

Abstract
Lateral organization and mobility of adhesion molecules play a significant role in determining the avidity with which cells can bind to target cells or surfaces. Recently, we have shown that the lateral mobility of the principal adhesion molecules on neutrophils is lower for rolling associated adhesion molecules (RAAMs: L-selectin and PSGL-1) than for β2 integrins (LFA-1 and Mac-1). Here we report that all four adhesion molecules exhibit distinct punctate distributions that are mobile on the cell surface. Using uniform illumination image correlation microscopy (UI-ICM), we measure the lateral mobility of these topologically distinct domains. For all four molecules, we find that diffusion coefficients calculated from domain mobility agree with measurements we made previously using fluorescence recovery after photobleaching (FRAP). This agreement indicates that the transport of receptors on the surface of the resting neutrophil is dominated by the lateral movement of domains rather than individual molecules. The diffusion of pre-assembled integrin domains to zones of neutrophil/endothelial contact may provide a mechanism to facilitate high avidity adhesion during the earliest stages of firm arrest.
Keywords: Luekocytes, inflammation, adhesion cascade, fluorescence microscopy
The adhesion of a cell to another cell or substrate depends on an array of factors. The intrinsic molecular affinity of an adhesion molecule for its counter-receptor is clearly important, but the distribution of molecules relative to the surface topography within a region of contact, the lateral mobility of adhesion molecules, and the formation of clusters of adhesion molecules, all can have profound effects both on the probability that an adhesive bond will form and on the strength of the resulting interaction. Human neutrophils employ multiple adhesion molecules to mediate rolling, firm adhesion and migration on endothelium (Andrian et al. 1991), and the distribution and mobility of these molecules are key determinants of neutrophil-endothelial interactions. Few studies have examined these attributes in the native cell, largely because of technical challenges in dealing with the highly reactive neutrophil and the paucity of methods for assessing these properties in living cells.
Two past reports from our laboratories bear on these issues. In the first, we developed a novel approach for measuring lateral mobility of membrane proteins on spherical cells using patterned fluorescence recovery after photobleaching (FRAP) (Gaborski et al. 2008). This procedure was applied to resting, spherical neutrophils, and revealed that the lateral mobility of rolling associated adhesion molecules (RAAMs: L-selectin and PSGL-1) is significantly lower than the mobility of integrins, LFA-1 (αLβ2) and Mac-1 (αMβ2), in the resting cell. In the second report, we applied total internal reflectance fluorescence microscopy (TIRFM) to examine the localization of these same molecules relative to the surface topography (Hocdé et al. 2009a; Hocdé et al. 2009b). Because the excitation of fluorophores during TIRFM falls off exponentially with distance from the surface with a characteristic dimension of ∼200 nm, a number that closely resembles the dimensions of surface ruffles and microvilli on the neutrophil surface, TIRFM provides a measure of the relative distribution of molecules at or away from the microvilli tips. It was found that of the four adhesion molecules examined, L-selectin had the highest TIRFM signal when normalized to epi-fluorescence images of molecules on cell body and the microvillus tips. The β2 integrins had a much lower TIRFM signal suggesting they are primarily localized away from the tips of the microvilli. These results for L-selectin and the β2 integrins are consistent with earlier electron microscopy studies that showed L-selectin staining with immunogold primarily on the tips of microvilli, while the β2 integrins were labeled only in between the microvilli on the cell body (Andrian et al. 1991; Bruehl et al. 1996; Ehringer et al. 1996). Surprisingly, we measured PSGL-1 to be closer in TIFRM intensity to the β2 integrins rather than L-selectin (Hocdé et al. 2009a; Hocdé et al. 2009b), which contradicted earlier impressions that PSGL-1 was located on the tips of microvilli alongside L-selectin (Moore et al. 1995). Work by Sundd et. al. has shown that PSGL-1 in mice is localized to discrete patches on newly discovered membrane slings, while LFA-1 distribution is more uniform during cell rolling (Sundd et al. 2013). This discovery is consistent with PSGL-1 having different membrane dynamics from LFA-1. While quantitative TIRFM data suggests PSGL-1 is more closely related to LFA-1, FRAP and our new ICM measurements show PSGL-1 membrane mobility is much different from LFA-1 and almost identical to L-selectin. The similarity in domain mobility between L-selectin and PSGL-1 is not surprising as they are believed to mediate neutrophil rolling, while the β2 integrins follow with firm arrest. In this report we show that receptor mobility on the cell surface may be dominated by domain movement or ultrastructural rearrangement rather than individual molecule diffusion. These dynamics may provide a mechanism for the transition from neutrophil rolling to early stages of firm arrest.
Distinct topological localization of neutrophil adhesion molecules is consistent with our observation that all four molecules present a non-uniform, punctate distribution when examined in standard epi-fluorescence images (Figure 1 and see Figure S1 for results using Fab fragments). In these experiments, cells were labeled with monoclonal antibodies (mAb) to one of the principal adhesion molecules on the cell, L-selectin (anti-CD62L) PSGL-1 (anti-CD162), LFA-1 (anti-αL or CD11a) or Mac-1 (anti-αM or CD11b). Standard high magnification epi-fluorescence provides a larger depth of focus (∼ 2 microns) compared to confocal microscopy, allowing the simultaneous visualization of molecules on both ruffles and the cell body.
Figure 1.
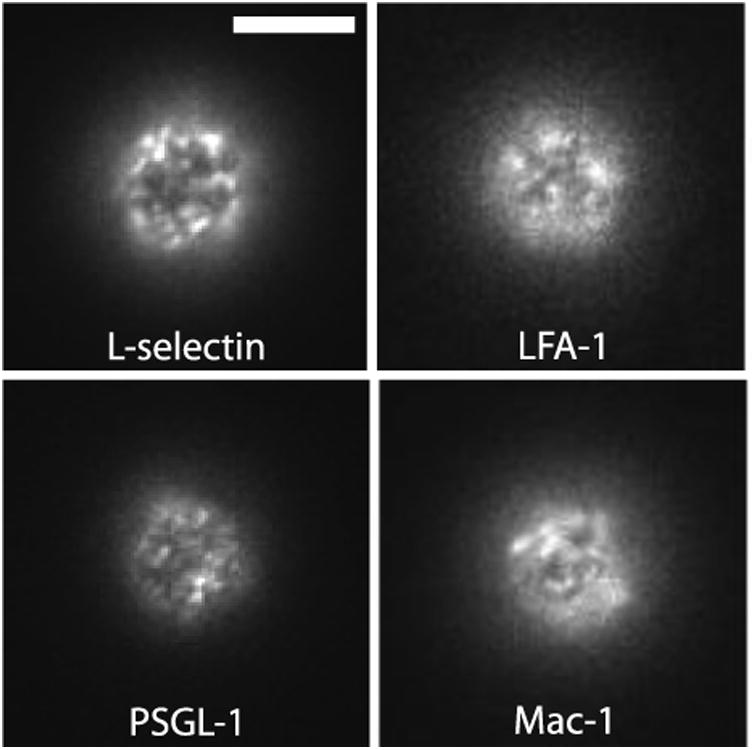
Adhesion molecules are found in domains on resting human neutrophils. Cells were labeled with primary fluorescent monoclonal antibodies against one of four adhesion molecules: L-selectin, LFA-1, PSGL-1 and Mac-1. White bar is 5 microns.
In order to observe the mobility of these receptor domains, epi-fluorescent time-lapse movies were collected. We confirmed that the receptors were in spatially distinct domains by co-labeling cells with AlexaFluor 488 anti-CD62L and AlexaFluor 546 anti-CD11a (Figure 2A). Single channel, higher temporal resolution image sequences revealed that the fluorescent domains associated with each type of adhesion molecule are mobile on the neutrophil surface (Figure 2B; time-lapse movie available online). In these experiments, images were recorded every 250 ms for 25 seconds and focused at the bottom of the cell. When RAAMs and β2 integrins were co-labeled on the same cell, it was evident that β2 integrin domains move more rapidly over the neutrophil surface than RAAM domains. Because FRAP data also revealed RAAMs to be less mobile than β2 integrins, we developed uniform-illumination image correlation microscopy (UI-ICM) (Gaborski et al. 2010) to calculate the mobilities of receptor domains in epi-fluorescent time-lapsed sequences and compare them to lateral mobilities determined using FRAP.
Figure 2.
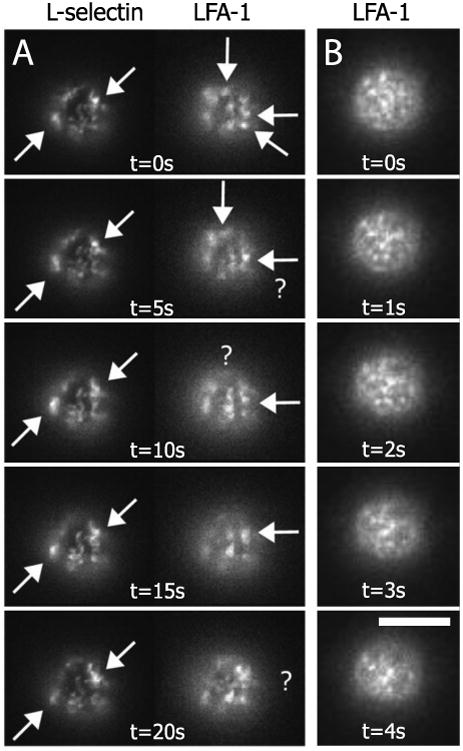
Time-lapsed epi-fluorescence images on the bottom surface of a human neutrophi. (A) CD62L and CD11a appear as distinct fluorescent patterns on the cell. CD62L features are more persistent in the image series compared to CD11a when imaged every 5 seconds. Arrows highlight specific domains that minimally diffuse. Domains that disappear between images are highlighted with a ‘?’. White bar is 5 microns. (B) CD11a imaged with greater temporal resolution on a different cell. Time-lapse movie available on-line.
Traditional image correlation microscopy uses a scanning laser beam to investigate the mobility of molecular clusters on cellular membranes (Petersen et al. 1993; Srivastava and Petersen 1998; Wiseman et al. 2000). Fluctuations in fluorescence intensities occur as molecules diffuse in and out of the confined excitation of the laser. These fluctuations can then be related to the mobility of receptor clusters using modifications of classic theory for fluorescence correlation spectroscopy in homogeneous solutions (Elson 2011). UI-ICM differs from traditional ICM in that it determines the mobility of molecules in uniform illumination. The spatial dimension over which diffusion must occur for decorrelation is the feature size in the image itself, which is determined by spatial autocorrelation of the fluorescence image (Gaborski et al. 2010). With the diffusion length scale defined, cluster diffusion coefficients are then determined from the decay of the autocorrelation function with time.
The diffusion coefficients determined by UI-ICM were ∼ 0.65 × 10-10 cm2/s for PSGL-1 (n=11) and L-selectin (n=9) and ∼ 1.3 × 10-10 cm2/s for both LFA-1 (n=9) and Mac-1 (n=9) (Figure 4). Interestingly, the mobility of the domains correlated with the function of the molecule (rolling vs. firm adhesion) and not with their topographical localization. Thus, the mobility of PSGL-1 domains closely resembled that of L-selectin domains, and both were approximately half as mobile as the integrin domains. The values of the diffusion coefficients were very close to those we determined previously by FRAP for these same molecules (Gaborski et al. 2008), suggesting that the fluorescence recovery seen in FRAP experiments can be explained in large part by the diffusion of clusters of receptors rather than individual molecules.
It has long been recognized that the lateral mobility of adhesion molecules can be rate limiting for bond formation between cells and other surfaces (Bell 1978). In cells, adhesion molecule mobility can be affected by a variety of factors, including protein-protein interactions, direct cytoskeletal tethering, or so-called cytoskeletal fences located just beneath the plasma membrane (Srivastava and Petersen 1998). The fact that the adhesion molecules on neutrophils appear to reside in clusters or domains suggests that some mechanism such as these may be limiting the dispersal of these molecules over the cell surface. The big surprise from these measurements was that the domains themselves diffuse over the cell surface and that this appears to account for the long-range diffusion observed previously using FRAP. In the case of L-selectin domains, which are known to reside at microvilli tips, they can be detected and tracked over periods of several minutes. This implies that the microvilli themselves are remodeling and relocating over the cell surface. These observations may also resolve the surprising result that even though these molecules reside in domains, the immobile fraction of the adhesion molecules on neutrophils was found to be zero over a time scales of minutes (Gaborski et al. 2008).
Measurements of lateral mobility of β2 integrins (LFA-1) on leukocytes have been previously performed on cells using both single particle tracking (SPT) and FRAP (Cairo et al. 2006). In the FRAP studies, a significant fraction of the molecules were found to be immobile, and in the SPT studies, a high percentage of molecules appeared to be confined to sub-micron sized regions. This contrasts sharply with our own observations of diffusion in neutrophils, where the population mobility was somewhat slower, but where no immobile fraction was detectable (Gaborski et al. 2008). There are important differences between the two studies. First, they deal with a different cell types. Cairo and colleagues (Cairo et al. 2006) worked with Jurkat cells and peripheral blood lymphocytes, which could exhibit substantially different behaviors than neutrophils. Second, the cells in those experiments were allowed to spread onto glass slides to immobilize them and attain a more or less planar geometry. The neutrophils in our studies were spherical and maintained in the resting state, thus avoiding possible artifacts of spreading, such as cell activation or immobilization of molecules against the glass slide. Of particular interest in the present context is the possibility that cell spreading may have significant effects on cell surface dynamics, and the motions of microvilli evident in the current report may be reduced or eliminated.
The present results support an emerging view of surface receptors existing in dynamic clusters on cell surface (Majstoravich et al. 2004; Welf et al. 2012). Even molecules that are linked to the cytoskeleton may “diffuse” long distances over the cell surface as the surface-associated cytoskeleton itself undergoes substantial lateral reorganization (Toplak et al. 2012). The pre-packaging of β2-integrins in mobile domains in resting neutrophils should increase the effectiveness of integrin-based adhesion during the transition from rolling to firm arrest. Because strong adhesions benefit from both high integrin density and activation to high affinity conformations (Carman and Springer 2003), the diffusion of domains into zones of contact with endothelium provides a mechanism for these components of high avidity binding to occur simultaneously.
Methods
Neutrophil Preparation
Neutrophils were labeled and prepared for microscopy as previously described (Gaborski et al. 2010; Gaborski et al. 2008). Briefly, cells were labeled by diluting whole blood into Hank's Balanced Salt Solution (HBSS) with 10 mM HEPES containing with fluorescently labeled for L-selectin (anti-CD62L; DREG-56, eBioscience, San Diego, CA) PSGL-1 (anti-CD162; PL1/3E2-25-5; Ancell, Bayport, MN), LFA-1 (anti-αL or CD11a; HI111; eBioscience, San Diego, CA) or Mac-1 (anti-αM or CD11b; ICRF44; eBioscience, San Diego, CA). Antibodies were fluorescenty labeled with AlexaFluor antibody labeling kits (Life Technologies, Carlsbad, CA). Fab fragments were enzymatically cleaved from the monoclonal antibodies using the Pierce Fab Preparation Kit (ThermoFisher, Rockford, IL). Cells were labeled at room temperature for fifteen minutes and washed three times by centrifugation. Cells were then re-suspended in HBSS plus 10mM HEPES and 4% fetal bovine serum (FBS) to prevent cell adhesion to glass cover slips. Experiments were performed with 60 minutes of labeling to minimize receptor internalization during imaging.
Fluorescence and image correlation microscopy
All fluorescence microscopy was performed at room temperature on an inverted Zeiss Axiovert 200M microscope equipped with a 100× 1.45 NA oil objective (Zeiss, Thornwood, NY). The microscope was also setup to collect TIRFM images using a solid-state 488 nm laser and the methods described by Axelrod (Axelrod 2001). Uniform Illumination Image Correlation Microscopy (UI-ICM) was performed and analyzed as previously described (Gaborski et al. 2008). Briefly, 100 images were captured every 250 ms for 25 seconds using a SensiCam EM cooled CCD camera with electron multiplication (Cooke, Romulus, MI). The sequence of 50 consecutive images with the least amount of lateral translation was analyzed. If the mean translation per image was more than 50 nm, the data was not further analyzed due to movement artifacts. Images were collected in wide-field epi-fluorescence. Measurement of TIRFM domains with UI-ICM is not reliable since minor cell wobble will result fluorescent fluctuations due to exponential decay of the evanescent wave. UI-ICM measurements were performed on cells labeled with mAb because Fab signal is significantly less and required longer exposure times. The upper limit of UI-ICM dynamic range is a function of exposure time and domain size. Fab domain sizes were approximately the same (Figure S1), but the longer exposure times precluded accurate measurement of their diffusive motion. The temporal decay of the image autocorrelation function was calculated using established algorithms (Petersen et al. 1993; Wiseman et al. 2000). The time constant for autocorrelation decay was used in combination with a length scale calculated from the spatial autocorrelation function (Gaborski et al. 2010).
Supplementary Material
Figure 3.

Diffusion coefficients for ICM and FRAP for RAAM's and β2 integrins. Using ICM, CD62L and CD162 have similar lateral mobility to one another and to the measurements made with FRAP. Likewise, CD1 1a and CD11b have similar lateral mobilities with ICM and FRAP. Error bars are +/- SEM (FRAP data from Gaborski 2008).
Acknowledgments
This work was supported under funding from the National Institutes of Health under Program Project Grant number PO1HL018208.
Reference List
- Andrian von U, Chambers J, McEvoy L, et al. Two-step model of leukocyte-endothelial cell interaction in inflammation: distinct roles for LECAM-1 and the leukocyte beta 2 integrins in vivo. Proc Natl Acad Sci U S A. 1991;88:7538–7542. doi: 10.1073/pnas.88.17.7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- Bell G. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- Bruehl RE, Springer TA, Bainton DF. Quantitation of L-selectin distribution on human leukocyte microvilli by immunogold labeling and electron microscopy. J Histochem Cytochem. 1996;44:835–844. doi: 10.1177/44.8.8756756. [DOI] [PubMed] [Google Scholar]
- Cairo C, Mirchev R, Golan D. Cytoskeletal regulation couples LFA-1 conformational changes to receptor lateral mobility and clustering. Immunity. 2006;25:297–308. doi: 10.1016/j.immuni.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Carman C, Springer T. Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr Opin Cell Biol. 2003;15:547–556. doi: 10.1016/j.ceb.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Ehringer WD, Edwards MJ, Miller FN. Mechanisms of alpha-thrombin, histamine, and bradykinin induced endothelial permeability. J Cell Physiol. 1996;167:562–569. doi: 10.1002/(SICI)1097-4652(199606)167:3<562∷AID-JCP20>;3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Elson EL. Fluorescence correlation spectroscopy: past, present, future. Biophys J. 2011;101:2855–2870. doi: 10.1016/j.bpj.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaborski TR, Clark A, Waugh RE, McGrath JL. Membrane mobility of beta2 integrins and rolling associated adhesion molecules in resting neutrophils. Biophys J. 2008;95:4934–4947. doi: 10.1529/biophysj.108.132886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaborski TR, Sealander MN, Ehrenberg M, et al. Image correlation microscopy for uniform illumination. J Microsc. 2010;237:39–50. doi: 10.1111/j.1365-2818.2009.03300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocdé SA, Hyrien O, Waugh RE. Cell adhesion molecule distribution relative to neutrophil surface topography assessed by TIRFM. Biophys J. 2009a;97:379–387. doi: 10.1016/j.bpj.2009.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocdé SA, Hyrien O, Waugh RE. Molecular accessibility in relation to cell surface topography and compression against a flat substrate. Biophys J. 2009b;97:369–378. doi: 10.1016/j.bpj.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majstoravich S, Zhang J, Nicholson-Dykstra S, et al. Lymphocyte microvilli are dynamic, actin-dependent structures that do not require Wiskott-Aldrich syndrome protein (WASp) for their morphology. Blood. 2004;104:1396–1403. doi: 10.1182/blood-2004-02-0437. [DOI] [PubMed] [Google Scholar]
- Moore KL, Patel KD, Bruehl RE, et al. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J Cell Biol. 1995;128:661–671. doi: 10.1083/jcb.128.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen N, Hoddelius P, Wiseman P, et al. Quantitation of membrane receptor distributions by image correlation spectroscopy: concept and application. Biophys J. 1993;65:1135–1146. doi: 10.1016/S0006-3495(93)81173-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava M, Petersen N. Diffusion of transferrin receptor clusters. Biophys Chem. 1998;75:201–211. doi: 10.1016/s0301-4622(98)00206-3. [DOI] [PubMed] [Google Scholar]
- Sundd P, Pospieszalska MK, Ley K. Neutrophil rolling at high shear: flattening, catch bond behavior, tethers and slings. Mol Immunol. 2013;55:59–69. doi: 10.1016/j.molimm.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toplak T, Pandzic E, Chen L, et al. STICCS reveals matrix-dependent adhesion slipping and gripping in migrating cells. Biophys J. 2012;103:1672–1682. doi: 10.1016/j.bpj.2012.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welf ES, Naik UP, Ogunnaike BA. A spatial model for integrin clustering as a result of feedback between integrin activation and integrin binding. Biophys J. 2012;103:1379–1389. doi: 10.1016/j.bpj.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman PW, Squier JA, Ellisman MH, Wilson KR. Two-photon image correlation spectroscopy and image cross-correlation spectroscopy. J Microsc. 2000;200:14–25. doi: 10.1046/j.1365-2818.2000.00736.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.