
Fig. 1.
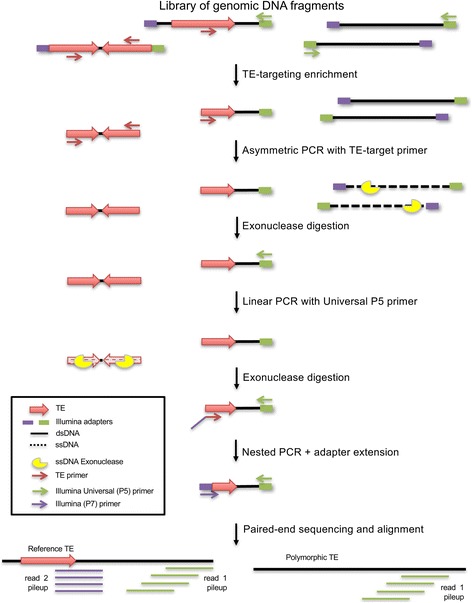
TE-NGS sequencing workflow. Enrichment for genomic fragments spanning active TEs and their unique flanking sequence is achieved by several enzymatic steps as described in the main text. First, genomic DNA is sheared, and adapters for sequencing are ligated to the genomic fragments following standard library preparation protocols. Next, a small aliquot (10 ng) of library is used as template for targeted amplification with primers complementary to TE subfamily-specific sequences and to the Illumina Universal PCR (P5) primer. Remaining genomic background fragments and inverted TEs in head-to-head orientation are removed by ssDNA exonuclease digestion after linear PCR amplification with TE-target primers or Illumina Universal primer, respectively. Last, amplification with nested primers targeting TE diagnostic bases, and containing Illumina i7 index and P7 primer sequences generates full double-stranded dual-adapter libraries containing unique indices for each sample and each TE subfamily, allowing for downstream pooling and multiplexing of many samples simultaneously. High throughput sequencing followed by alignment to the reference genome demarcates the TE insertion site by its 3′ end (read 2) and unique flanking sequence (read 1). TE insertions present in the reference genome can be identified by clustering of read pairs, whereas read 2 generated from polymorphic or novel TE insertions absent from the reference will map with lower quality and/or not at all; these TE can be identified by clusters of read 1 alone (see Methods; Supplemental Material for detailed procedures)