, PM2, PP4 | NA |
| P25 | SLC9A6 | XD | NM_006359.2 | c.316_325 + 28del | splice site | Heterozygosity | Pathogenic | PVS1, PS2, PM2, PP4 | de novo |
AD autosomal dominant, XD X dominant, AR autosomal recessive, VUS variant of unknown significance, PVS pathogenic very strong, PS pathogenic strong, PM pathogenic moderate, PP pathogenic supporting, NA not available
Table 2.
Results of mutation analysis in 3 patients with pathogenic or likely pathogenic copy number variations
| Patient | Affected region | Duplication/Deletion | Size | Major genes involved in the region | Additional study | Result of the additional study | Zygosity | Suspected syndrome | Phenotypic correlation |
|---|---|---|---|---|---|---|---|---|---|
| P26 | 14q11.2-q12 | Duplication | 12 Mb | FOXG1, CHD8, SUPT16H | array CGH | arr 14q11.2(20,528,528–32,297,926)×3 | Heterozygosity | Not availablea | Early onset of infantile spasms,Developmental delay |
| P27 | 15q11.2 | Deletion | 3.5 Mb | UBE3A, GABRB3, SNRPN | MLPA | Deletion of maternal allele | Heterozygosity | Angelman syndrome | Distinctive electro-encephalography patternc |
| P28 | 19p13.3 | Duplication | 2.5 Mb | Not specific epilepsy-associated genes | array CGH | arr 19p13.3(3,462,574–6,583,781)×3 | Heterozygosity | 19p13.3 microduplication syndromeb | Distinctive facial dysmorphism |
Fig. 2.
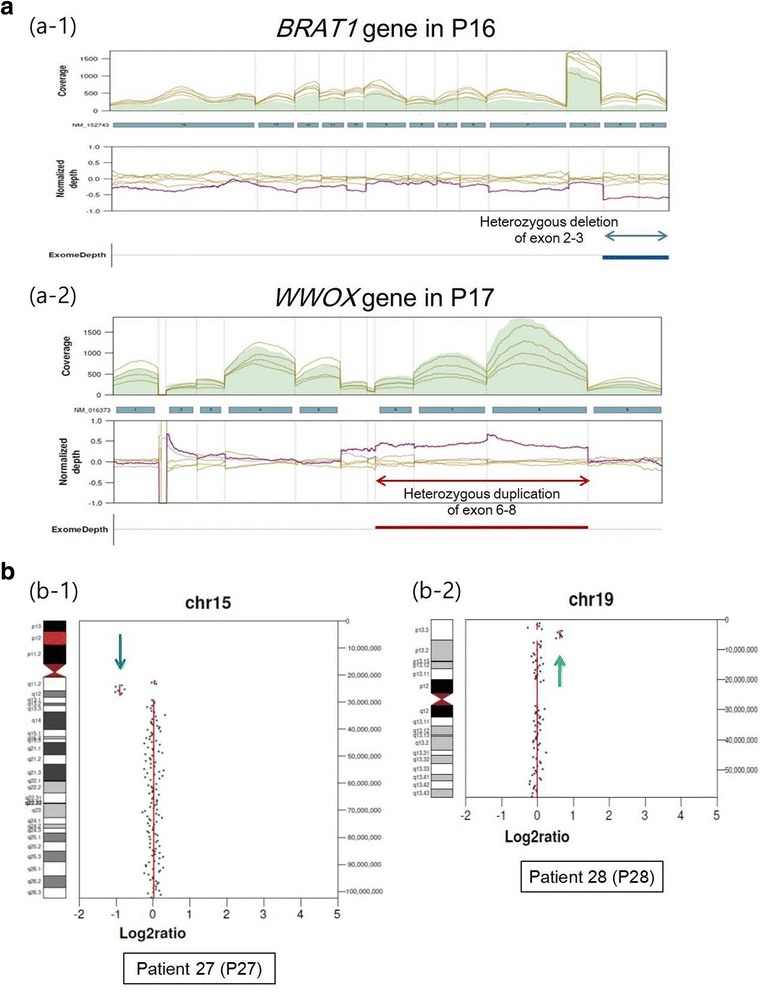
Profiles of mutations detected in our study. a Exonic deletions and duplications detected by relative-depth comparisons. a-1 Heterozygous deletion involving exons 2−3 in the BRAT1 gene of patient 16 (P16), (a-2) heterozygous duplication involving exons 6−8 in the WWOX gene in patient 17 (P17). b Examples of chromosomal copy number alterations detected by off-target analysis of targeted next-generation sequencing results. b-1 Heterozygous deletion of the 15q11.2 region (downward arrow) observed in patient 27. b-2 Heterozygous duplication of the 19p13.3 region (upward arrow) observed in patient 28
For CNVs, abnormalities were found in heterozygote form for three different chromosomes in three patients (Table 2, Fig. 2b). Two patients had seizure type of epileptic spasms, whereas the seizure type of the other patient was unknown. The affected segment sizes varied from 2.5 to 12 Mb, and all CNVs were confirmed using methylation-specific MLPA or array comparative genomic hybridization. In patient P27, methylation-specific MLPA confirmed deletion of maternal alleles in the 15q11.2 region, which contains SNRPN, UBE3A, and GABRB3. The phenotype and distinctive EEG pattern of patient P27 were compatible with Angelman syndrome, which is prolonged runs of high amplitude rhythmic 2–3 Hz activity predominantly over the frontal regions with superimposed interictal epileptiform discharges and high amplitude rhythmic 4–6 Hz activity prominent in the occipital regions with spikes.
Overall, 16/51 (31.4%) patients with IS and 8/19 (42.1%) patients with unknown EOE could be genetically diagnosed with our epilepsy NGS panel (online Additional file 1: Table S4). The most common type of mutation in IS was missense in STXBP1. Both patients with Dravet syndrome harbored frameshift mutations in SCN1A and one patient with Doose syndrome had a CHD2 mutation.
Clinical factors associated with genetic abnormalities
When all patients with EOE were assessed together, we did not find statistically significant differences in gender, age of seizure onset, and history of neonatal seizures between the patients with sufficient genetic cause group identified by NGS and the patients with negative results group, except for history of febrile seizures (28.6% vs. 6.5%) (online Additional file 1: Table S5). However, when only patients who had IS were assessed, severe clinical outcomes were more prevalent in patients with genetic cause than in patients without identified genetic cause. Patients with an identified genetic cause were more likely to never develop eye contact (50.0% vs. 17.1%, P = 0.02) (Table 3). Also more patients had an earlier seizure onset than in patients without a detected pathogenic mutation, suggesting more severe epilepsy in this group of patients with genetic causes (3.2 ± 3.0 months vs. 6.2 ± 4.4 months, P = 0.02).
Table 3.
Clinical and demographic information of the patients with infantile spasm
| Total number | Group 1 (Patients with positive genetic cause, % within group 1) | Group 2 (Patients without positive genetic cause, % within group 2) | P-value | |
|---|---|---|---|---|
| Number of subjects | 51 | 16 | 35 | |
| Female | 23 | 10 (62.5) | 13 (37.1) | 0.09 |
| Seizure onset (months) | 1.00 | |||
| 0 < n ≤ 12 months | 49 | 16 (100) | 33 (94.3) | |
| 12 months < n ≤ 24 months | 2 | 0 | 2 (5.7) | |
| 24 months < n ≤ 36 months | 0 | 0 | 0 | |
| mean age (± standard deviation) | 3.2 (± 3.0) | 6.2 (± 4.4) | 0.02 | |
| Global developmental delay | 41 | 14 (87.5) | 27 (77.1) | 0.47 |
| Absence of eye contact | 14 | 8 (50.0) | 6 (17.1) | 0.02 |
| Neonatal seizures | 9 | 5 (31.3) | 4 (11.4) | 0.12 |
| Febrile seizures | 2 | 2 (12.5) | 0 | 0.09 |
| Status epilepticus | 1 | 0 | 1 (2.9) | 1.00 |
| Premature death | 2 | 1 (6.3) | 1 (2.9) | 0.53 |
| NICU stay | 20 | 7 (43.8) | 8 (22.9) | 0.19 |
Discussion
Using a customized NGS panel, more than one-third of unknown EOEs could be diagnosed genetically. Given the high diagnostic yield, NGS panel could be a powerful diagnostic tool for clinical practices with EOE patients. We developed a diagnostic process that pediatric epileptologists and bioinformaticians can easily implement in a clinical setting. Furthermore, we provide a simplified flowchart that other epilepsy centers can follow by showing benefits acquired at each step sequentially. All analytical steps were carefully controlled, and diagnostic rates at all stages were reviewed.
In this study, more than 170 genes were examined to include as many genes as possible in order not to underestimate the genetic contribution to epilepsy. As anticipated, a few well-known genes accounted for the majority of observed abnormalities. Single-gene mutations accounted for 89.2% of all abnormalities detected; these included 16 mutations (57.1%) in major affected genes in our study (STXBP1, CDKL5, KCNQ2, SCN1A, KCNT1, SYNGAP1, MECP2, and GNAO1) and 9 mutations (32.1%) in various genes (BRAT1, WWOX, ZEB2, CHD2, PRICKLE2, COL4A1, DNM1, SCN8A, and SLC9A6). Although PRICKLE2 is controversial to be defined as epilepsy-associated gene based on previous studies, our case might serve as an additional evidence for pathogenicity. Overall, 90.2% (156) of the 172 genes in our panel showed no abnormalities. These results suggest that the currently recognized causative genes of EOE can only account for a very small fraction of the total causes of EOE, and more genes can cause EOE than have been previously reported.
Our results are consistent with those of previous studies [2, 7]. A recent Chinese study used an NGS panel for EOE that included 17 genes, and successfully diagnosed 32% (56/175) of patients [10]. A recent Japanese study examined 35 genes and diagnosed 23% (12/53) of patients [11]. Except for a few commonly mutated genes, most pathogenic variants in other genes with minor frequency seem to be rare causes of EOE. The diagnostic yield of NGS panel appeared to be determined by the number of commonly mutated genes included in the panel rather than the total number of genes in the panel [12]. This result might indicate that specific genes which have been repeatedly reported by multiple centers in multiple patients who share similar clinical phenotypes including epilepsy deserve more cautious and detailed analysis than other genes. Clinical use of NGS panel for EOE will be maximized when the incidence of all causative genes is determined and considered, rather than blindly increasing the number of genes in a panel with a hope to proportionally increase the diagnostic rate.
Patient inclusion criteria play a critical role in determining the diagnostic rate of NGS panel. The diagnostic rate increases when the study includes only narrowly classified patients with severe clinical phenotypes. In our EOE study, patients with febrile seizures and poor eye contact were more likely to exhibit genetic abnormalities. This phenomenon was more clearly demonstrated in the IS patient subgroup, and patients were more likely to exhibit genetic abnormalities if they had early onset epileptic spasms or poor eye contact (P < 0.05). These results suggest that patients with unexplained IS are more likely to exhibit abnormalities in the NGS panel if they experience seizures at younger ages and have more phenotypic traits, such as absence of eye contact.
Although intractable epilepsy is a condition that can occur at any age, it occurs most frequently in infancy and early childhood. In this study, we included only patients who met the following criteria: (1) seizures before 3 years of age and (2) progressive refractory EOE of unknown origin or well-known refractory epilepsy syndrome. We believe that our study was performed with the strict inclusion criteria, including age limits. In previous studies, the diagnostic rate of NGS panel tended to negatively correlate with age limits. The diagnostic rates increased to 32% when only patients under 6 months of age were included [10], and declined to 12% when all patients under 18 years of age were included [13]. However, it is possible that selection bias of patients with severe phenotypes might have caused relatively high diagnotic yield in this study.
Many studies have proved the clinical usefulness of NGS panel, and more epilepsy centers will use NGS panel for the management of EOE. An objective and standardized interpretation of data is critical for academic communication and validation [14]. Here, we show the importance of analyzing the genomic data systematically using a standardized approach. We found that accurate interpretation of gene abnormalities requires the following three crucial components: (1) population data and database review; (2) computational data, allelic data, and literature review; and (3) detailed clinical review, family study, and consensus discussions. Our analysis process presented the diagnostic yield of the NGS panel to be 37.8%. It should also be emphasized that consensus discussions can occur only when bioinformaticians, geneticists, and clinical neurologists closely communicate. We conducted multi-disciplinary team meetings every month, supported by frequent online and offline communications. Our study emphasizes the importance of collaborative work for correct interpretation of NGS results. The benefits of team interpretations of genomic data in rare diseases have been previously emphasized [15]. In this study, we demonstrate that team interpretation can be particularly beneficial for managing patients with EOE whose known genes and genetic inheritance patterns are diverse.
In our study, exonic and chromosomal CNVs were identified in 2.7% (2/74) and 4.1% (3/74) of patients, respectively. The importance of CNVs in epilepsy has been confirmed in previous studies [16–18]. With the rapid evolution of NGS technologies and algorithms, CNV detection using NGS could replace traditional methods. Exonic CNVs can be detected using relative depth comparisons, and whole chromosomal CNVs can be detected using off-target analysis. All identified CNVs were confirmed by MLPA or chromosomal microarrays, as well as phenotype comparison with patients of similar affected regions such as distinctive facial dysmorphism (P28 in Table 2). We also concluded that off-target analysis could be an effective and practical way to detect chromosomal CNVs, and targeted NGS testing may serve as a candidate first-line genomic test to replace chromosomal microarrays.
Unfortunately, we failed to identify causal mutations for approximately two-thirds of patients. Unknown genes may be causative factors in these patients [19, 20]. The fact that several NGS or WES studies have reported similar percentages of negative results [5, 10, 21] indicate that other genetic mechanisms including epigenetics and somatic mutation may play a role.
Conclusions
In conclusion, this study demonstrated that the targeted NGS panel has excellent diagnostic performance and great utility for managing patients with EOE. Accurate and appropriate interpretation with extensive CNV detection strategies increased the diagnostic yield of targeted NGS.
Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C1601) and a grant of the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (grant number: NRF-2015R1D1A1A01059210).
Funding
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C1601) and a grant of the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (grant number: NRF-2015R1D1A1A01059210).
Availability of data and materials
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACMG
American College of Medical Genetics and Genomics
- AMP
Association for Molecular Pathology
- bp
Base pair
- CNV
Copy number variation
- EEG
Electroencephalography
- EOE
Early-onset epilepsy
- ExAC
Exome Aggregation Consortium
- FATHMM
Functional Analysis through Hidden Markov Models
- GATK
Genome analysis toolkit
- HGMD
Human Gene Mutation Database
- MAF
Minor allele frequency
- MLPA
Multiplex ligation-dependent probe amplification
- MMFI
Malignant migrating focal seizures in infancy
- MRI
Magnetic resonance imaging
- NGS
Next generation sequencing
- NICU
Neonatal intensive care unit
- OMIM
Online Mendelian Inheritance in Man
- PolyPhen-2
Polymorphism Phenotyping v2
- SIFT
Sorting Tolerant from Intolerant
- SNV
Small nucleotide variant
- VUS
Variant of unknown significance
- WES
Whole-exome sequencing
Additional file
List of 172 targeted genes included in the epilepsy panel. Table S2. Clinical and demographic information of the patients. Table S3. Quality control matrices of NGS test results for all patients in this study. Table S4. Diagnostic yield of targeted NGS according to types of epilepsy syndrome. Table S5. Clinical factors associated with genetic abnormalities. (XLSX 31 kb)
Authors’ contributions
DHK, JRC, STL and HK conceived and designed the project. JHR, SHK, ISH, SSK, HWK, JK, SL and HK coordinated the project. Participants were recruited and phenotyped by SHK, MJC, AK, SEY, YML, HJC, JSL, HDK and HK. ISH, HWK, and JK facilitated sample archiving and performed the molecular analysis. JHR, SSK, JK, JRC and SL analysed the data. JHR and SHK drafted the manuscript with input from HDK, JRC, SL and HK. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of Severance Hospital (IRB 4–2016-0080). Informed consent was obtained from patients or their parents.
Consent for publication
Informed consent was obtained from patients or their parents.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12920-018-0320-7) contains supplementary material, which is available to authorized users.
Contributor Information
John Hoon Rim, Email: johnhoon1@yuhs.ac.
Se Hee Kim, Email: SEHEEKIM@yuhs.ac.
In Sik Hwang, Email: ISHWANG@yuhs.ac.
Soon Sung Kwon, Email: TNSTNSDLEK@yuhs.ac.
Jieun Kim, Email: jkim1220@gmail.com.
Hyun Woo Kim, Email: PINEBLUE77@yuhs.ac.
Min Jung Cho, Email: MJCHO84@yuhs.ac.
Ara Ko, Email: CHEREARA@yuhs.ac.
Song Ee Youn, Email: YOUNSE71@yuhs.ac.
Jihun Kim, Email: JIHUN264@yuhs.ac.
Young Mock Lee, Email: YMLEEMD@yuhs.ac.
Hee Jung Chung, Email: AGATHAC@yuhs.ac.
Joon Soo Lee, Email: JOONSL96@yuhs.ac.
Heung Dong Kim, Phone: +82-2-2228-2061, Email: HDKIMMD@yuhs.ac.
Jong Rak Choi, Phone: +82-2-2228-2441, Email: CJR0606@yuhs.ac.
Seung-Tae Lee, Phone: +82-2-2228-2450, Email: LEE.ST@yuhs.ac.
Hoon-Chul Kang, Phone: +82-2-2228-2075, Email: hipo0207@yuhs.ac.
References
- 1.Gaily E, Lommi M, Lapatto R, Lehesjoki AE. Incidence and outcome of epilepsy syndromes with onset in the first year of life: a retrospective population-based study. Epilepsia. 2016;57(10):1594–1601. doi: 10.1111/epi.13514. [DOI] [PubMed] [Google Scholar]
- 2.Trump N, McTague A, Brittain H, Papandreou A, Meyer E, Ngoh A, et al. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J Med Genet. 2016;53(5):310–317. doi: 10.1136/jmedgenet-2015-103263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lemke JR, Riesch E, Scheurenbrand T, Schubach M, Wilhelm C, Steiner I, et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia. 2012;53(8):1387–1398. doi: 10.1111/j.1528-1167.2012.03516.x. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Gotway G, Pascual JM, Park JY. Diagnostic yield of clinical next-generation sequencing panels for epilepsy. JAMA Neurol. 2014;71(5):650–651. doi: 10.1001/jamaneurol.2014.405. [DOI] [PubMed] [Google Scholar]
- 5.Della Mina E, Ciccone R, Brustia F, Bayindir B, Limongelli I, Vetro A, et al. Improving molecular diagnosis in epilepsy by a dedicated high-throughput sequencing platform. Eur J Hum Genet. 2015;23(3):354–362. doi: 10.1038/ejhg.2014.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Kong W, Gao Y, Liu X, Gao K, Xie H, et al. Gene mutation analysis in 253 Chinese children with unexplained epilepsy and intellectual/developmental disabilities. PLoS One. 2015;10(11):e0141782. doi: 10.1371/journal.pone.0141782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moller RS, Larsen LH, Johannesen KM, Talvik I, Talvik T, Vaher U, et al. Gene panel testing in epileptic encephalopathies and familial epilepsies. Mol Syndromol. 2016;7(4):210–219. doi: 10.1159/000448369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE commission on classification and terminology, 2005-2009. Epilepsia. 2010;51(4):676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- 9.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Q, Li J, Zhao Y, Bao X, Wei L, Wang J. Gene mutation analysis of 175 Chinese patients with early-onset epileptic encephalopathy. Clin Genet. 2016; 10.1111/cge.12901. [DOI] [PubMed]
- 11.Kodera H, Kato M, Nord AS, Walsh T, Lee M, Yamanaka G, et al. Targeted capture and sequencing for detection of mutations causing early onset epileptic encephalopathy. Epilepsia. 2013;54(7):1262–1269. doi: 10.1111/epi.12203. [DOI] [PubMed] [Google Scholar]
- 12.Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45(7):825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. 2015;56(5):707–716. doi: 10.1111/epi.12954. [DOI] [PubMed] [Google Scholar]
- 14.Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;98(6):1067–1076. doi: 10.1016/j.ajhg.2016.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarailo-Graovac M, Shyr C, Ross CJ, Horvath GA, Salvarinova R, Ye XC, et al. Exome sequencing and the Management of Neurometabolic Disorders. N Engl J Med. 2016;374(23):2246–2255. doi: 10.1056/NEJMoa1515792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olson H, Shen Y, Avallone J, Sheidley BR, Pinsky R, Bergin AM, et al. Copy number variation plays an important role in clinical epilepsy. Ann Neurol. 2014;75(6):943–958. doi: 10.1002/ana.24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheffer IE, Mefford HC. Epilepsy: beyond the single nucleotide variant in epilepsy genetics. Nat Rev Neurol. 2014;10(9):490–491. doi: 10.1038/nrneurol.2014.146. [DOI] [PubMed] [Google Scholar]
- 18.Fry AE, Rees E, Thompson R, Mantripragada K, Blake P, Jones G, et al. Pathogenic copy number variants and SCN1A mutations in patients with intellectual disability and childhood-onset epilepsy. BMC Med Genet. 2016;17(1):34. doi: 10.1186/s12881-016-0294-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dyment DA, Tetreault M, Beaulieu CL, Hartley T, Ferreira P, Chardon JW, et al. Whole-exome sequencing broadens the phenotypic spectrum of rare pediatric epilepsy: a retrospective study. Clin Genet. 2015;88(1):34–40. doi: 10.1111/cge.12464. [DOI] [PubMed] [Google Scholar]
- 20.Veeramah KR, Johnstone L, Karafet TM, Wolf D, Sprissler R, Salogiannis J, et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia. 2013;54(7):1270–1281. doi: 10.1111/epi.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. 2016;18(9):898–905. doi: 10.1038/gim.2015.186. [DOI] [PubMed] [Google Scholar]
- 22.Bertossi C, Cassina M, De Palma L, Vecchi M, Rossato S, Toldo I, et al. 14q12 duplication including FOXG1: is there a common age-dependent epileptic phenotype? Brain and Development. 2014;36(5):402–407. doi: 10.1016/j.braindev.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 23.Smyk M, Poluha A, Jaszczuk I, Bartnik M, Bernaciak J, Nowakowska B. Novel 14q11.2 microduplication including the CHD8 and SUPT16H genes associated with developmental delay. Am J Med Genet A. 2016;170a(5):1325–1329. doi: 10.1002/ajmg.a.37579. [DOI] [PubMed] [Google Scholar]
- 24.Orellana C, Rosello M, Monfort S, Mayo S, Oltra S, Martinez F. Pure duplication of 19p13.3 in three members of a family with intellectual disability and literature review. Definition of a new microduplication syndrome. Am J Med Genet A. 2015;167(7):1614–1620. doi: 10.1002/ajmg.a.37046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.