
Figure 1.
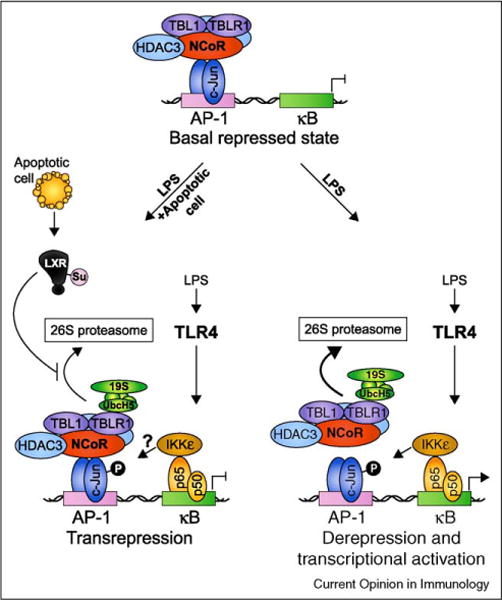
Ligand-dependent transrepression as a possible mechanism for apoptotic cell-mediated suppression of TLR-induced inflammatory gene expression. In the repressed ‘off’ state, LPS-induced genes such as the well characterized iNOS gene are actively suppressed by a multisubunit complex containing NCoR, HDAC3, TBL1, and TBLR1. When TLR4 signaling is initiated, IκB is degraded and NF-κB p50-p65 subunits enter the nucleus and bind to κB elements in the promoter. Phosphorylation of p65 on S536 creates a docking site for IκB Kinase ε (IKKε) allowing it to phosphorylate adjacent c-Jun/NCoR complexes and initiate corepressor clearance through TBLR1-mediated recruitment of UbcH5 and the 19S proteasome, ubiquitination, and subsequent degradation by the 26S proteasome. Binding of NF-κB allows recruitment of coactivators resulting in derepression and transcriptional activation. In this way, coupled AP-1/κB elements have been suggested to serve as ‘integrated circuits’ for switching promoters from a repressed to an activated state in response to an inflammatory trigger [28••]. When both TLR ligands and nuclear receptor ligands are present (depicted here as ‘LPS + apoptotic cell’ where the ligands for LXR are derived from apoptotic cells), ligand binding to LXR results in its SUMOylation by the E2 ligase Ubc9 and the E3 ligase HDAC4 and subsequent ability to interfere with NCoR corepressor clearance [27]. The result is ligand-dependent transrepression of inflammatory-response gene transcription.