

Chan et al. show that epithelial cells respond to bacterial components in the environment by releasing subunits of the keratin 6a (K6a) filament network to the cytosol for degradation by the ubiquitin–proteasome system. This generates antimicrobial peptides from K6a that are important for innate defense of the mucosal surface.
Abstract
Skin and mucosal epithelia deploy antimicrobial peptides (AMPs) to eliminate harmful microbes. We reported that the intermediate filament keratin 6a (K6a) is constitutively processed into antimicrobial fragments in corneal epithelial cells. In this study, we show that K6a network remodeling is a host defense response that directly up-regulates production of keratin-derived AMPs (KAMPs) by the ubiquitin–proteasome system (UPS). Bacterial ligands trigger K6a phosphorylation at S19, S22, S37, and S60, leading to network disassembly. Mutagenic analysis of K6a confirmed that the site-specific phosphorylation augmented its solubility. K6a in the cytosol is ubiquitinated by cullin-RING E3 ligases for subsequent proteasomal processing. Without an appreciable increase in K6a gene expression and proteasome activity, a higher level of cytosolic K6a results in enhanced KAMP production. Although proteasome-mediated proteolysis is known to produce antigenic peptides in adaptive immunity, our findings demonstrate its new role in producing AMPs for innate immune defense. Manipulating K6a phosphorylation or UPS activity may provide opportunities to harness the innate immunity of epithelia against infection.
Introduction
Skin and mucosal epithelia of digestive, genitourinary, respiratory, and ocular systems comprise the largest surface area of the body. They are in direct contact with the external environment and therefore exposed to microorganisms that are potentially pathogenic, including bacteria, fungi, parasites, and viruses. These surfaces produce and deploy an array of antimicrobial molecules as one of their first lines of defense. Antimicrobial peptides (AMPs) belong to diverse families of oligopeptides (~10–50 amino acid residues) that are evolutionarily conserved and widespread across all kingdoms of life (Wang et al., 2016; Zhang and Gallo, 2016). The general mechanism of action of AMPs includes killing of the pathogens by membrane disruption or inactivation of their life cycle and growth (Wang et al., 2014). In addition, many AMPs function in the host as potent regulators of innate and adaptive immune responses and as agonists of angiogenesis and wound healing (Lai and Gallo, 2009; Mahlapuu et al., 2016). Collectively, these processes effectively control microbial infections and maintain tissue health and homeostasis.
Keratin is the most abundant class of intermediate filament protein in the skin and mucosal epithelium (Windoffer et al., 2011). Humans have >50 keratins, which are classified as either acidic type I keratins (K9–K10, K12–K28, and K31–K40) or as basic type II keratins (K1–K8 and K71–K86; Moll et al., 2008). Both types are expressed in keratinocytes and epithelial cells and are characterized by a central conserved α-helical rod domain containing coiled-coil motifs for dimerization and spanned by divergent short non–α-helical head and tail domains for protein–protein interactions (Herrmann et al., 2009). Type I and type II keratins form cytosolic heterodimers and oligomers at cell focal adhesion sites (Windoffer et al., 2006). These subunits are incorporated into filamentous keratin networks at the cell periphery, which in turn coalesce into fibrous bundles at the perinuclear space, providing mechanical support and protection to the cell (Kölsch et al., 2010; Windoffer et al., 2011). Conversely, the disassembly of the filamentous keratin network to yield soluble subunits reusable for the next round of assembly is equally important, as this allows the filamentous keratin network to adapt to changing functional needs of the cell (Kölsch et al., 2010; Windoffer et al., 2011; Pan et al., 2013).
Dynamic assembly and disassembly of the keratin network are driven by posttranslational modifications (PTMs) including site-specific phosphorylation (Snider and Omary, 2014; Sawant and Leube, 2017). In general, phosphorylation at serine and threonine residues promotes disassembly of the keratin filamentous network. Keratin members are often modified by PTMs to regulate nonmechanical functional effects as well (Snider and Omary, 2014; Sawant and Leube, 2017). For example, K8 can be heavily phosphorylated to absorb excess ATP that would otherwise be used by other pathways to activate proapoptotic factors leading to uncontrolled liver damage (Ku and Omary, 2006; Ku et al., 2007). Hyperphosphorylation of K8 and its heterotypic partner K18 are biomarkers for chronic hepatitis C virus infection of the liver (Toivola et al., 2004). Individuals who carry a rare K8 G61C mutation are prone to develop liver diseases such as hepatic cirrhosis (Ku et al., 2001). This mutation sterically obstructs K8 phosphorylation at S73 by stress-activated kinases, reducing its solubility and availability as a phosphate sponge during hepatotoxic assault (Ku and Omary, 2006; Ku et al., 2007). This phenotype has been confirmed in transgenic mice models harboring either the human K8 G61C or S73A mutation (Ku and Omary, 2006). Aberrant expression and phosphorylation of these and other type I or type II keratin members also occur during acute viral infection, although the pathological consequence is unclear (Pugh et al., 1998; Murata et al., 2002; McIntosh et al., 2010).
We previously reported a series of naturally occurred and overlapping short peptides (13–26 amino acids) from the carboxyl terminal region (residues 515–559) of keratin 6a (K6a), termed keratin-derived AMPs (KAMPs; Tam et al., 2012; Lee et al., 2016). KAMPs are constitutively expressed in human corneal epithelial cells and display potent antibacterial and cytoprotective effects against multiple bacterial pathogens. Whereas canonical AMPs such as defensins and cathelicidin are salt sensitive and disrupt bacterial membrane through electrostatic interaction and defined secondary structure determinants (α-helix and/or β-sheet; Wang et al., 2014), KAMPs are salt tolerant and mediate membrane disruption through a distinct and different mechanism involving flexible glycine-rich sequences (Tam et al., 2012). KAMPs are the only human example of a structurally unique class of non-αβ AMPs (Rozek et al., 2000; Lee et al., 2016), which represent a new class of antimicrobial agents that can potentially address the public health requirement to tackle antibiotic-resistant bacteria (Fox, 2013; Mahlapuu et al., 2016).
Although K6a is constitutively expressed in many stratified epithelial tissues, including the cornea (Moll et al., 2008; Dyrlund et al., 2012), and is induced by inflammatory cytokines during tissue injury and repair (Komine et al., 2001; Rotty and Coulombe, 2012), the regulation of filamentous K6a remodeling and the mechanism of K6a processing into KAMPs remain elusive. In this study, we demonstrate the roles of PTMs and the ubiquitin–proteasome system (UPS) in these processes in the context of corneal innate immune defense. The K6a filament network depolymerizes in response to bacterial cell wall products and flagellin. Although K6a gene expression is not up-regulated under these conditions, cytosolic K6a and KAMP levels are significantly increased, suggesting that the filamentous form of K6a acts as a reserve pool of KAMP precursors. Furthermore, we report that S19, S22, S37, and S60 are the phosphorylation sites on K6a that enhance its solubility and that cytosolic K6a is targeted for ubiquitination by cullin–RING E3 ligases (CRLs) followed by proteasomal processing to produce KAMPs. These data provide novel mechanistic insights into the antimicrobial roles of cytokeratins and UPS in epithelial innate defense.
Results
Bacterial ligands trigger filamentous K6a network reorganization
We first tested the hypothesis that the filamentous keratin network in epithelial cells undergoes reorganization in response to bacterial products. We used a telomerase-immortalized human corneal epithelial cell line (hTCEpi) that undergoes differentiation, stratification, and desquamation similar to the native corneal epithelium (Robertson et al., 2005, 2008, 2011). It is well established that human corneal epithelial cells express functional Toll-like receptors (TLRs) that recognize distinct pathogen-associated molecular patterns (Zhang et al., 2003; Johnson et al., 2005, 2008; Ueta et al., 2005; Kumar et al., 2006; Roy et al., 2011); thus, we treated hTCEpi cells with purified bacterial surface structures from gram-positive and gram-negative bacteria, namely flagellin, lipopolysaccharide (LPS), or lipoteichoic acid (LTA), which activate TLR-5, TLR-4 and TLR-2, respectively, and compared the filamentous K6a organization in these cells by immunocytochemistry. To induce expression of the TLR-4 coreceptor MD-2 and thereby enable LPS/MD-2/TLR-4 signaling, these cells were pretreated briefly (2 h) with IFN-γ (Roy et al., 2011, 2014). Using confocal microscopy, we observed that the filamentous K6a network in hTCEpi cells became more diffused in response to bacterial ligands compared with untreated control cells and that the reduction of filament density at the perinuclear region was particularly evident (Fig. 1 A). Similar observations were made for telomerase-immortalized human mammary epithelial cells and, to some extent, for SV40-immortalized human ectocervical epithelial cells (Fig. S1). In contrast, the K6a network in hTCEpi cells not primed with IFN-γ (deficient in MD-2) did not respond to LPS and showed no appreciable difference from untreated control cells (Fig. S2). We further quantified the fluorescence signal intensity of these filaments in hTCEpi cells along the distance from the nucleus to the plasma membrane and confirmed our observation (Fig. 1 B). As keratin filaments are known to disassemble and release reusable subunits at the perinuclear region (Windoffer et al., 2011), the reduced filament density in bacteria ligand–treated hTCEpi cells suggests disassembly of K6a cytoskeleton as an epithelial response to bacterial pathogens.
Figure 1.

Bacterial ligands trigger depolymerization of the filamentous K6a network. (A) Confocal microscopic images of hTCEpi cells treated with vehicle, flagellin (FliC; 0.5 µg/ml), LPS (1 µg/ml), or LTA (1 µg/ml) for 16 h followed by immunostaining for K6a (green) and counterstaining for nuclei (red). For cells treated with LPS, a 2-h pretreatment with IFN-γ (250 ng/ml) was used to induce LPS responsiveness. The intense filamentous K6a network staining surrounding the nucleus in the control cell (insets) became diffused and weak after stimulation with bacterial ligands. Bars, 10 µm. (B) Quantification of fluorescence signal intensity of K6a filament as a function of the distance (μm) from the nucleus toward the plasma membrane using ImageJ. Signal intensity of each cell was measured along three lines at 0.06 µm steps to obtain a mean. The intensity values were normalized to the nuclear signal. Means ± SEM of 11–14 cells pooled from three to four independent experiments are shown. K6a filament intensity of treated cells was reduced significantly compared with control (P < 0.0001).
Bacterial ligands promote solubility of K6a and production of its antimicrobial fragments (KAMPs)
In an effort to understand the significance of K6a network reorganization, we assessed the levels of cytosolic K6a and its antimicrobial fragments (KAMPs) in hTCEpi organotypic culture treated with bacterial ligands. Immunoblotting of NP-40–soluble proteins in the cell lysates showed that flagellin, LPS (with IFN-γ preincubation), or LTA stimulation led to a significant increase in the level of cytosolic K6a protein without detectable changes in its filament level (Fig. 2, A–C). In conjunction, the level of KAMPs in the same samples showed marked increase after stimulation (Fig. 2, A–C). To determine whether K6a gene expression was affected by exposure to these bacterial ligands, we used real-time PCR to quantify K6a mRNA and found no significant differences between control and stimulated cells (Fig. 2 D), thereby eliminating the possibility that the increased availability of cytosolic K6a is caused by elevated gene expression. Collectively, these data suggest that a slight shift of K6a from the filamentous to cytosolic form is sufficient to up-regulate KAMPs production without negatively impacting the mechanical properties of the cells.
Figure 2.

Bacterial ligands promote K6a solubility and production of KAMPs. (A–C) Immunoblot analysis of NP-40–soluble and -insoluble lysate fractions of hTCEpi organotypic culture treated with vehicle control or flagellin (FliC; 0.5 µg/ml; A), LPS (1 µg/ml; B), or LTA (1 µg/ml; C) for 16 h. For cells treated with LPS, a 2-h pretreatment with IFN-γ (250 ng/ml) was used to induce LPS responsiveness. Cytosolic K6a (60 kD) in the NP-40–soluble lysate fractions (10 µg total protein) and filamentous K6a (60 kD) in the insoluble fractions (1 µg total protein) were resolved in 12% Bis-Tris gels followed by immunoblotting. Same soluble lysate fractions (40 µg total protein) were also resolved in 16% tricine gel and immunoblotted for KAMPs (∼3 kD). Actin (42 kD) was used as a loading control. Quantification of band intensity was performed with Bio-Rad Laboratories Image Lab software and normalized with actin. Means ± SEM are shown. n = 4–8 independent experiments. **, P < 0.01; ***, P < 0.001. (D) Quantitative RT-PCR assessment of K6a gene expression in hTCEpi cells treated with vehicle control, flagellin (FliC; 0.5 µg/ml), LPS (1 µg/ml), or LTA (1 µg/ml) for 3 and 6 h. K6a gene expression was normalized to actin. Compared with control cells, K6a gene expression in treated cells was unaffected under the indicated conditions (P > 0.05). Means ± SD are shown. Experiments were performed three times.
Serine phosphorylation of K6a augments its solubility in response to bacterial ligands
As PTMs of intermediate filaments regulate their organization, assembly and disassembly dynamics, and, importantly, their functions (Snider and Omary, 2014; Sawant and Leube, 2017), we investigated whether K6a is posttranslationally modified in response to bacterial ligands. Immunoprecipitation of K6a from the cytosolic extracts of hTCEpi organotypic culture in tandem with mass spectrometric analysis revealed four major phosphorylation sites of K6a at S19, S22, S37, and S60 (Figs. 3 A, S3, and S4). Either flagellin (Fig. 3 B) or LTA (Fig. 3 D) induced K6a double phosphorylation at S19 and S22 (∼4-fold and ∼1.5-fold of the basal level, respectively), whereas LTA also caused a modest increase of S60 phosphorylation (∼1.25-fold). In contrast, as shown in Fig. 3 C, LPS induced S37 phosphorylation (∼1.5-fold). Overall, the data demonstrate that bacterial ligands induce changes in serine phosphorylation of K6a.
Figure 3.

Serine phosphorylation increases K6a solubility. (A) Cytosolic K6a was immunoprecipitated from hTCEpi organotypic culture treated with various bacterial ligands followed by LC-MS analysis. Four different phosphopeptides were identified. (B–D) The degree of modification (abundance of phosphoform/abundance of unmodified form) was determined for each phosphopeptide. The fold amount of each modification after treatment relative to unstimulated control (basal levels = 1) is presented. (E) Phosphorylation of K6a positively correlates with its cytosolic level in hTCEpi organotypic culture. hTCEpi cells were treated with DMSO or 200 nM phosphatase inhibitor calyculin A for 5 h before they were harvested and immunoprecipitated (IP) with preimmune serum or anti-K6a antiserum. Samples were immunoblotted (IB) by anti-K6a antiserum or antiphosphoserine antibody. (F) Dephosphorylation of K6a by CIP. Remaining eluates from calyculin A–treated samples from E were divided into three fractions, resolved, transferred to polyvinylidene difluoride membrane, and incubated with CIP (fraction 3), without CIP (fraction 2), or CIP buffer only (fraction 1). Membranes were immunoblotted for K6a (fraction 1) or phosphorylated K6a (fractions 2 and 3).
To substantiate the importance of serine phosphorylation of K6a, we treated hTCEpi organotypic culture with the phosphatase inhibitor calyculin A to inhibit the activity of protein phosphatase 1 and 2A and thereby induce hyperphosphorylation (Takuma et al., 1993). Immunoblotting using antiserum against K6a showed that the level of cytosolic K6a was drastically elevated in the presence of calyculin A (Fig. 3 E). As the level of filamentous K6a was concomitantly reduced, calyculin A caused a significant shift of K6a from the filamentous form to the cytosolic form. Furthermore, immunoprecipitation of cytosolic K6a followed by immunoblotting with an antibody against phosphoserine proteins indicated that a significant portion of cytosolic K6a was serine phosphorylated, which was confirmed by calf intestine phosphatase digestion (Fig. 3, E and F). These results demonstrate that phosphorylation of K6a at serine residues regulates its solubility.
Phosphorylation at ser-19, -22, -37, and -60 increases K6a solubility
Next, hTCEpi cells were transfected with plasmid constructs expressing HA-tagged WT K6a or K6a mutants with alanine substitutions at each of the four serine residues 19, 22, 37, and 60 that were identified by our mass spectrometric analysis as differentially phosphorylated in response to bacterial ligands (Fig. 4 A). Immunoblot analysis of the cytosolic fractions of these cells with an antibody against the HA tag showed that solubility of K6a with quadruple serine mutations (S19A/S22A/S37A/S60A) was substantially lower compared with the WT form (Fig. 4 B), supporting that phosphorylation at these serine sites mediates bacterial ligand-induced solubilization of K6a and in turn, KAMP production. Moreover, compared with the quadruple mutant, the solubility of the K6a triple mutant (S19A/S22A/S37A) was slightly improved, and the solubility of the double mutant (S19A/S22A) was improved further but remained lower than the WT (Fig. 4 B), suggesting that phosphorylation at S60 is insufficient and requires phosphorylation at the other three sites for optimal solubility. Likewise, as shown in Fig. 4 C, single mutations at S19, S22, S37, or S60 reduced the levels of cytosolic K6a to different extents, although the S37A mutation exhibited the strongest effect. Collectively, these data indicate that phosphorylation of any of these four serine residues promotes K6a solubility, whereas phosphorylation of all four serines maximizes solubility.
Figure 4.
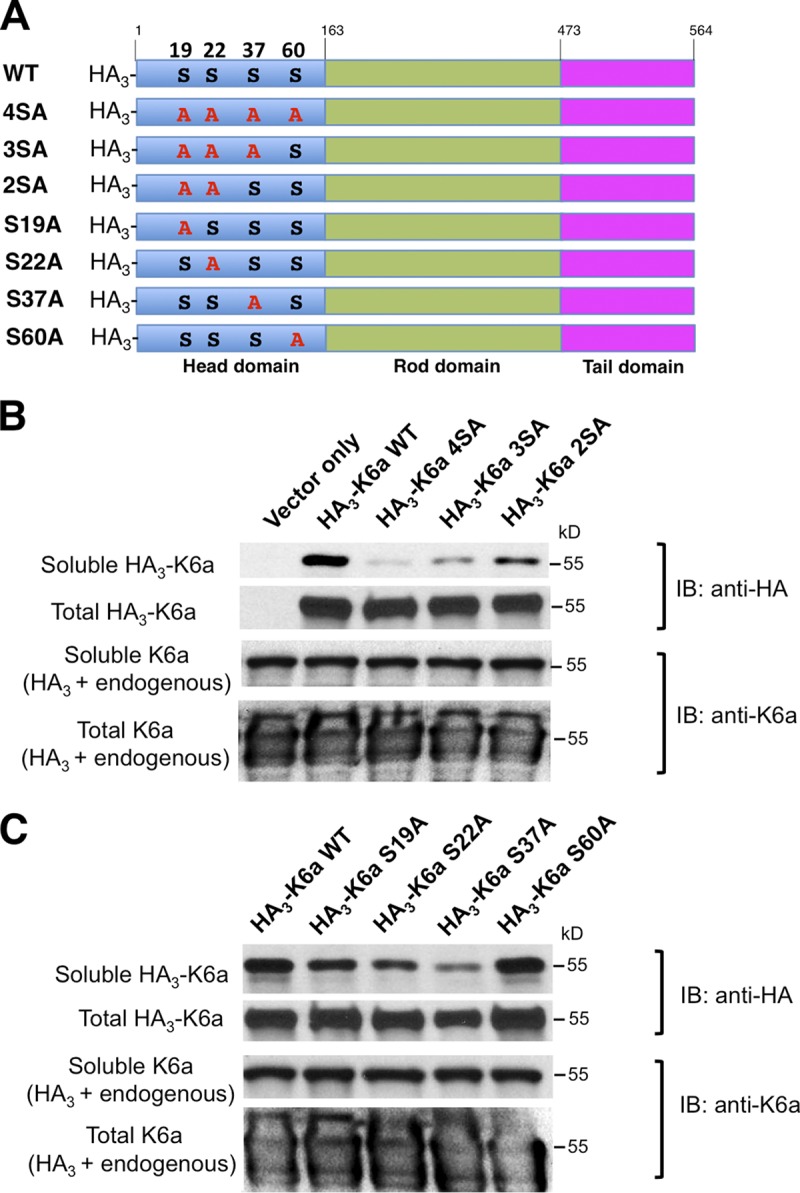
Site-specific serine-to-alanine K6a mutants are less soluble. (A) hTCEpi cells were nucleofected with empty plasmid or plasmids encoding HA-tagged WT K6a or HA-tagged K6a mutants with various Ser-to-Ala mutations. After harvest, cells were disrupted by NP-40 lysis buffer to dissolve soluble K6a or by RIPA buffer supplemented with 8 M urea to dissolve both soluble and filamentous K6a into total K6a. (B) Solubilities of K6a mutants with multiple serine-to-alanine mutations. Note that the solubilities of the quadruple (S19A, S22A, S37A, and S60A) and triple mutants (S19A, S22A, and S37A) were drastically reduced compared with WT, whereas the double mutant (S19A and S22A) had noticeable improvement. (C) Solubilities of K6a single serine-to-alanine mutants. Note that the single S19A, S22A and S37A mutants displayed markedly reduced solubilities compared with WT, with the S37A mutant being the least soluble. In contrast, the solubility of the S60A mutant was comparable to WT. Experiments were performed three times. IB, immunoblot.
The UPS degrades cytosolic K6a to generate KAMPs
The UPS is reported to degrade excess K8/K18 subunits released from the intermediate filament network in simple epithelial cells in response to shear stress and as a normal protein turnover mechanism (Ku and Omary, 2000; Jaitovich et al., 2008). These observations prompted us to investigate whether K6a in the cytosolic pool is processed by the UPS to produce KAMPs. We treated hTCEpi organotypic culture with 200 nM of the proteasome inhibitor epoxomicin, which is a potent and specific inhibitor of all three peptidase activities of both constitutive and immune proteasomes (Meng et al., 1999). We found that epoxomicin inhibited 80% of proteasomal trypsin-like peptidase activity without compromising cell viability (Fig. 5 A). We also found that under basal conditions and LTA stimulation, total ubiquitinated proteins in the cytosol were markedly increased as a result of proteasome inactivation (Fig. 5 B). Accumulation of ubiquitinated K6a was confirmed by blotting K6a immunoprecipitates with antiubiquitin antibody (Fig. 5 B). The immunoprecipitates were further analyzed by mass spectrometry. We detected that K6a was ubiquitinated at K180, K194, and K204 (Figs. 5 C and S5) and that damage-specific DNA binding protein 1 (DDB1), a substrate adapter protein for cullin 4 (CUL4; Higa et al., 2006) of the CRL complexes, was coimmunoprecipitated with K6a. To determine whether CRLs are involved in K6a ubiquitination, we treated hTCEpi cells with 200 nM of MLN4924, which blocks cullin neddylation and thereby activation. It was evident that, especially after proteasome inhibition by epoxomicin, MLN4924 treatment reduced the levels of ubiquitinated K6a (Fig. 5 D), suggesting that K6a ubiquitination and subsequent processing by the proteasome is mediated in part by CRL family.
Figure 5.

The UPS degrades soluble K6a to generate KAMPs. (A) Effects of proteasome inhibition to the cell viability of hTCEpi organotypic culture. Cells pretreated with 0, 50, 100, or 200 nM of epoxomicin for 2 h followed by cotreatment with the same dosage of epoxomicin and LTA (1 µg/ml) for 16 h were incubated with MTT substrate solution or proteasome-Glo cell-based reagent. Cell viability across all epoxomicin concentrations was maintained ∼80–100% in contrast with a dose-dependent decrease in proteasome activities to as low as 20% at the highest concentration of epoxomicin used. Means ± SD (n = 4) are shown. (B) Effect of proteasome inhibition on the level of polyubiquitinated K6a. hTCEpi organotypic culture were treated with DMSO (lanes 1, 3, and 5) or epoxomicin (200 nM; lanes 2, 4, and 6) for 2 h followed by continual treatment with DMSO (vehicle control) or epoxomicin in the absence (lanes 1, 2, and 5) or presence (lanes 3, 4, and 6) of LTA (1 µg/ml) for 18 h. Cells were harvested and disrupted by RIPA lysis buffer, and lysates were immunoprecipitated (IP) with anti-K6a antiserum (lanes 1–4) or preimmune serum (lanes 5 and 6). Ubiquitinated K6a and total K6a were immunoblotted (IB) with antiubiquitin antibody (topmost panel) and anti-K6a antiserum (second panel) respectively. Epoxomicin promotes the accumulation of polyubiquitinated K6a (lanes 2 and 4). (C) Cytosolic K6a was immunoprecipitated from hTCEpi organotypic culture treated with various bacterial ligands in the presence or absence of epoxomicin. LC-MS analysis identified three different ubiquitination sites on K6a. (D) Effect of CRL inhibitor MLN4924 on the level of polyubiquitinated K6a. hTCEpi cells were pretreated with MLN4924 (200 nM) for 1 h followed by DMSO (lanes 1–3) or epoxomicin (lanes 4–6) treatment for 18 h. Cell lysates were immunoprecipitated with preimmune serum (lanes 1 and 4) or anti-K6a antiserum (lanes 2 and 3 and lanes 5 and 6). Note the decrease of polyubiquitinated K6a level when CRL was inhibited. (E) Effect of proteasome inhibition on the level of KAMPs. hTCEpi cells were treated with DMSO (lane 1) or epoxomicin (200 nM; lane 2) for 2 h followed by continual treatment with DMSO and epoxomicin in the presence of LTA (1 µg/ml) for 16 h before cell harvest and disruption by NP-40 lysis buffer to collect cytosolic fractions. The level of KAMPs decreased in the presence of epoxomicin. (F) Trypsin-like proteasome activity of hTCEpi cells was unaffected by bacterial ligand challenge (means ± SD). Experiments were performed at least three times. RLU, relative light unit.
We next sought to assess the effect of proteasome inactivation on the level of cytosolic KAMPs in hTCEpi organotypic culture and found that the level of KAMPs was reduced in LTA-stimulated cells that were cotreated with epoxomicin (Fig. 5 E). To assess the physiological impact of proteasome inactivation and reduced KAMP production, we first tested the bactericidal activity of the <10-kD cytosolic lysate fractions against Pseudomonas aeruginosa, which is an important cause of corneal infections and blindness. As shown in Fig. 6 A, the viable bacterial count after 3 h incubation was significantly higher in the lysate fraction from epoxomicin-treated hTCEpi organotypic culture compared with the untreated cell lysate fraction, indicating that proteasome activity is critical for endogenous antimicrobial activity. Furthermore, local administration of epoxomicin to the corneas of live mice drastically increased bacterial adherence to the intact corneal surface ex vivo (Fig. 6 B) and reduced bacterial clearance from the ocular surface in vivo (Fig. 6 C). These results are in agreement with our previous observation that knocking down K6a markedly reduced the bactericidal activity of hTCEpi cell lysates in vitro and increased bacterial adherence on murine corneas ex vivo (Tam et al., 2012). Collectively, these data indicate that the UPS processes cytosolic K6a and generates KAMPs. Furthermore, although bacterial ligands triggered disassembly of the K6a filament network, leading to an increased cytosolic pool of K6a and KAMPs in hTCEpi cells (Figs. 1 and 2), ligand stimulation did not affect the level of proteasomal trypsin-like activity in these cells (Fig. 5 F). These observations indicate that the availability of cytosolic K6a (substrate) is the major determinant for rapid and timely production of KAMPs (product) that is required particularly during the early stages of infection to efficiently eliminate pathogenic microbes.
Figure 6.

The physiological impact of proteasome inactivation on corneal defense against P. aeruginosa. (A) In vitro antibacterial activity. Organotypic culture of hTCEpi cells was stimulated with LTA for 16 h in the presence or absence of epoxomicin (200 nM). Cells were lysed and processed to obtain the <10-kD lysate fractions. P. aeruginosa was added to the lysate fractions and incubated for 3 h at 37°C. Viable bacteria counts were determined after incubation. (B) Ex vivo antibacterial activity. Epoxomicin (80 pmol) or vehicle control was subconjunctivally delivered and topically administered to intact mouse cornea in vivo followed by exposure to bacterial culture supernatant. Enucleated eye balls were submerged in P. aeruginosa inoculum (1011 CFU/ml) for 3 h at 35°C. After extensive rinsing with PBS, corneas were dissected and homogenized to quantify bacterial adherence. (C) In vivo bacterial clearance activity. Mice were treated with epoxomicin and bacterial culture supernatant as described in B. P. aeruginosa (107 CFUs total) was inoculated to mouse eyes in vivo. After 3 h, viable bacteria counts in the ocular surface washes were determined. Means ± SD are shown. **, P < 0.01; ***, P < 0.001. Experiments were performed three times.
Discussion
In this study, we demonstrate that PTM and the UPS regulate filamentous K6a network remodeling and AMP production in response to bacterial ligands in human corneal epithelial cells. Our data show that host cells respond to both gram-positive (i.e., LTA) and gram-negative (i.e., LPS and flagellin) bacterial ligands by depolymerizing the keratin cytoskeleton, which in turn increases the availability of soluble K6a and KAMPs in the cytosol. Using high-resolution mass spectrometry, we identified four serine residues (S19, S22, S37, and S60) in the K6a head domain that are phosphorylated under basal conditions. Importantly, phosphorylation of these sites is increased after stimulation with bacterial ligands and correlates with increased levels of cytosolic K6a. Mutating a single site or a combination of these sites from serine to alanine blocked this activity and reduced K6a solubility. Furthermore, cytosolic K6a is a ubiquitination substrate, which is targeted by the proteasome machinery for processing into KAMPs. The de novo synthesized KAMPs can then kill bacterial pathogens by disrupting their cell envelope via the flexible glycine-rich motif as we described previously (Tam et al., 2012; Lee et al., 2016). A major role of the proteasome is to generate antigenic peptides for MHC I presentation (McCarthy and Weinberg, 2015); however, our findings reveal an essential role for the proteasome in production of KAMPs, which are an effective antimicrobial defense of epithelial cells.
TLRs located on the epithelial cell surface play a critical role in the very first line of defense against harmful microbes. The bacterial ligands used in this study, namely LTA, LPS, and flagellin, are recognized by TLR-2, TLR-4–MD-2 complex, and TLR-5 respectively. It is important to note that (a) activation of LPS/TLR-4 signaling requires a coreceptor MD-2 (Park et al., 2009), (b) epithelial cells, including those from the cornea, conjunctiva, gut, and lung, do not constitutively express this TLR-4 coreceptor and are hyporesponsive to LPS, and (c) IFN-γ has been shown to induce cell surface expression of MD-2 in these cells and confer LPS responsiveness as indicated by NF-κB activation and production of proinflammatory cytokines (Abreu et al., 2002; Suzuki et al., 2003; Jia et al., 2004; Talreja et al., 2005; Zhang et al., 2009; Vamadevan et al., 2010; Roy et al., 2011). In our study, we showed that K6a network reorganization and up-regulation of cytosolic K6a and KAMPs do not occur in corneal epithelial cells that are not primed with IFN-γ, supporting the notion that the K6a-mediated antimicrobial response to LPS is TLR driven. Nevertheless, we did not exclude the possibility that other cell surface receptors, notably the scavenger receptors (SRs) and EGFR, are involved. The SR family recognizes microbial ligands, and several members have been shown to cooperate with TLRs to activate immune responses (Canton et al., 2013). For instance, defective expression of CD36 (a class B SR) in bronchial epithelial cells weakens TLR-2 response to LTA (Mayer et al., 2007). The interplay of TLRs with EGFR has also been well established (Shaykhiev et al., 2008; Chattopadhyay et al., 2015; De et al., 2015). In the case of flagellin stimulation, EGFR can be activated without TLR-5 and NF-κB activation, potentially through an unidentified cell surface receptor, to produce AMPs LL-37 and hBD-2 and promote wound healing (Gao et al., 2010). It is unlikely that purified flagellin used in this study was recognized by intracellular receptors such as NOD-like receptors (NLRs) because live bacteria or specialized vehicles, such as pore-forming toxins (Molofsky et al., 2006) and lipid-based/liposome formulations (Franchi et al., 2006; Miao et al., 2006), are required to deliver flagellin to the cytosol.
Accumulating evidence reveals that keratins regulate diverse cellular functions, including gene and protein expression (Kim et al., 2006; Hobbs et al., 2015), subcellular targeting of protein and organelles (Petrosyan et al., 2015), and cell growth, survival, and differentiation (Ku et al., 2010; Fu et al., 2014; Petrosyan et al., 2015; Hobbs et al., 2016). PTMs are crucial to the functions of keratins (Sawant and Leube, 2017), and our data reveal new functional significance of keratin phosphorylation in innate immunity (Geisler and Leube, 2016), which may provide exciting insights into the association of K8/18 hyperphosphorylation and network remodeling with virus infection in hepatocytes, pancreatic acinar cells, and intestinal epithelial cells (Liao et al., 1995; Toivola et al., 2004, 2009). Similar to K8, K17, and K18 phosphorylation (Liao and Omary, 1996; Ku et al., 2002; Kim et al., 2006), K6a phosphorylation might enhance its interaction with the cytoplasmic adapter and scaffolding 14-3-3 proteins. These interactions might also limit cytosolic K6a from recycling back to the insoluble filamentous pool and thus promote its association with and ubiquitination by its E3 ligase for proteasomal processing to KAMPs. Consistent with this possibility, a motif scan of the K6a sequence indicates that S22 is a potential phosphobinding site for 14-3-3 (Obenauer et al., 2003). In addition, phosphorylation of K6a might enhance its interaction with ubiquitin ligases and therefore downstream processing to KAMPs. Indeed, CRLs often prefer or even require substrate phosphorylation for binding (Petroski and Deshaies, 2005; Nakayama and Nakayama, 2006; Deshaies and Joazeiro, 2009; Leung-Pineda et al., 2009; Skaar et al., 2009). Overall, K6a phosphorylation at distinct serine residues likely stimulates several mechanisms that contribute to KAMP production.
Intriguingly, although the flagellin-induced S19 and S22 double phosphorylation on K6a is more evident than the LPS-induced S37 phosphorylation and the LTA-induced S60 phosphorylation, all three bacterial ligands we tested were effective at triggering both solubilization and processing of K6a to increase production of KAMPs. It is possible that other PTMs, especially the labile ones such as O-linked glycosylation, were not detected in this study. Phosphorylation, sumoylation, acetylation, and glycosylation alone or in combination can modulate keratin solubility (Snider and Omary, 2014). Indeed, we observed mono- and dimethylation of K6a arginine 16 after bacterial ligand stimulation (unpublished data). Further experiments are underway to determine whether arginine methylation also regulates K6a solubility as well as its role together with serine phosphorylation.
Mammalian cells use at least two pathways to generate bioactive AMPs, including the classical pathway of proteolytic processing of precursor peptides such as defensins and cathelicidins (Lehrer and Ganz, 2002; Ganz, 2003) and autophagic sequestration of cytosolic proteins such as ubiquitin and ribosomal protein rpS30 precursor Finkel-Biskis-Reilly murine sarcoma virus (FBR-MuSV) associated ubiquitously (FAU) followed by lysosomal degradation producing small fragments (cryptides) that have antimicrobial properties (Ponpuak et al., 2010; Ponpuak and Deretic, 2011). Macrophages use the latter pathway against mycobacteria (Ponpuak et al., 2010; Ponpuak and Deretic, 2011). In this study, we uncovered an unexpected role of UPS in human corneal epithelial cells as a third pathway to generating AMPs. By inhibiting proteasome activity, we demonstrated an essential role for the proteasome in generating KAMPs and regulating the antimicrobial activity of these epithelial cells and the ocular defense against bacterial colonization. Interestingly, the extent of the increase in KAMPs (product) was greater than that of cytosolic K6a (substrate) under LPS (Fig. 2 B), but not flagellin or LTA stimulation (Fig. 2, A and C), suggesting that a pathway or pathways in addition to the UPS may be involved in generating KAMPs. Although suppression of proteasome activity can also trigger activation of autophagy as a compensatory effect (Tang et al., 2014; Yakoub and Shukla, 2015), our finding that epoxomicin significantly inhibits KAMP production (Fig. 5, B and E) demonstrates that the UPS plays a more dominant role than the potential autophagy pathway in bacterial ligand–induced KAMP production. Future studies will identify which E3 ligase ubiquitinates and targets K6a for proteasomal processing and assess the contribution of autophagy to KAMP production. Also, as KAMPs are an example of cryptides generated by the UPS, it would be of great interest to investigate whether more cryptides are generated by this system to enrich the arsenal of mucosal tissues for rapid antimicrobial protection.
In conclusion, our work identifies a novel cellular defense strategy involving finely regulated keratin cytoskeleton remodeling responses to extracellular bacterial ligands and the subsequent ubiquitin–proteasomal processing that produces KAMPs. As K6a gene expression is increased by IL-1, TNF-α, EGF, and TGF-α (Bernerd et al., 1993; Jiang et al., 1993; Komine et al., 2001) and proteasome activity is increased by IFN-γ and type I IFN (McCarthy and Weinberg, 2015), we believe that cytosolic K6a level and KAMP production may be further enhanced during infection and wound healing. Therapeutic stimulation of K6a expression and phosphorylation status as well as proteasome activity may prove to be beneficial strategies for strengthening the natural immune system to prevent and fight infections.
Materials and methods
Cell culture and treatments
hTCEpi cells were maintained at 37°C and 5% CO2 with standard keratinocyte growth medium 2 (KGM-2) containing 0.15 mM CaCl2 (low Ca–KGM-2; Lonza; Robertson et al., 2005). Except for confocal microscopy and plasmid nucleofection, all experiments were performed in hTCEpi cells grown as multilayered cultures (organotypic culture) by seeding onto 3-µm polyester transwell membrane supports inserted into a six-well plate (Costar; Augustin et al., 2011). In brief, cells were maintained in low Ca–KGM-2 until confluence and then differentiated in KGM-2 containing 1.15 mM CaCl2 (high Ca–KGM-2) for 4 d, with media present both in the inserts and six-well compartments. Cells were then airlifted to become multilayered by exposing to high Ca–KGM-2 present only in the lower six-well compartment for 7 d and in the absence of antibiotics for the last 2 d. Cells were treated with the following reagents for various durations and dosages as indicated in figure legends, which included recombinant human IFN-γ (285-IF-100/CF; R&D Systems), purified flagellin (tlrl-pafla; from P. aeruginosa; InvivoGen), LTA (L2515; from Staphylococcus aureus; Sigma-Aldrich), LPS (L8643; from P. aeruginosa; Sigma-Aldrich), calyculin A (sc-24000; Santa Cruz Biotechnology, Inc.), epoxomicin (324800; EMD Millipore), and MLN4924 (505477; EMD Millipore). For confocal microscopy, human telomerase–immortalized mammary epithelial cells (hTERT-HME1 cells; Takara Bio Inc.; a gift from G. Stark, Cleveland Clinic, Cleveland, OH) and HPV-16 E6/7–immortalized human ectocervical epithelial cells (CRL-2614; ATCC) were also used. The two cell lines were maintained in Medium 171 with mammary epithelial growth supplement (Thermo Fisher Scientific) and keratinocyte–serum-free media with 0.1 ng/ml human recombinant EGF, 0.05 mg/ml bovine pituitary extract, and additional calcium chloride 44.1 mg/liter (final concentration 0.4 mM; Thermo Fisher Scientific), respectively.
Antibodies
Rabbit polyclonal antiserums were raised against 10-mer KAMP (GGLSSVGGGS; Pacific Immunology) or 19-mer KAMP (RAIGGGLSSVGGGSSTIKY; New England Peptide), which correspond with amino acid residues 537–546 and 533–551, respectively, of the carboxyl terminal region of K6a. Both antibodies recognize full-length K6a and KAMPs. Other antibodies used were all mouse monoclonal, which included antiphosphoserine antibody (clone 4A4; EMD Millipore), antimonoubiquitination/antipolyubiquitin antibody (clone FK2; Enzo Life Sciences), anti-HA antibody (clone 16B12; BioLegend), and antiactin antibody (clone 8D10D10; Cell Signaling Technology).
Immunocytochemistry and confocal microscopy
hTCEpi cells at 90% confluence on Millicell EZ slides (EMD Millipore) were fixed with 4% paraformaldehyde in PBS for 15 min and permeabilized with 0.1% Triton X-100 in PBS supplemented with 5% BSA (wt/vol) for 15 min. Cells were blocked with 5% BSA for 1 h to reduce nonspecific binding of antibodies. K6a was stained with Alexa Fluor 488–labeled anti–19-mer KAMP antibody in 5% BSA at 4°C overnight. Nuclei were stained by propidium iodide or DAPI in mounting medium. Confocal images were captured with a TCS SP8 microscope using a 40× oil immersion objective (1.30 NA) and a zoom factor 2 or 3 (Leica Microsystems).
Western blot detection of K6a and KAMPs
hTCEpi cells were stimulated with bacterial ligands for 16 h. Bacterial ligands used were 0.5 µg/ml flagellin (FliC), 1 µg/ml LTA, or 1 µg/ml LPS. Before LPS stimulation, cells were pretreated with 250 ng/ml of IFN-γ for 2 h. After stimulation, cells were trypsinated by 1× trypLE (Thermo Fisher Scientific), washed with PBS, and lysed with NP-40 lysis buffer (1% [vol/vol] NP-40 in 20 mM Hepes, pH 7.4, 120 mM NaCl, 1 mM EDTA, and 1× protease and phosphatase inhibitor cocktail) for 15 min on ice. Soluble fractions containing cytosolic K6a and KAMPs were collected by centrifugation at 15,000 g for 10 min at 4°C. Insoluble pellets containing filamentous K6a were dissolved in 2% SDS buffer. Protein concentrations were determined by BCA assay (Thermo Fisher Scientific). Samples were resolved in 12% Bis-Tris protein gels (NuPAGE) and immunoblotted with anti–19-mer KAMPs antiserum to detect cytosolic and filamentous K6a, or they were resolved in 16% tricine gels (Thermo Fisher Scientific) and immunoblotted for KAMPs. Actin served as the loading control. Gels were transferred to 0.1-µm nitrocellulose membrane (GE Healthcare) using Trans-Blot Turbo Transfer System (Bio-Rad Laboratories). VisiGlo Select HRP chemiluminescent substrate (Amresco) and Pierce ECL Western blotting substrate (Thermo Fisher Scientific) were used to detect KAMPs and K6a/actin respectively.
Real-time quantitative RT-PCR
hTCEpi cells were stimulated with bacterial ligands for 3 or 6 h as described for Western blot detection of KAMPs and K6a. After treatment, cells were lysed by TRI reagent (Ambion), and total RNA was extracted with Direct-Zol RNA miniprep kit (Zymo Research). Purified RNA was reverse-transcribed with oligo-dT reverse primers and synthesized into cDNA with a Maxima H minus first-strand cDNA synthesis kit (Thermo Fisher Scientific). Real-time PCR was performed with a Luminaris color probe quantitative PCR master mix (Thermo Fisher Scientific) on a LightCycler 480 system (Roche) with predesigned TaqMan probe-primer sets (Integrated DNA Technologies) for K6a (Hs.PT.58.26132549.g), β-actin (Hs.PT.39a.22214847), and GAPDH (Hs.PT.39a.22214836) mRNAs. Thermal cycling reactions were consisted of 10 min of denaturation at 95°C followed by 40 cycles of 15 s at 95°C and 60 s at 60°C. Standard curves of housekeeping genes and K6a were generated to validate their equal efficiencies of amplification. Gene expression was normalized to β-actin and expressed as 2-ΔΔCT relative to mock-treated cells.
Liquid chromatography–tandem mass spectrometry (LC-MS) analysis of phospho- and ubiquitinated peptides
For mass spectrometry analysis of K6a PTMs, hTCEpi cells were stimulated with bacterial ligands and then lysed with NP-40 lysis buffer. Cytosolic extract (500 µg total protein) was precleared and then incubated overnight with 20 µl of anti–10-mer KAMP-conjugated agarose beads. Washed beads were eluted in 20 µl of Laemmli’s buffer at 95°C for 10 min, resolved in 12% Bis-Tris protein gels (NuPAGE), and stained by Coomassie Blue dye. K6a-containing gel bands (∼60 kD) were excised from the gel, digested with trypsin or chymotrypsin, and analyzed by a capillary column LC-MS system. The LC-MS system consisted of an Acclaim PepMap 100 Å–pore-size C18 reversed phase capillary chromatography column (Thermo Fisher Scientific) that was programmed to run at standard liquid chromatography gradient from 2–70% acetonitrile in 110 min and an LTQ-Orbitrap Elite hybrid mass spectrometer (Thermo Fisher Scientific) with the microelectrospray ion source operated at 2.5 kV. Collision-induced dissociation spectra were collected and searched against the K6a sequence using the Sequest algorithm (Eng et al., 1994) for K6a PTMs. The parameters used in this search included a peptide mass accuracy of 10 ppm, fragment ion mass accuracy of 0.6 D, carbamidomethylated cysteines as a constant modification, and oxidized methionine, lysine acetylation, and phosphorylation at S, T, and Y as a dynamic modification. The results were filtered based on Mascot ion scores and Sequest XCorr scores. All positively identified phosphopeptides were manually validated. The degree of phosphorylation of each peptide was determined by plotting chromatograms for the unmodified and phosphorylated forms and calculating the peak area ratios (phosphorylated/unmodified form; Willard et al., 2003; Waitkus et al., 2014).
Immunoprecipitation
For phospho-K6a and Ub-K6a analyses, hTCEpi cells were lysed by radioimmunoprecipitation assay (RIPA) buffer with 1× Halt protease and phosphatase inhibitor cocktail (Cell Signaling Technology) or in addition 10 mM N-ethylmaleimide (Sigma-Aldrich) to inhibit the action of deubiquitinases. Lysates were collected and precleared by centrifugation at 14,000 rpm for 20 min at 4°C. Precleared lysate (500 µg total protein) was mixed with 30 µl of anti–19-mer KAMP antiserum or preimmune serum for 2 h at 4°C followed by incubation with protein A/G plus agarose beads (Santa Cruz Biotechnology, Inc.) overnight. After six washes in RIPA buffer, immunoprecipitated proteins were eluted from the beads by incubating in 50 µl of Laemmli’s buffer at 95°C for 10 min. Eluted samples were resolved in Mini-Protean TGX 7.5% precast gel (Bio-Rad Laboratories) and immunoblotted with anti–19-mer KAMP antiserum, antiphosphoserine antibody, and antiubiquitin antibody for total K6a, phospho-K6a, and Ub-K6a, respectively. HRP-conjugated goat anti–mouse (Bio-Rad Laboratories) and conformation-specific mouse anti–rabbit (clone L27A9; Cell Signaling Technology) IgG (H+L) secondary antibodies were used accordingly. Dephosphorylation of the transferred K6a was performed on the immunoblot membrane using calf alkaline phosphatase (CIP; Cell Signaling Technology) in a buffer containing 100 mM Tris-HCl, pH 7.9, 50 mM NaCl, 10 mM MgCl2, and 1 mM dithiothreitol for 1 h at 37°C.
Plasmids and site-directed mutagenesis
To construct the mammalian expression vector encoding HA3-K6a, we used pCMV3-His-K6a (Sino Biological Inc.) as the PCR template, 5′-ATATGAATTCGCCACCATGGCCTACCCATACGATGTTCCTGACTATGCGGGCTATCCCTATGACGTCCCGGACTATGCAGGATATCCATATGACGTTCCAGATTACGCTGGTGCCAGCACATCCACCACCATC-3′ as the EcoRI site–containing forward primer, and 5′-ATATGCGGCCGCTTAGTGCTTGTAGCTCTTCCTGCTGGAG-3′ as the NotI site–containing reverse primer. Cloning of the K6a cDNAs into EcoRI- and NotI-digested pEGFP-N1 (Takara Bio Inc.) resulted in the removal of the EGFP sequence. Single or tandem site-directed mutagenesis of HA3-K6a to generate single or multiple serine-to-alanine mutations was performed using the QuikChange II XL site-directed mutagenesis kit (Promega). Primer sequences are shown in Table 1.
Table 1. Primer sequences for site-directed mutagenesis of HA3-K6a.
| Site | Strand | Mutagenic primer sequence (5′–3′) |
|---|---|---|
| S19A | sense | GCCGCCGGGGTTTCGCTGCCAACTCAGCC |
| S19A | antisense | GGCTGAGTTGGCAGCGAAACCCCGGCGGC |
| S22A | sense | GTTTCAGTGCCAACGCAGCCAGGCTCCCT |
| S22A | antisense | AGGGAGCCTGGCTGCGTTGGCACTGAAAC |
| S19A and S22A | sense | CGCCGGGGTTTCGCTGCCAACGCAGCCAGGCTCC |
| S19A and S22A | antisense | GGAGCCTGGCTGCGTTGGCAGCGAAACCCCGGCG |
| S37A | sense | CTTCAGCAGCGTCGCCGTGTCCCGCTC |
| S37A | antisense | GAGCGGGACACGGCGACGCTGCTGAAG |
| S60A | sense | CCAGGCCATACAGAGCGCGGCTGCCAAAGC |
| S60A | antisense | GCTTTGGCAGCCGCGCTCTGTATGGCCTGG |
Plasmid nucleofection
Mammalian expression vectors encoding HA3-tagged WT K6a or serine-to-alanine mutants were introduced to hTCEpi cells using the AMAXA Basic Nucleofector kit for Primary Mammalian Epithelial Cells (Lonza). Cells at a concentration of 2 × 106 cells/100 µl of nucleofector solution containing 2 µg of plasmid were loaded into the transfection cuvette and electroporated using program U-20. Nucleofected cells were recovered in 600 µl of low Ca–KGM-2 in 2-ml Eppendorf tubes and were placed at 37°C for 30 min. Cells were plated at the desired density into 24-well plates and maintained in low Ca–KGM-2 at 37°C and 5% CO2 for 72 h before harvest.
Cell viability and proteasome activity assays
hTCEpi cells were treated with 0, 50, 100, or 200 nM of epoxomicin in high Ca–KGM-2 for 2 h, cotreated with the same amounts of epoxomicin and LTA at 1 µg/ml for 16 h, and washed by PBS. Cells were assessed for viability and proteasome activity using an MTT growth assay (EMD Millipore) and a proteasome-Glo trypsin-like cell-based assay (Promega), respectively, according to the manufacturers’ instructions. A Wallac microplate reader (PerkinElmer) was used to measure absorbance or bioluminescence.
In vitro antimicrobial assay
hTCEpi cells were trypsinated, washed, and lysed with sterile saline by three cycles of freeze and thaw. Preparation of <10-kD cytosolic lysate fractions and antimicrobial assays against P. aeruginosa (cytotoxic strain 6206) were performed as described previously (Tam et al., 2012). In brief, lysates (1 mg/ml) normalized by BCA assays were fractionated at 4°C using a series of water-prerinsed Vivaspin protein concentrators with membrane cutoff at 100, 30, or 10 kD (GE Healthcare). All filtrates, including the <10-kD fractions, were normalized to the initial volume with saline. The inoculum was consisted of a P. aeruginosa culture at 108 colony-forming units (CFUs)/ml (OD650 reading at 0.1) in keratinocyte basal medium 2 (KBM-2; Lonza) diluted 100-fold in <10-kD lysate fractions. The mixtures were incubated for 3 h at 37°C followed by 10-fold serial dilutions by PBS and plated onto tryptic soy agar plates. After incubation overnight, the number of colonies in the agar plates was counted and adjusted for the dilution factor to derive viable bacterial counts (CFU/ml).
Murine models of bacterial clearance from the ocular surface
All procedures were conducted in compliance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic. Bacterial culture supernatant was prepared by growing P. aeruginosa strain PAO1 overnight with aeration in tryptic soy broth at 37°C followed by centrifugation at 16,000 g for 30 min and sterilization with a 0.22-µm filter. Epoxomicin (5 µl of 16 µM per eye) or vehicle control was injected subconjunctivally and applied topically (once each) to 8–10-wk-old sex-matched C57BL/6J mice (The Jackson Lab). After 2 h, the eyes were inoculated three times within an hour with bacterial culture supernatant (5 µl per eye) to up-regulate expression of defense factors (Maltseva et al., 2007; Augustin et al., 2011; Sullivan et al., 2015). The procedure of topical administration of epoxomicin and bacterial culture supernatant was repeated once after 24 h. Slit-lamp examination of mouse eyes was used to confirm that the physical barrier (i.e., tight junctions) of the corneas remained intact after epoxomicin treatment, as indicated by the absence of corneal staining with fluorescein. To assess the susceptibility of corneas to bacterial adhesion in the absence of other confounding host factors, mice were sacrificed by lethal injection of anesthetic cocktail (25 mg/ml ketamine and 0.2 mg/ml dexmedetomidine), and whole eyes were enucleated and rinsed with PBS to exclude tear fluid followed by incubation with 200 µl carbenicillin-resistant GFP-expressing P. aeruginosa strain PAO1 (PAO1-GFP; 1011 CFU/ml) for 3 h at 35°C as previously described (Alarcon et al., 2011; Tam et al., 2012). After extensive rinsing with PBS, corneas were excised and homogenized in PBS to collect viable bacteria adhered to the cornea followed by quantification by plating on tryptic soy agar supplemented with 300 µg/ml carbenicillin. To assess bacterial clearance from the ocular surface in vivo as previously described (Mun et al., 2009; Heimer et al., 2013), mice were inoculated with 107 CFU (in 5 µl) of PAO1-GFP after i.p. injection of anesthetic cocktail. At 3 h after inoculation, 4 µl of PBS was added to the ocular surface and then collected immediately using a 10-µl-volume glass Drummond capillary tube placed at the lateral canthus. The number of viable bacteria within the ocular surface washes was quantified by plating on tryptic soy agar.
Statistics
All experiments except the animal experiments were analyzed by the unpaired two-tailed Student’s t test to determine p-values. The nonparametric Mann-Whitney U test was used to analyze animal experiments. All experiments were performed at least three times. P < 0.05 was considered significant.
Online supplemental material
Fig. S1 contains confocal images of K6a filament network reorganization in human mammary epithelial cells and ectocervical epithelial cells in response to bacterial ligands. Fig. S2 contains confocal images of K6a filament network in human corneal epithelial cells showing that priming the cells with IFN-γ is required for LPS-induced network reorganization. Figs. S3 and S4 contain supporting data for Fig. 3 A showing the tandem mass spectrometry spectra for K6a peptides with a single phosphorylation site at serines 22, 37, or 60 or double phosphorylation sites at serines 19 and 22. Fig. S5 contains supporting data for Fig. 5 C showing the tandem mass spectrometry spectra for K6a peptides ubiquitinated at lysines 180, 194, and 204.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health/National Eye Institute grant R01EY023000 and the Cleveland Clinic research fund (to C. Tam). Mass spectrometry was performed at the Cleveland Clinic Lerner Research Institute Proteomics and Metabolomics Core supported by National Institutes of Health shared instrument grant (1S10RR031537-01). We also acknowledge support from National Institutes of Health/National Eye Institute P30 Core Grant (P30EY025585-01A1) and an unrestricted grant from Research to Prevent Blindness, Inc. awarded to the Cole Eye Institute.
C. Tam is listed as a coinventor on US Patent issued November 17, 2015, No. 9,187,541 B2 entitled “Anti-Microbial Peptides and Methods of Use Thereof.” The author has no additional competing financial interests. All authors except C. Tam declare no competing financial interests.
Author contributions: J.K.L. Chan, D. Yuen, P.H.-M. Too, Y. Sun, and D. Man performed the experiments. B. Willard performed mass spectrometric analysis. C. Tam and J.K.L. Chan designed the experiments analyzed and interpreted the data, and wrote the manuscript. C. Tam conceived and supervised the project.
References
- Abreu M.T., Arnold E.T., Thomas L.S., Gonsky R., Zhou Y., Hu B., and Arditi M.. 2002. TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J. Biol. Chem. 277:20431–20437. 10.1074/jbc.M110333200 [DOI] [PubMed] [Google Scholar]
- Alarcon I., Tam C., Mun J.J., LeDue J., Evans D.J., and Fleiszig S.M.. 2011. Factors impacting corneal epithelial barrier function against Pseudomonas aeruginosa traversal. Invest. Ophthalmol. Vis. Sci. 52:1368–1377. 10.1167/iovs.10-6125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin D.K., Heimer S.R., Tam C., Li W.Y., Le Due J.M., Evans D.J., and Fleiszig S.M.. 2011. Role of defensins in corneal epithelial barrier function against Pseudomonas aeruginosa traversal. Infect. Immun. 79:595–605. 10.1128/IAI.00854-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernerd F., Magnaldo T., Freedberg I.M., and Blumenberg M.. 1993. Expression of the carcinoma-associated keratin K6 and the role of AP-1 proto-oncoproteins. Gene Expr. 3:187–199. [PMC free article] [PubMed] [Google Scholar]
- Canton J., Neculai D., and Grinstein S.. 2013. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 13:621–634. 10.1038/nri3515 [DOI] [PubMed] [Google Scholar]
- Chattopadhyay S., Veleeparambil M., Poddar D., Abdulkhalek S., Bandyopadhyay S.K., Fensterl V., and Sen G.C.. 2015. EGFR kinase activity is required for TLR4 signaling and the septic shock response. EMBO Rep. 16:1535–1547. 10.15252/embr.201540337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S., Zhou H., DeSantis D., Croniger C.M., Li X., and Stark G.R.. 2015. Erlotinib protects against LPS-induced endotoxicity because TLR4 needs EGFR to signal. Proc. Natl. Acad. Sci. USA. 112:9680–9685. 10.1073/pnas.1511794112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies R.J., and Joazeiro C.A.. 2009. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78:399–434. 10.1146/annurev.biochem.78.101807.093809 [DOI] [PubMed] [Google Scholar]
- Dyrlund T.F., Poulsen E.T., Scavenius C., Nikolajsen C.L., Thøgersen I.B., Vorum H., and Enghild J.J.. 2012. Human cornea proteome: identification and quantitation of the proteins of the three main layers including epithelium, stroma, and endothelium. J. Proteome Res. 11:4231–4239. 10.1021/pr300358k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng J.K., McCormack A.L., and Yates J.R.. 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5:976–989. 10.1016/1044-0305(94)80016-2 [DOI] [PubMed] [Google Scholar]
- Fox J.L. 2013. Antimicrobial peptides stage a comeback. Nat. Biotechnol. 31:379–382. 10.1038/nbt.2572 [DOI] [PubMed] [Google Scholar]
- Franchi L., Amer A., Body-Malapel M., Kanneganti T.D., Ozören N., Jagirdar R., Inohara N., Vandenabeele P., Bertin J., Coyle A., et al. . 2006. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 7:576–582. 10.1038/ni1346 [DOI] [PubMed] [Google Scholar]
- Fu D.J., Thomson C., Lunny D.P., Dopping-Hepenstal P.J., McGrath J.A., Smith F.J.D., Irwin McLean W.H., and Leslie Pedrioli D.M.. 2014. Keratin 9 is required for the structural integrity and terminal differentiation of the palmoplantar epidermis. J. Invest. Dermatol. 134:754–763. 10.1038/jid.2013.356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T. 2003. Defensins: antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3:710–720. 10.1038/nri1180 [DOI] [PubMed] [Google Scholar]
- Gao N., Kumar A., Jyot J., and Yu F.S.. 2010. Flagellin-induced corneal antimicrobial peptide production and wound repair involve a novel NF-kappaB-independent and EGFR-dependent pathway. PLoS One. 5:e9351 10.1371/journal.pone.0009351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler F., and Leube R.E.. 2016. Epithelial Intermediate Filaments: Guardians against Microbial Infection? Cells. 5:29 10.3390/cells5030029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimer S.R., Evans D.J., Mun J.J., Stern M.E., and Fleiszig S.M.. 2013. Surfactant protein D contributes to ocular defense against Pseudomonas aeruginosa in a murine model of dry eye disease. PLoS One. 8:e65797 10.1371/journal.pone.0065797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann H., Strelkov S.V., Burkhard P., and Aebi U.. 2009. Intermediate filaments: primary determinants of cell architecture and plasticity. J. Clin. Invest. 119:1772–1783. 10.1172/JCI38214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa L.A., Wu M., Ye T., Kobayashi R., Sun H., and Zhang H.. 2006. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 8:1277–1283. 10.1038/ncb1490 [DOI] [PubMed] [Google Scholar]
- Hobbs R.P., DePianto D.J., Jacob J.T., Han M.C., Chung B.M., Batazzi A.S., Poll B.G., Guo Y., Han J., Ong S., et al. . 2015. Keratin-dependent regulation of Aire and gene expression in skin tumor keratinocytes. Nat. Genet. 47:933–938. 10.1038/ng.3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs R.P., Batazzi A.S., Han M.C., and Coulombe P.A.. 2016. Loss of Keratin 17 induces tissue-specific cytokine polarization and cellular differentiation in HPV16-driven cervical tumorigenesis in vivo. Oncogene. 35:5653–5662. 10.1038/onc.2016.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitovich A., Mehta S., Na N., Ciechanover A., Goldman R.D., and Ridge K.M.. 2008. Ubiquitin-proteasome-mediated degradation of keratin intermediate filaments in mechanically stimulated A549 cells. J. Biol. Chem. 283:25348–25355. 10.1074/jbc.M801635200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H.P., Kline J.N., Penisten A., Apicella M.A., Gioannini T.L., Weiss J., and McCray P.B. Jr. 2004. Endotoxin responsiveness of human airway epithelia is limited by low expression of MD-2. Am. J. Physiol. Lung Cell. Mol. Physiol. 287:L428–L437. 10.1152/ajplung.00377.2003 [DOI] [PubMed] [Google Scholar]
- Jiang C.K., Magnaldo T., Ohtsuki M., Freedberg I.M., Bernerd F., and Blumenberg M.. 1993. Epidermal growth factor and transforming growth factor alpha specifically induce the activation- and hyperproliferation-associated keratins 6 and 16. Proc. Natl. Acad. Sci. USA. 90:6786–6790. 10.1073/pnas.90.14.6786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A.C., Heinzel F.P., Diaconu E., Sun Y., Hise A.G., Golenbock D., Lass J.H., and Pearlman E.. 2005. Activation of toll-like receptor (TLR)2, TLR4, and TLR9 in the mammalian cornea induces MyD88-dependent corneal inflammation. Invest. Ophthalmol. Vis. Sci. 46:589–595. 10.1167/iovs.04-1077 [DOI] [PubMed] [Google Scholar]
- Johnson A.C., Li X., and Pearlman E.. 2008. MyD88 functions as a negative regulator of TLR3/TRIF-induced corneal inflammation by inhibiting activation of c-Jun N-terminal kinase. J. Biol. Chem. 283:3988–3996. 10.1074/jbc.M707264200 [DOI] [PubMed] [Google Scholar]
- Kim S., Wong P., and Coulombe P.A.. 2006. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature. 441:362–365. 10.1038/nature04659 [DOI] [PubMed] [Google Scholar]
- Kölsch A., Windoffer R., Würflinger T., Aach T., and Leube R.E.. 2010. The keratin-filament cycle of assembly and disassembly. J. Cell Sci. 123:2266–2272. 10.1242/jcs.068080 [DOI] [PubMed] [Google Scholar]
- Komine M., Rao L.S., Freedberg I.M., Simon M., Milisavljevic V., and Blumenberg M.. 2001. Interleukin-1 induces transcription of keratin K6 in human epidermal keratinocytes. J. Invest. Dermatol. 116:330–338. 10.1046/j.1523-1747.2001.01249.x [DOI] [PubMed] [Google Scholar]
- Ku N.O., and Omary M.B.. 2000. Keratins turn over by ubiquitination in a phosphorylation-modulated fashion. J. Cell Biol. 149:547–552. 10.1083/jcb.149.3.547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N.O., and Omary M.B.. 2006. A disease- and phosphorylation-related nonmechanical function for keratin 8. J. Cell Biol. 174:115–125. 10.1083/jcb.200602146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N.O., Gish R., Wright T.L., and Omary M.B.. 2001. Keratin 8 mutations in patients with cryptogenic liver disease. N. Engl. J. Med. 344:1580–1587. 10.1056/NEJM200105243442103 [DOI] [PubMed] [Google Scholar]
- Ku N.O., Michie S., Resurreccion E.Z., Broome R.L., and Omary M.B.. 2002. Keratin binding to 14-3-3 proteins modulates keratin filaments and hepatocyte mitotic progression. Proc. Natl. Acad. Sci. USA. 99:4373–4378. 10.1073/pnas.072624299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku N.O., Strnad P., Zhong B.H., Tao G.Z., and Omary M.B.. 2007. Keratins let liver live: Mutations predispose to liver disease and crosslinking generates Mallory-Denk bodies. Hepatology. 46:1639–1649. 10.1002/hep.21976 [DOI] [PubMed] [Google Scholar]
- Ku N.O., Toivola D.M., Strnad P., and Omary M.B.. 2010. Cytoskeletal keratin glycosylation protects epithelial tissue from injury. Nat. Cell Biol. 12:876–885. 10.1038/ncb2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Zhang J., and Yu F.S.. 2006. Toll-like receptor 2-mediated expression of beta-defensin-2 in human corneal epithelial cells. Microbes Infect. 8:380–389. 10.1016/j.micinf.2005.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y., and Gallo R.L.. 2009. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 30:131–141. 10.1016/j.it.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.T., Wang G., Tam Y.T., and Tam C.. 2016. Membrane-Active Epithelial Keratin 6A Fragments (KAMPs) Are Unique Human Antimicrobial Peptides with a Non-αβ Structure. Front. Microbiol. 7:1799 10.3389/fmicb.2016.01799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrer R.I., and Ganz T.. 2002. Cathelicidins: a family of endogenous antimicrobial peptides. Curr. Opin. Hematol. 9:18–22. 10.1097/00062752-200201000-00004 [DOI] [PubMed] [Google Scholar]
- Leung-Pineda V., Huh J., and Piwnica-Worms H.. 2009. DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res. 69:2630–2637. 10.1158/0008-5472.CAN-08-3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J., and Omary M.B.. 1996. 14-3-3 proteins associate with phosphorylated simple epithelial keratins during cell cycle progression and act as a solubility cofactor. J. Cell Biol. 133:345–357. 10.1083/jcb.133.2.345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J., Lowthert L.A., and Omary M.B.. 1995. Heat stress or rotavirus infection of human epithelial cells generates a distinct hyperphosphorylated form of keratin 8. Exp. Cell Res. 219:348–357. 10.1006/excr.1995.1238 [DOI] [PubMed] [Google Scholar]
- Mahlapuu M., Håkansson J., Ringstad L., and Björn C.. 2016. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 6:194 10.3389/fcimb.2016.00194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltseva I.A., Fleiszig S.M., Evans D.J., Kerr S., Sidhu S.S., McNamara N.A., and Basbaum C.. 2007. Exposure of human corneal epithelial cells to contact lenses in vitro suppresses the upregulation of human beta-defensin-2 in response to antigens of Pseudomonas aeruginosa. Exp. Eye Res. 85:142–153. 10.1016/j.exer.2007.04.001 [DOI] [PubMed] [Google Scholar]
- Mayer A.K., Muehmer M., Mages J., Gueinzius K., Hess C., Heeg K., Bals R., Lang R., and Dalpke A.H.. 2007. Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J. Immunol. 178:3134–3142. 10.4049/jimmunol.178.5.3134 [DOI] [PubMed] [Google Scholar]
- McCarthy M.K., and Weinberg J.B.. 2015. The immunoproteasome and viral infection: a complex regulator of inflammation. Front. Microbiol. 6:21 10.3389/fmicb.2015.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh P.B., Laskey P., Sullivan K., Davy C., Wang Q., Jackson D.J., Griffin H.M., and Doorbar J.. 2010. E1--E4-mediated keratin phosphorylation and ubiquitylation: a mechanism for keratin depletion in HPV16-infected epithelium. J. Cell Sci. 123:2810–2822. 10.1242/jcs.061978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L., Mohan R., Kwok B.H., Elofsson M., Sin N., and Crews C.M.. 1999. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA. 96:10403–10408. 10.1073/pnas.96.18.10403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao E.A., Alpuche-Aranda C.M., Dors M., Clark A.E., Bader M.W., Miller S.I., and Aderem A.. 2006. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7:569–575. 10.1038/ni1344 [DOI] [PubMed] [Google Scholar]
- Moll R., Divo M., and Langbein L.. 2008. The human keratins: biology and pathology. Histochem. Cell Biol. 129:705–733. 10.1007/s00418-008-0435-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky A.B., Byrne B.G., Whitfield N.N., Madigan C.A., Fuse E.T., Tateda K., and Swanson M.S.. 2006. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203:1093–1104. 10.1084/jem.20051659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun J.J., Tam C., Kowbel D., Hawgood S., Barnett M.J., Evans D.J., and Fleiszig S.M.. 2009. Clearance of Pseudomonas aeruginosa from a healthy ocular surface involves surfactant protein D and is compromised by bacterial elastase in a murine null-infection model. Infect. Immun. 77:2392–2398. 10.1128/IAI.00173-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T., Goshima F., Nishizawa Y., Daikoku T., Takakuwa H., Ohtsuka K., Yoshikawa T., and Nishiyama Y.. 2002. Phosphorylation of cytokeratin 17 by herpes simplex virus type 2 US3 protein kinase. Microbiol. Immunol. 46:707–719. 10.1111/j.1348-0421.2002.tb02755.x [DOI] [PubMed] [Google Scholar]
- Nakayama K.I., and Nakayama K.. 2006. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer. 6:369–381. 10.1038/nrc1881 [DOI] [PubMed] [Google Scholar]
- Obenauer J.C., Cantley L.C., and Yaffe M.B.. 2003. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31:3635–3641. 10.1093/nar/gkg584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X., Hobbs R.P., and Coulombe P.A.. 2013. The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr. Opin. Cell Biol. 25:47–56. 10.1016/j.ceb.2012.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B.S., Song D.H., Kim H.M., Choi B.S., Lee H., and Lee J.O.. 2009. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 458:1191–1195. 10.1038/nature07830 [DOI] [PubMed] [Google Scholar]
- Petroski M.D., and Deshaies R.J.. 2005. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6:9–20. 10.1038/nrm1547 [DOI] [PubMed] [Google Scholar]
- Petrosyan A., Ali M.F., and Cheng P.W.. 2015. Keratin 1 plays a critical role in golgi localization of core 2 N-acetylglucosaminyltransferase M via interaction with its cytoplasmic tail. J. Biol. Chem. 290:6256–6269. 10.1074/jbc.M114.618702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponpuak M., and Deretic V.. 2011. Autophagy and p62/sequestosome 1 generate neo-antimicrobial peptides (cryptides) from cytosolic proteins. Autophagy. 7:336–337. 10.4161/auto.7.3.14500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponpuak M., Davis A.S., Roberts E.A., Delgado M.A., Dinkins C., Zhao Z., Virgin H.W. IV, Kyei G.B., Johansen T., Vergne I., and Deretic V.. 2010. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 32:329–341. 10.1016/j.immuni.2010.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh J.C., Guo J.T., Aldrich C., Rall G., Kajino K., Tennant B., England J.M., and Mason W.S.. 1998. Aberrant expression of a cytokeratin in a subset of hepatocytes during chronic WHV infection. Virology. 249:68–79. 10.1006/viro.1998.9326 [DOI] [PubMed] [Google Scholar]
- Robertson D.M., Li L., Fisher S., Pearce V.P., Shay J.W., Wright W.E., Cavanagh H.D., and Jester J.V.. 2005. Characterization of growth and differentiation in a telomerase-immortalized human corneal epithelial cell line. Invest. Ophthalmol. Vis. Sci. 46:470–478. 10.1167/iovs.04-0528 [DOI] [PubMed] [Google Scholar]
- Robertson D.M., Ho S.I., and Cavanagh H.D.. 2008. Characterization of DeltaNp63 isoforms in normal cornea and telomerase-immortalized human corneal epithelial cells. Exp. Eye Res. 86:576–585. 10.1016/j.exer.2007.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson D.M., Kalangara J.P., Baucom R.B., Petroll W.M., and Cavanagh H.D.. 2011. A reconstituted telomerase-immortalized human corneal epithelium in vivo: a pilot study. Curr. Eye Res. 36:706–712. 10.3109/02713683.2011.582662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotty J.D., and Coulombe P.A.. 2012. A wound-induced keratin inhibits Src activity during keratinocyte migration and tissue repair. J. Cell Biol. 197:381–389. 10.1083/jcb.201107078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S., Sun Y., and Pearlman E.. 2011. Interferon-gamma-induced MD-2 protein expression and lipopolysaccharide (LPS) responsiveness in corneal epithelial cells is mediated by Janus tyrosine kinase-2 activation and direct binding of STAT1 protein to the MD-2 promoter. J. Biol. Chem. 286:23753–23762. 10.1074/jbc.M111.219345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S., Karmakar M., and Pearlman E.. 2014. CD14 mediates Toll-like receptor 4 (TLR4) endocytosis and spleen tyrosine kinase (Syk) and interferon regulatory transcription factor 3 (IRF3) activation in epithelial cells and impairs neutrophil infiltration and Pseudomonas aeruginosa killing in vivo. J. Biol. Chem. 289:1174–1182. 10.1074/jbc.M113.523167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozek A., Friedrich C.L., and Hancock R.E.. 2000. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry. 39:15765–15774. 10.1021/bi000714m [DOI] [PubMed] [Google Scholar]
- Sawant M.S., and Leube R.E.. 2017. Consequences of Keratin Phosphorylation for Cytoskeletal Organization and Epithelial Functions. Int. Rev. Cell Mol. Biol. 330:171–225. 10.1016/bs.ircmb.2016.09.005 [DOI] [PubMed] [Google Scholar]
- Shaykhiev R., Behr J., and Bals R.. 2008. Microbial patterns signaling via Toll-like receptors 2 and 5 contribute to epithelial repair, growth and survival. PLoS One. 3:e1393 10.1371/journal.pone.0001393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar J.R., D’Angiolella V., Pagan J.K., and Pagano M.. 2009. SnapShot: F Box Proteins II. Cell. 137:1358. [DOI] [PubMed] [Google Scholar]
- Snider N.T., and Omary M.B.. 2014. Post-translational modifications of intermediate filament proteins: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 15:163–177. 10.1038/nrm3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan A.B., Tam K.P., Metruccio M.M., Evans D.J., and Fleiszig S.M.. 2015. The importance of the Pseudomonas aeruginosa type III secretion system in epithelium traversal depends upon conditions of host susceptibility. Infect. Immun. 83:1629–1640. 10.1128/IAI.02329-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M., Hisamatsu T., and Podolsky D.K.. 2003. Gamma interferon augments the intracellular pathway for lipopolysaccharide (LPS) recognition in human intestinal epithelial cells through coordinated up-regulation of LPS uptake and expression of the intracellular Toll-like receptor 4-MD-2 complex. Infect. Immun. 71:3503–3511. 10.1128/IAI.71.6.3503-3511.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma T., Ichida T., Okumura K., and Kanazawa M.. 1993. Protein phosphatase inhibitor calyculin A induces hyperphosphorylation of cytokeratins and inhibits amylase exocytosis in the rat parotid acini. FEBS Lett. 323:145–150. 10.1016/0014-5793(93)81467-E [DOI] [PubMed] [Google Scholar]
- Talreja J., Dileepan K., Puri S., Kabir M.H., Segal D.M., Stechschulte D.J., and Dileepan K.N.. 2005. Human conjunctival epithelial cells lack lipopolysaccharide responsiveness due to deficient expression of MD2 but respond after interferon-gamma priming or soluble MD2 supplementation. Inflammation. 29:170–181. 10.1007/s10753-006-9014-y [DOI] [PubMed] [Google Scholar]
- Tam C., Mun J.J., Evans D.J., and Fleiszig S.M.. 2012. Cytokeratins mediate epithelial innate defense through their antimicrobial properties. J. Clin. Invest. 122:3665–3677. 10.1172/JCI64416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B., Cai J., Sun L., Li Y., Qu J., Snider B.J., and Wu S.. 2014. Proteasome inhibitors activate autophagy involving inhibition of PI3K-Akt-mTOR pathway as an anti-oxidation defense in human RPE cells. PLoS One. 9:e103364 10.1371/journal.pone.0103364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivola D.M., Ku N.O., Resurreccion E.Z., Nelson D.R., Wright T.L., and Omary M.B.. 2004. Keratin 8 and 18 hyperphosphorylation is a marker of progression of human liver disease. Hepatology. 40:459–466. 10.1002/hep.20277 [DOI] [PubMed] [Google Scholar]
- Toivola D.M., Ostrowski S.E., Baribault H., Magin T.M., Ramsingh A.I., and Omary M.B.. 2009. Keratins provide virus-dependent protection or predisposition to injury in coxsackievirus-induced pancreatitis. Cell Health Cytoskelet. 1:51–65. 10.2147/CHC.S5792 [DOI] [Google Scholar]
- Ueta M., Hamuro J., Kiyono H., and Kinoshita S.. 2005. Triggering of TLR3 by polyI:C in human corneal epithelial cells to induce inflammatory cytokines. Biochem. Biophys. Res. Commun. 331:285–294. 10.1016/j.bbrc.2005.02.196 [DOI] [PubMed] [Google Scholar]
- Vamadevan A.S., Fukata M., Arnold E.T., Thomas L.S., Hsu D., and Abreu M.T.. 2010. Regulation of Toll-like receptor 4-associated MD-2 in intestinal epithelial cells: a comprehensive analysis. Innate Immun. 16:93–103. 10.1177/1753425909339231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waitkus M.S., Chandrasekharan U.M., Willard B., Tee T.L., Hsieh J.K., Przybycin C.G., Rini B.I., and Dicorleto P.E.. 2014. Signal integration and gene induction by a functionally distinct STAT3 phosphoform. Mol. Cell. Biol. 34:1800–1811. 10.1128/MCB.00034-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Mishra B., Epand R.F., and Epand R.M.. 2014. High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochim. Biophys. Acta. 1838:2160–2172. 10.1016/j.bbamem.2014.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Li X., and Wang Z.. 2016. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44(D1):D1087–D1093. 10.1093/nar/gkv1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard B.B., Ruse C.I., Keightley J.A., Bond M., and Kinter M.. 2003. Site-specific quantitation of protein nitration using liquid chromatography/tandem mass spectrometry. Anal. Chem. 75:2370–2376. 10.1021/ac034033j [DOI] [PubMed] [Google Scholar]
- Windoffer R., Kölsch A., Wöll S., and Leube R.E.. 2006. Focal adhesions are hotspots for keratin filament precursor formation. J. Cell Biol. 173:341–348. 10.1083/jcb.200511124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windoffer R., Beil M., Magin T.M., and Leube R.E.. 2011. Cytoskeleton in motion: the dynamics of keratin intermediate filaments in epithelia. J. Cell Biol. 194:669–678. 10.1083/jcb.201008095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakoub A.M., and Shukla D.. 2015. Autophagy stimulation abrogates herpes simplex virus-1 infection. Sci. Rep. 5:9730 10.1038/srep09730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.J., and Gallo R.L.. 2016. Antimicrobial peptides. Curr. Biol. 26:R14–R19. 10.1016/j.cub.2015.11.017 [DOI] [PubMed] [Google Scholar]
- Zhang J., Xu K., Ambati B., and Yu F.S.. 2003. Toll-like receptor 5-mediated corneal epithelial inflammatory responses to Pseudomonas aeruginosa flagellin. Invest. Ophthalmol. Vis. Sci. 44:4247–4254. 10.1167/iovs.03-0219 [DOI] [PubMed] [Google Scholar]
- Zhang J., Kumar A., Wheater M., and Yu F.S.. 2009. Lack of MD-2 expression in human corneal epithelial cells is an underlying mechanism of lipopolysaccharide (LPS) unresponsiveness. Immunol. Cell Biol. 87:141–148. 10.1038/icb.2008.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.