

Abstract
Mutations in the gene tafazzin (TAZ) result in Barth syndrome (BTHS). Patients present with hypotonia, cyclic neutropenia, 3-methyglutaconic aciduria, and cardiomyopathy, which is the major cause of mortality. The recessive, X-linked TAZ gene encodes a mitochondrial membrane-associated phospholipid modifying enzyme, which adds unsaturated fatty acid species to monolysocardiolipin to generate mature cardiolipin in the mitochondrial membrane that is essential for mitochondrial morphology and function. To identify intrinsic mitochondrial localization sequences in the human TAZ protein, we made sequential TAZ peptide-eGFP fusion protein expression constructs and analyzed the localization of eGFP fluorescence by confocal microscopy. We assessed these fusion proteins for mitochondrial localization through cotransfection of H9c2 cells with plasmids encoding organellar markers linked to TdTomato. We have identified two peptides of TAZ that are independently responsible for mitochondrial localization. Using CRISPR-generated TAZ knock out cell lines, we found that these peptides are able to direct proteins to mitochondria in the absence of endogenous TAZ. These peptides are not located within the predicted enzymatic clefts of TAZ, implying that some BTHS disease causing mutations may affect mitochondrial localization without affecting transacylase activity. These novel peptides improve our understanding of TAZ intracellular trafficking, provide insight into the molecular basis of BTHS and provide molecular reagents for developing targeted mitochondrial therapies.
Keywords: Tafazzin, Barth Syndrome, Orphan Disease, Mitochondrial Localization, Organellar Markers, TAZ-eGFP
Graphical Abstract
1. Introduction
Barth syndrome is an X-linked recessive disorder that presents clinically with 3-methylglutaconic aciduria, neutropenia, hypotonia and cardiomyopathy. Causative genetic mutations have been mapped to the tafazzin (TAZ) gene, which encodes a phospholipid transacylase that regulates the maturation of cardiolipin (CL) [1, 2]. The TAZ gene lies on the short arm of chromosome X and is encoded by 11 exons. Four splice variants have been identified and validated: full length (TAZ FL), a variant lacking exon 5 (TAZ Δ5), one lacking exon 7 (TAZ Δ7) and one variant lacking both exon 5 and exon 7 (TAZ Δ5Δ7). Studies show that although all four variants localize to mitochondria, only the former two have transacylase activity, with TAZ Δ5 being predominant. TAZ FL is present only in primates, whereas TAZ Δ5 is highly conserved [3, 4].
While the function of the non-enzymatically active isoforms of TAZ has yet to be determined, TAZ FL and TAZ Δ5 catalyze the final maturation step of the ubiquitous mitochondrial phospholipid, CL [5, 6]. TAZ is localized to mitochondrial membranes in mammals, where it acts by exchanging acyl chains between CL and other phospholipids, the final result being a CL molecule with predominantly unsaturated fatty acyl chains [7]. It has been postulated that TAZ activity is promiscuous and the observed specificity of its action is conferred by the restraints of the membrane space where it is most active [8].
CL is a unique phospholipid that is exclusively synthesized and functions in mitochondrial membranes [7, 9]. It consists of a diphosphatidylglycerol molecule that binds four acyl chains. TAZ remodels CL by exchanging its initially saturated acyl chains for unsaturated acyl chains, making the molecule more thermodynamically favorable for membranes with a high degree of curvature [8]. Tetralinoleoyl CL (L4CL) is the most abundant mature species in mammalian muscle. In BTHS patient tissues, mature CL species are decreased and the immature monolysocardiolipin (MLCL), a CL molecule with only three acyl chains, accumulates [10], CL interacts with protein complexes involved in the formation and maintenance of mitochondrial inner membrane cristae, as well as with respiratory supercomplexes in the inner mitochondrial membrane (IMM) [11, 12]. Failure to properly remodel CL ultimately results in defects of mitochondrial morphology and function, both of which have been implicated in cardiovascular disease by numerous studies [13, 14].
Critically, as CL is only synthesized in mitochondria, correct targeting of TAZ must occur in order for CL to undergo its final step of remodeling. Previous work in yeast has identified a 28 amino acid peptide in the C-terminus of the TAZ is necessary for mitochondrial targeting. However, the exact mitochondrial targeting sequence for human TAZ is yet unknown [15]. Since heart failure is known to be associated with various types of mitochondrial dysfunction [16], identification of mitochondrial targeting reagents will facilitate development of mitochondrially-targeted therapies for heart failure.
In this study, we investigated the internal sequences that are responsible for targeting TAZ to the mitochondria. Using various regions of TAZ fused with eGFP, we found two distinct fragments that were capable of localizing to mitochondria independently. TAZ (84–95) confers exclusive targeting to mitochondria of eGFP, while TAZ (185–220) results in partial targeting to mitochondria along with other subcellular compartments.
2. Materials and Methods
2.1 DNA constructs
The tdTomato open reading frame (ORF) was isolated by PCR from pCAG-ChromsonR-tdTomato (a gift from Edward Boyden, Addgene plasmid # 59169, [17]) and cloned into peGFP-N1 to replace the eGFP ORF. To construct expression plasmids encoding organelle-specific fluorescent markers that localize to lysosomes, peroxisomes, mitochondrial inner and outer membranes, the ORFs of LAMP2b (NP_054701.1), PXMP2 (NP_061133.1), TIMM23 (NP_006318.1), TOMM20 (NP_055580.1), and TAZ (isoform 1, NP_000107.1 and isoform 2, NP_851828.1), respectively, were isolated from 293T cell cDNA by PCR with corresponding primers. The ORFs of organelle-specific markers were cloned in-frame at the N-terminus of tdTomato at Bgl II and Sal I sites, driven by the CMV early promoter. The ORF of wild type TAZ was cloned into peGFP-N1 (Clontech, Mountain View, CA) at Bgl II and Sal I sites to generate the TAZ full length phTAZv1-eGFP and the TAZ Δ5 isoform 2 phTAZ-eGFP.
For constructing the N-terminal deletion series of human TAZ-eGFP fusion proteins, we selected endogenous codons encoding methionines as initiation codons to create in-frame fusions with eGFP at Bgl II and Sal I sites. The C-terminal TAZ deletion series were generated by PCR using hTAZ-eGFP as template and a reverse primer complementary to TAZ to create TAZ C-terminal mutations; the forward primer encoded a linker peptide (Gly-Gly-Gly) between the TAZ fragment and eGFP. The PCR products were isolated, digested with Sal I and ligated for transformation into STBL3 bacteria. For generating the single point mutations of hTAZ-eGFP, recombinant DNAs were generated by PCR of plasmid phTAZ-eGFP with the mutated nucleotides in primers according to the BTHS human TAZ gene variants database (Barth Syndrome Foundation, available at http://www.barthsyndrome.org/english/view.asp?x=1 (Accessed July 15, 2017), [4]).
To generate a plasmid for clustered, regularly interspaced, short palindromic repeat RNA-guided (CRISPR) targeting of TAZ in female rat cardiomyoblast H9c2 cells, two sets of primers for gRNA expression were used to target TAZ exon 3: 5′-TTTCAGGATCCCTACGAAAA-3′ and 5′-CTGAAGTTGATGCGTTGGTG-3′ and were both cloned into pSQT1313 for the gRNA multiplex expression.
Plasmid constructs were confirmed by sequencing (Eurofins, Louisville, KY).
2.2 Cell culture and cell transfection
Rat cardiomyoblast H9c2 cells were cultured in 10% FBS/DMEM on a cover slide. For fluorescence colocalization experiments, H9c2 cells were seeded at 25,000 cells/well on a 12 mm diameter coverslide in a 24-well plate and allowed to attach overnight. Equal amounts of plasmid DNAs, encoding mutated- or full length hTAZ- eGFP and organelle-specific tdTomato, were used to transfect cells with Lipofectamine 3000 (ThermoFisher Scientific, Waltham, MA) according to the manufacturer’s instructions. Cells were harvested and fixed with 4% paraformaldehyde/PBS and mounted with DAPI-Vectashield (Vector Laboratories, Burlingame, CA) or DAPI Fluoromount-G (Southern Biotech, Birmingham, AL).
2.3 Tafazzin CRISPR knockout cell lines
To generate female rat cardiomyoblast H9c2 Tafazzin (NM_001025748) knockout lines, we used a dimeric CRISPR RNA-guided FokI nuclease approach for genome editing [18]. This strategy reduces the potential for off-target effects by fusing the dimerization-dependent FokI nuclease to a catalytically inactive Cas9 (dCas9) to induce double stranded DNA breaks. This approach requires two separate guide RNAs on opposing strands of DNA. The H9c2 cells were plated in a 24-well plate at a density of 25,000 cells/well and transfected with 250 ng of the plasmid pSQT-rTAZ-exon3 and 750 ng of pSQT1601-FokI-dCAS9 to create indels, and co-transfected with 80 ng pIRES-hrGFP-Neo or with 120 ng pLKO-scramble for cell selection, using lipofectamine 3000 (ThermoFisher Scientific, Waltham, MA). Single colonies resistant to 500 μg/mL G418 for 7-days or 1 μg/mL puromycin for 2-days were screened by PCR with primers flanking rTAZ exon 3 in introns 2 and 3 (Table 1) which generated a 120 bp PCR product in wild type cells. To verify the TAZ alleles, the PCR products were resolved in 8% polyacrylamide (19:1 acrylamide:bisacrylaminde) in 0.5x TBE buffer. The PCR products from each colony were cloned into pJET1.2 (ThermoFisher Scientific, Waltham, MA) and verified by sequencing (Eurofins, Louisville, KY). The candidate clones were further tested for the 10 most probable predicted off-targets (http://crispr.mit.edu) by PCR with 10 sets of primers (Table 1).
Table 1.
Primers to Assess for CRISPR Off-Target Mutations in Rat H9c2 Cells.
| chr | Position | Off-target sequence | Primer |
|---|---|---|---|
| 3 | 158982241 | 5′-CTGGAGTTGGTGTGTTGGTGAGG-3′ | 5′-ATCTGGGAGAAGATTTGCC-3′ 5′-CAATCCAGTCATCATCCCC-3′ |
| 4 | 157813433 | 5′-TTTCAGGATGATTACGAAAAAAG-3′ | 5′-GCACACTTCCAGGTCTTG-3′ 5′-TTCCTAGTCCTGGGGTTG-3′ |
| 4 | 159638475 | 5′-TTTCAGGATGATTACGAAAAAAG-3′ | 5′-GGAGGCAGAGATGTGCTC-3′ 5′-GAAGTTGTGGCAGAAGG-3′ |
| 5 | 159545402 | 5′-CAGCAGCTGGTGCGTTGGTGGAG-3′ | 5′-GGCCCACCGGATTCCAG-3′ 5′-GCGAATGTTTGCAAGACC-3′ |
| 5 | 170642529 | 5′-CTGAGGCTCATGCGTTGGTGTGG-3′ | 5′-CCCAGGCTCCACTTTTAG-3′ 5′-GCTGGTACTCTGGGAGG-3′ |
| 7 | 27695381 | 5′-ATGAGGTGGATGTGTTGGTGAGG-3′ | 5′-TTATTGGCAACGTTTTAGGG-3′ 5′-AGAACTCTCGATGGCCAC-3′ |
| 11 | 50427060 | 5′-TTGAAGGAAACCTACGAAAAAGG-3′ | 5′-AATGCAGTGCTAGCCAAG-3′ 5′-CAGCAACATCATCTAGGG-3′ |
| 11 | 78928855 | 5′-TATCAGGATGCCTATGAAAAGGG-3′ | 5′-CAAGAACTTGACAAACATCAAC-3′ 5′-CACAATTACATCTTTGAGG-3′ |
| 14 | 47547999 | 5′-TACCAAGATTCCTACGAAAATAG-3′ | 5′-AGATGTCATGATAATGGC-3′ 5′-TTGATCCAAAATGCATAGTTG-3′ |
| 14 | 76188503 | 5′-TAGCAGGATACATACGAAAAGAG-3′ | 5′-TCTCCCACATCCCATGGTG-3′ 5′-GCAAAGATTTGAACAGATAAATTG-3′ |
2.4 Confocal microscopy
The confocal images were taken with a 60x oil lens under a Nikon A1R confocal mounted on a Nikon TiE inverted microscope. The images are shown with maximal intensity projection of z-planes.
2.5. Western Blotting
H9c2 cells were cultured to confluency in 15 centimeter plates. Cells were then collected by scraping and centrifuged at 600 g for 5 minutes and the pellet resuspended in mitochondria isolation buffer (250 mM sucrose, 10 mM Tris pH 7.4, 0.1 mM EDTA). Cells were homogenized in a glass dounce homogenizer until 90% of cells showed trypan blue staining (70 strokes). Homogenates were then spun at 600 g for 10 minutes to pellet nuclei and cell debris and then at 7,600 g to pellet the crude mitochondrial fraction. The pellet was re-suspended in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 1% Triton X-100, 0.25% SDS) with protease inhibitor cocktail (Sigma P8340) added. Protein concentration was measured by Bradford assay and 20 micrograms of protein from each sample were resolved by SDS-PAGE (10%). Proteins were transferred onto nitrocellulose membranes, which were then probed with antibodies against succinate dehydrogenase (SDHA, MitoSciences) and TAZ [19], generously shared by S. M. Claypool. The SDHA antibody was used at a dilution of 1:10,000 and the TAZ antibody was used at a dilution of 1:1,000. The membrane was then blotted with secondary antibodies horse anti-mouse and goat anti-rabbit IgG HRP-conjugates (Cell Signaling Technologies, both at 1:5,000 dilution). The membranes were incubated with enhanced chemiluminescence agents (Pierce) and analyzed by a ChemiDoc Image System (Bio-Rad).
3. Results
3.1 Full length TAZ localizes to mitochondria
Initial efforts to colocalize TAZ-eGFP fusion proteins with MitoTracker Red dye were unsatisfactory, due to bleeding of red signal into other organelles that likely resulted from toxicity (data not shown). To investigate further the TAZ mitochondrial localization signal, organellar marker-tdTomato fusion proteins and TAZ-eGFP fusion protein were co-expressed. Control studies with eGFP or tdTomato alone did not affect Tom20 or TAZ-eGFP fusion protein localization to mitochondria (Figure 1A and B). A previous study reports that TAZ protein is embedded in mitochondrial membranes, facing the intermembrane space [19]. Both TAZ isoforms tested, the 292 amino acid TAZ full length isoform 1 and the 262 amino acid TAZ Δ5 (isoform 2), localized to mitochondria when co-expressed with the fusion proteins Tim23-tdTomato and Tom20-tdTomato, mitochondrial markers of the inner or outer membrane, respectively (Figure 1C, D, G and H). These data are consistent with a previous report using HA-tagged TAZ [20]. Neither TAZ variant-eGFP fusion protein colocalized with peroxisomes or lysosomes when co-expressed with the organelle-specific markers PXMP2- and LAMP2b-tdTomato fusion proteins, respectively (Figure 1E and F, data not shown). In agreement with previous studies, exon 5 does not affect the mitochondrial localization of TAZ [20]. We therefore used isoform 2 to investigate the amino acid sequences necessary for mitochondrial localization, however, amino acid numbers and mutation positions are in accordance with isoform 1 length, as described in the BTHS human TAZ gene variants database (Barth Syndrome Foundation, available at http://www.barthsyndrome.org/english/view.asp?x=1 (Accessed July 15, 2017), [4]).
Figure 1.

Mitochondrial localization of human tafazzin isoforms in rat cardiomyoblast H9c2 cells. The H9c2 cells were transfected with EGFP and tdTomato native and fusion protein expression plasmids and fixed with 4% paraformaldehyde 24 hours after transfection and photographed with a confocal microscope with a 60x oil objective lens. The maximal intensity projection of z-stack images are shown for the co-expression of hTAZ isoform 1 and 2 with organelle markers. A. eGFP and mitochondrial outer membrane marker hTom20-tdTomato, B. TAZ isoform 2 hTAZ-eGFP and tdTomato, C. hTAZ-eGFP and mitochondrial outer membrane hTom20-tdTomato, D. hTAZ-eGFP and hTim23-tdTomato, E. hTAZ-eGFP and lysosomal hLamp2b-tdTomato, F. hTAZ-eGFP and peroxisome hPxmp2-tdTomato, G. TAZ isoform 1 hTAZv1-eGFP and hTom20-tdTomato, H. hTAZv1-eGFP and hTim23-tdTomato. Cells showing mitochondrial localization of hTAZ-eGFP with mitochondrial markers are indicated by yellow arrows and cells in which tdTomato is more strongly expressed are indicated by orange arrows. All images were taken with a 60 x oil lens and the white bar in panel B represents 50 μm.
3.2 Mitochondrial Localization of N- and C-terminus truncated TAZ fragments
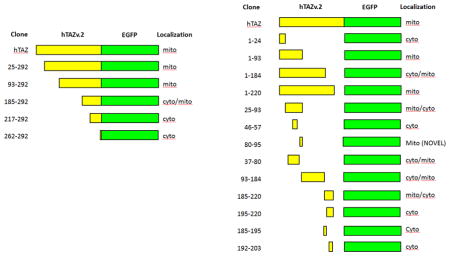
To identify TAZ mitochondrial localization peptides, we made a series of N- and C-terminus deletions of TAZ, in-frame with eGFP. For N-terminus deletions, the TAZ mutants were translated from endogenous methionines at M25, M93, M185, M217, and M262 as start codons (Figure 2A). The N-terminus deletion mutants TAZ (25–292) and TAZ (93–292) showed strong mitochondrial localization (Figure 2B and C), whereas TAZ (185–292) showed partial mitochondrial signal, along with some cytosolic signal (Figure 2D). Both TAZ (217–292) and TAZ (262–292) showed cytosolic signal only (Figure 2E and F), suggesting a potential mitochondrial targeting sequence between amino acids 185 and 217.
Figure 2.

Mitochondrial localization of hTAZ isoform 2 serial N′-terminal deletion mutants. A. Schematic diagram of hTAZ isoform 2 serial N′-terminal deletion mutants, which have no exon 5 encoding amino acids 124–154 (broken line). The amino acid numbers assigned by the full length hTAZ isoform 1 are listed on the left of each mutated TAZ fusion protein. The endogenous methionines in hTAZ were selected as initiation codons to create in-frame fusion proteins with eGFP. The maximal intensity projection of z-stack confocal images are shown for the mitochondrial outer membrane marker hTom20-tdTomato with hTAZ mutants B. hTAZ(25–292), C. hTAZ(93–292), D. hTAZ(185–292), E. hTAZ(217–292), and F. hTAZ(262–292). All images were taken with a 60 x oil lens and the white bar in panel D represents 50 μm.
We also investigated four TAZ C-terminus deletions (Figure 3A). TAZ (1–24) showed cytosolic eGFP signal (Figure 3B), whereas both TAZ (1–93) and (1–220) showed mitochondrial localization (Figure 3C and E). TAZ (1–184) showed some mitochondrial localization, but also significant cytosolic signal (Figure 3D). These results indicated that a TAZ mitochondrial localization signal lies between amino acids 25 and 220. Since both TAZ (1–93) and TAZ (185–292) showed some mitochondrial localization, we investigated whether TAZ contains two mitochondrial localization signals: one in between amino acids 25–93 and the other between amino acids 185–220.
Figure 3.

Mitochondrial localization of hTAZ isoform 2 serial C′-terminal deletion mutants. A. Schematic diagram of hTAZ isoform 2 serial C′-terminal deletion mutants, which have no exon 5 encoding amino acids 124–154 (broken line). The amino acid numbers assigned by the full length hTAZ isoform 1 are listed on the left of each mutated TAZ. The peptide sequence gly-gly-gly was inserted as a linker at the C′-terminal TAZ deletion site in each clone to create an in-frame fusion protein with eGFP. The maximal intensity projection of z-stack confocal images were are shown for the mitochondrial outer membrane marker hTom20-tdTomato with hTAZ mutants B. hTAZ(1–24), C. hTAZ(1–93), D. hTAZ(1–184), and E. hTAZ(1–220). All images were taken with a 60 x oil lens and the white bar in panel B represents 50 μm.
3.3 TAZ contains two distinct and independent mitochondrial targeting peptides
To identify the possible mitochondrial localization signals in TAZ, we isolated the fragments encoding amino acids 25–92, 93–184, and 185–220 to generate plasmids encoding fusion proteins with eGFP (Figure 4A) and co-transfected these plasmids with the hTom20-tdTomato plasmid into H9c2 cells. Both TAZ (25–93) and TAZ (185–220) showed mitochondrial localization, with some cytosolic signal (Figure 4B and D). TAZ (93–184) localization is primarily cytosolic with a small fraction of signal present in mitochondria (Figure 4C). These results indicated that TAZ possesses two mitochondrial localization signals and each can function independently.
Figure 4.
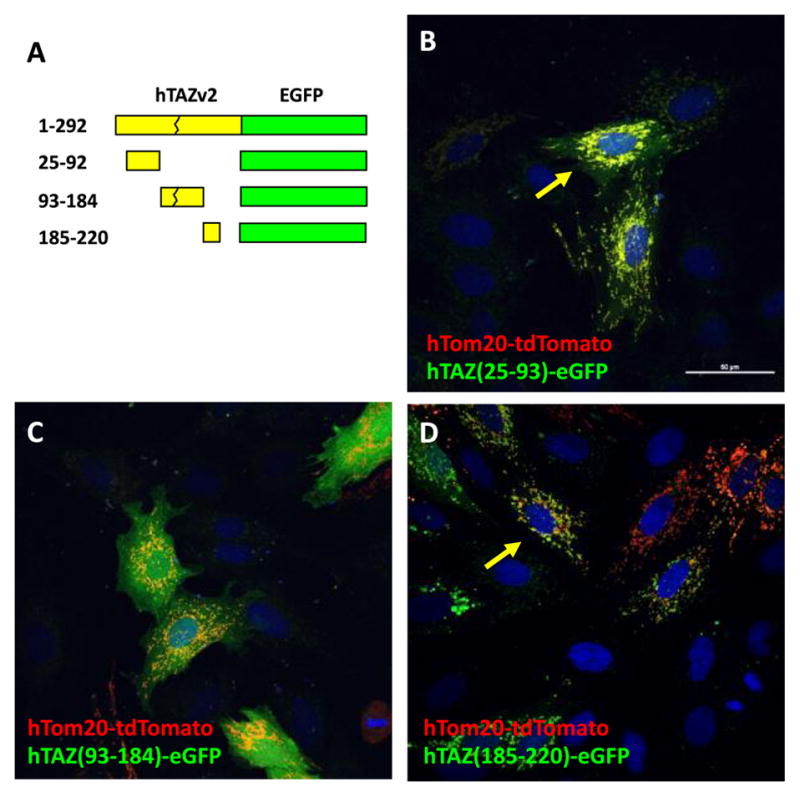
Mitochondrial localization of hTAZ isoform 2 subfragments. A. Schematic diagram of hTAZ isoform 2 subfragments fused to eGFP. The peptide sequence gly-gly-gly was inserted as a linker at the C′-terminal TAZ deletion site when needed to create an in-frame fusion protein with eGFP. The maximal intensity projection of z-stack (0.2 mm x 6 slices) confocal images are shown for the mitochondrial outer membrane marker hTom20-tdTomato with hTAZ mutants B. hTAZ(25–92), C. hTAZ(93–184), and D. hTAZ(185–220). All images were taken with a 60 x oil lens and the white bar in panel B represents 50 μm.
3.4 Fine mapping of essential TAZ fragments that confer mitochondrial localization
We further dissected the TAZ (185–220) into three smaller fragments: 185–195, 195–220 and 192–203 and generated eGFP fusion proteins (Figure 5A). Interestingly, all of these smaller fragments were unable to direct the fusion protein to the mitochondria and were found only in the cytosol (Figure 5B, C, and D). These results indicated that the amino acids 185–220 are undividable and necessary for mitochondrial localization.
Figure 5.
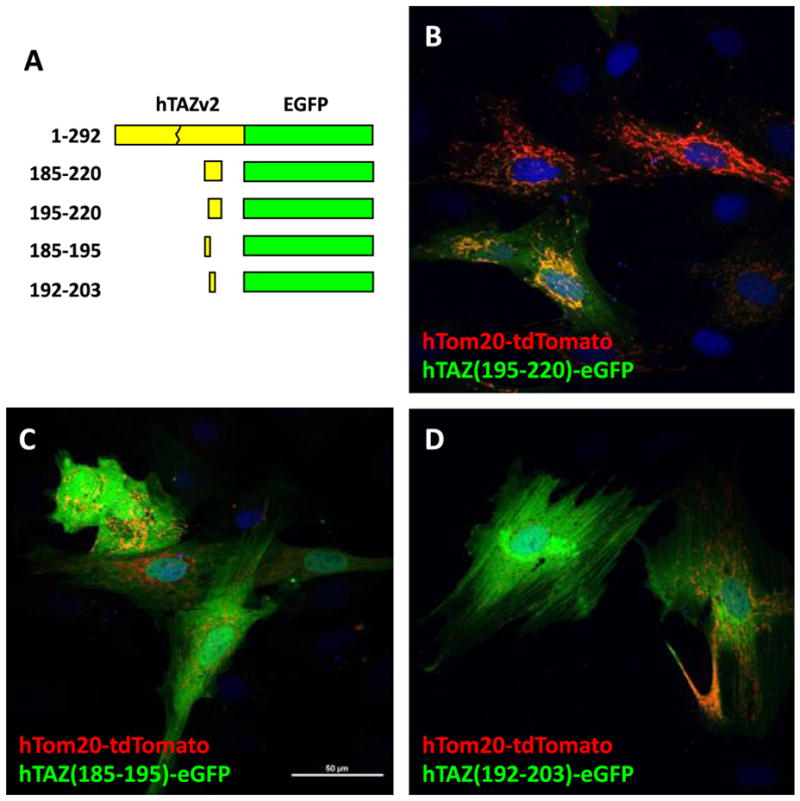
Mitochondrial localization of hTAZ isoform 2 subfragments derived from hTAZ(185–220). A. Schematic diagram of hTAZ isoform 2 subfragments derived from hTAZ(185–220) fused to eGFP, which contain amino acids encoded from part of exon 7 (185–194) and entire exon 8 (195–215) and part of exon 9 (216–220). The peptide sequence gly-gly-gly was inserted as a linker at the C′-terminal TAZ deletion site to create an in-frame fusion protein with eGFP. The maximal intensity projection of z-stack (0.2 mm x 6 slices) confocal images are shown for the mitochondrial outer membrane marker hTom20-tdTomato with hTAZ mutants B. hTAZ(195–220), C. hTAZ(185–195), and D. hTAZ(192–203). All images were taken with a 60 x oil lens and the white bar in panel C represents 50 μm.
To narrow down the essential amino acid sequence required for mitochondrial localization, the TAZ (25–93) fragment was divided into smaller fragments (Figure 6A). Co-localization of hTom20-tdTomato and mutated TAZ fragment–eGFP proteins were analyzed in H9c2 cells by confocal microscopy. The N-terminus of this TAZ fragment, amino acids 37–80, which is encoded by exon 2, was mostly in the cytosol (Figure 6B). A computer predicted helix structure [21] spanning amino acids 46–57 within this fragment does not confer mitochondrial localization (Figure 6C). In contrast, the other computer predicted helix (GOR4) at the C-terminus of this fragment, spanning amino acids 80–95, clearly possesses a mitochondrial targeting peptide (Figure 6D). To shorten the minimal amino acid sequence required to lead to mitochondrial localization of eGFP, we further narrowed down the sequence to: 82–95, 84–95 and 80–92. All three of these fragments still directed eGFP to mitochondria (Figure 6E, F, and G). We attempted to further reduce the fragment, however, amino acid sequence 82–92 was unable to localize to mitochondria (Figure 6H). Taken together, our results indicate that this region of TAZ contains an independent peptide responsible for mitochondrial localization, but the borders of this peptide appear to be inexact, which may be explained by secondary structure requirements. A basic local alignment search tool analysis against known proteins in the National Center for Biotechnology Information database (BLASTP) for the peptide sequence encompassing amino acids 80–95 revealed no homology to any known proteins other than tafazzin, demonstrating the novelty of this mitochondrial localization sequence (data not shown).
Figure 6.

Mitochondrial localization of hTAZ isoform 2 subfragments derived from hTAZ(25–92).. A. Schematic diagram of hTAZ isoform 2 subfragments derived from hTAZ(25–92) fused to eGFP, which contain amino acids encoded from part of exon 1, exon 2 (37–80) and exon 3 (80–93). The amino acid sequence of hTAZ exon 3 is also shown. The splicing junction encodes the amino acid R94, which is a site of common missense mutations in Barth syndrome patients. The W95 residue encoded in exon 4 is indicated (red letter). The peptide sequence gly-gly-gly was inserted as a linker at the C′-terminal TAZ deletion sites to create an in-frame fusion protein with eGFP. The maximal intensity projection of z-stack (0.2 mm x 6 slices) confocal images are shown for the mitochondrial outer membrane marker hTom20-tdTomato with hTAZ mutants B. hTAZ(37–80), C. hTAZ(46–57), D. hTAZ(80–95), E. hTAZ(82–95), F. hTAZ(84–95), G. hTAZ(80–92), H. hTAZ(82–92). All images were taken with a 60 x oil lens and the white bar in panel E represents 50 μm.
3.5 Mitochondrial localization of TAZ-eGFP fusion proteins containing Barth Syndrome missense mutations
Barth Syndrome missense mutations are located throughout the TAZ gene. To investigate whether known missense mutations in the identified localization peptides encoded by exon 3 or exon 8 would affect TAZ localization, we engineered selected TAZ mutations into the mitochondrially localizing eGFP fusion proteins and assessed their localization. We investigated the TAZ Barth Syndrome mutations located in exon 8 by introducing these mutations into the full length hTAZ-eGFP fusion protein (Figure 7A). One of the hot spots for TAZ mutations is at G197. However, TAZG197R mutation does not affect mitochondrial localization. Similarly, the other mutations in exon 8, I209N, L210R, L212P, and H214R, also do not affect mitochondrial localization (Figure 7B, C, D, E, and F).
Figure 7.

Mitochondrial localization of hTAZ-eGFP proteins containing Barth syndrome missense mutations. A. Amino acid sequence of hTAZ exon 8 (purple letters) and 9 (green letters). The TAZ missense mutations in Barth syndrome are labeled in red letters. Confocal images of of TAZ mutants are shown: B. G197R, C. I209N, D. L210R, E. L212P, F. H214R, and, G. R94S (located in exon 3). All images were taken with a 60 x oil lens and the white bar in panel D represents 50 μm.
Another hot spot for mutation, R94, is located at the end of exon 3. Although mutations in the first or second nucleotides in the codon do not affect RNA splicing, the known mutated amino acids are relatively diverse and include cysteine, serine, histidine, leucine, or glycine. TAZ with the R94S mutation is still able to localize to mitochondria (Figure 7G).
3.6 Localization of TAZ-eGFP in TAZ knockout H9C2 cells
The exact mechanism by which TAZ is imported into mitochondria is still unknown. To exclude the possibility of homodimerization, whereby the TAZ fragment-eGFP fusion proteins are carried into the mitochondria by normal, endogenously expressed TAZ interacting with the fusion protein, we verified localization of the previous constructs in TAZ knockout (KO) H9c2 cells. As we found exon 3 to be responsible for mitochondrial localization, we targeted it for deletion in the myoblasts using CRISPR/Cas9 technology (Figure 8A). We selected three separate lines: one line, CR38, does not contain a TAZ mutation and two lines that contain TAZ deletions that were verified by sequencing. The sequence alignments revealed that both deletion cell lines, CR22 and CR131, contain TAZ nonsense mutations by deletion of 11 and 29 nucleotides of exon 3, respectively (Figure 8B). To further confirm the protein expression in both cell lines, western blot analyses showed that TAZ proteins were detectable in the wild type and CR38 lines, but not in the nonsense clones CR22 and CR131 (Figure 8C).
Figure 8.

TAZ-eGFP mitochondrial localization in H9c2 TAZ Knockout cells. A. Strategy of TAZ knockout in H9c2 by two tracer-gRNAs. Anti-sense (red letters) and sense (blue letters) guide RNAs target the 5′- and 3′-end of exon 3, respectively. B. Sequence alignment of TAZ CRISPR mutations in targeted H9c2 cell lines derived from single colonies. The exon 3 encoded amino acids are indicated above the WT DNA sequence. Clone CR22 has deletions (dashed line) in both alleles: one with an 11 nt deletion and the other with a 12 nt deletion combined with a single nucleotide insertion (red letter). CR131 has a 29 nt deletion. Both KO clones contain out-of-frame nonsense mutations. C. TAZ western blot of WT H9c2 and H9c2-derived TAZ CRISPR knockout cells. Antibodies were used to detect TAZ and also the mitochondrial complex 2 marker SDHA. D–G. Mitochondrial localization of TAZ-eGFP proteins in WT and TAZ-KO cells containing a stable lentiviral-hTom20-tdtomato marker. D. hTAZ-eGFP in WT cells. E. hTAZ(84–95)-eGFP in WT cells. F. hTAZ-eGFP in KO cells, and G. hTAZ(84–95)-eGFP in KO cells. All images were taken with a 60 x oil lens and the white bar in panel D represents 50 μm.
To investigate whether TAZ isoform 2 or TAZ (84–95) targeting to mitochondria is mediated by endogenous wild type TAZ, we created cell lines derived from H9c2 wild type, CR22 and CR131 that constitutively expressed hTom20-tdTomato by lentivirus infection. Using these cell lines, we found that TAZ-eGFP and TAZ (84–95)-eGFP are still able to localize to mitochondria in both the TAZ KO lines and the WT myoblasts (Figure 8E, F, G, and H). We observed similar results with TAZ(185–220)-eGFP (data not shown). These data suggest that the short mitochondrial targeting sequences in TAZ are sufficient to target proteins to mitochondria in the absence of full length TAZ.
4. Discussion
The acyltransferase TAZ is responsible for cardiolipin maturation in the mitochondrial inner membrane. TAZ is encoded in the nucleus and then targeted to the mitochondrial inner membrane, where its substrate is exclusively remodeled. Therefore, mislocalization of this protein, even if fully enzymatically active, could have severe implications for CL remodeling. It is thus critical to understand how TAZ is targeted to mitochondria. Targeting of proteins to mitochondria can be achieved with different types of targeting sequences, depending on the final destination and function of the protein. Whereas matrix proteins contain a cleavable N-terminal presequence that is recognized by the translocase of the outer membrane 20 (Tom20) complex, membrane and intermembrane space (IMS) proteins typically contain non-cleavable internal targeting sequences that are recognized by the Tom70/71 complexes [22]. A subgroup of IMS and inner mitochondrial membrane (IMM) proteins are targeted to their correct location by further interactions with the mitochondrial intermembrane space assembly (MIA) complex, via a characteristic double cysteine (C) motif (typically in the form of CxnC) [23, 24]. Mutations in these internal sequences could lead to aberrant sorting within the mitochondria, as well as failure to be targeted to the mitochondria at all. Human tafazzin contains one Cx3C motif located downstream of the 80–95 localization domain.
In this report, we determined the mitochondrial localization sequences for TAZ and found that there are two discrete regions that can target it to the mitochondria. The two sequences, TAZ (84–95) and TAZ (185–220), are located in exons 3 and 7/8, respectively. While the exon 3 sequence targets TAZ exclusively to mitochondria, the exon 7/8 sequence does not confer precise localization. Both of these sequences are able to direct eGFP to mitochondria in the absence of wild type TAZ, in TAZ deficient cells. Although several BTHS-causing point mutations are located within these two regions, our data shows that many of these mutations do not disrupt mitochondrial localization of the protein. This is in accordance with previous studies showing that most BTHS point mutations modeled in yeast do not lead to protein mislocalization, but rather are either catalytically null, or more labile and predisposed to degradation [25].
4.1 TAZ contains two distinct mitochondria targeting regions
As the two mitochondrial localization sequences are able to independently target TAZ to the mitochondria, this suggests a redundant role for one of the peptides. TAZ (84–95) confers exclusive mitochondrial localization, indistinguishable from wild type TAZ, whereas TAZ (185–220) targets other sub-cellular compartments as well. The former sequence is unique and does not share homology with any other known proteins or mitochondrial localization sequences. The latter sequence may interact with other domains of the protein in the wild type molecule to form an exclusive mitochondrial targeting signal, or may be a remnant of an evolutionarily older signal. Of the four TAZ isoforms documented in humans, two lack exon 7, which encodes amino acids 181–195, part of the TAZ (185–220) peptide. Studies in 293 cells show these two isoforms are still able to localize to mitochondria, suggesting a redundant role for this peptide [20].
Our studies utilize laser scanning confocal microscopy to determine co-localization with mitochondrial markers, however this technique cannot distinguish between different sub-mitochondrial compartments due to the diffraction limit of light microscopy. This methodological limitation prevents us from excluding the possibility that TAZ(80–95)-eGFP could localize to a different mitochondrial compartment than the IMM. Overexpression might cause the TAZ(80–95)-eGFP, hTOM20-tdTomato or hTIM23-tdTomato constructs to localize to both membranes and possibly the mitochondrial matrix, and thus may also confound the results. Future studies using super resolution microscopy, when available, will likely be informative.
4.2 Exon 3 of TAZ plays an important role in mitochondrial localization
TAZ (84–95), the peptide responsible for exclusive mitochondrial localization, encompasses a large part of exon 3 of the TAZ gene. Recent in-silico prediction of TAZ structure showed that most of this region is found on an exposed part of the globular protein structure and contains part of a patch of positive residues that help anchor it to the IMM in conjunction with the actual membrane insertion domain, predicted to reside in exon 1 [21]. R94, one of the sites most commonly found to have single point mutations leading to BTHS, is one of the residues that helps anchor TAZ to the membrane. This led us to surmise a mutation in this spot could lead to de-stabilization and mislocalization of the protein. In our experiments, however, the TAZ R94S mutation did not affect mitochondrial localization of eGFP. This finding correlates with the ability of the TAZ (80–92) peptide to direct eGFP to the mitochondria. It is possible that TAZ R94S can still localize to mitochondria, but has difficulty remaining anchored to the IMM, where it needs to locate in order to interact with its substrate.
Intriguingly, both TAZ (80–92) and TAZ (84–95) are able to localize to the mitochondria, however, the overlapping sequence of 82–92 does not. The former two peptides contain a similar number of positively charged and hydrophobic amino acids. It has been proposed that mitochondrial targeting sequences consist of alpha helices enriched in positively charged and hydrophobic amino acids that bind the outer membrane translocases [23, 26]. It is therefore tempting to speculate that structure in the form of a minimal length amphipathic helix is also required, in addition to a specific amino acid sequence. This would explain why TAZ (82–92) is not able to localize eGFP to mitochondria, while the other two peptides are sufficient. It is also possible that the added amino acids provide essential post-translational modifications, which are not present in the shortened sequences.
4.3 In-silico modeling of human TAZ structure
Although the bulk of exon 3 is exposed, the in-silico model locates the G80 and L82 residues in a secondary structure within the core of the protein. While it could be intuitively expected that a mitochondrial targeting sequence would need to be exposed to the surface of the protein for interaction with the TOM complex, a large number of proteins have been shown to enter mitochondria in an incompletely folded state, accompanied by chaperones on the cytosolic side [27]. It is therefore possible that a region normally buried within the core of the protein can serve as an organellar localization domain. Similarly, I209, L210, L212, H214 and G216, part of the TAZ (185–220) peptide, are also predicted to locate in the core of the protein. Therefore, the predicted model of TAZ structure represents an enzymatic steady state, rather than a dynamic, structure-shifting, mitochondrial targeting state.
4.4 Correlation of human and yeast TAZ mitochondrial localization studies
Yeast studies have previously identified the membrane insertion domain to reside between amino acids 215–232 in Taz1p, corresponding to 201–219 in human TAZ [15]. This domain, in conjunction with an N-terminus flanking sequence, 204–232 in Taz1p (192–219 in human TAZ) was proposed to facilitate mitochondrial import. In agreement with the yeast TAZ study, we found that TAZ (185–220) is able to partially target eGFP to mitochondria. Any attempts to shorten this sequence, however, using either TAZ (185–195), TAZ (192–203), or TAZ (195–220), completely abolished mitochondrial targeting. Whereas in this study exon 3 of TAZ more precisely targets mitochondria, in the yeast studies, mitochondrial targeting of Taz1p lacking the 204–232 peptide, but containing exon 3 was completely abolished. These results indicate that Taz1p and human TAZ differ in their mitochondrial targeting mechanisms and lend support to the speculation that the 185–220 peptide in human TAZ is a remnant of an evolutionarily older signal.
Several other BTHS missense mutations (G197R, I209N, L210R, L212P, and H214R) are located within the TAZ (185–220) peptide. We investigated the role these mutations might play in the localization of the protein, however, none of these mutations affected mitochondrial localization of TAZ. This is unsurprising, as our experiments have shown that the TAZ (84–95) peptide is able to independently confer mitochondrial localization. In previous experiments, where I209, L210 and L212 mutations were modeled in yeast, they were shown to localize to the mitochondrial matrix rather than the IMM or OMM. Our data does not distinguish between these two submitochondrial compartments, and we cannot rule out the possibility that this occurs with human TAZ. Failure to localize to the IMM is expected to prevent proper remodeling of MLCL and thus affect membrane curvature and respiratory complex assembly [8, 28].
Herndon et al. [15] reported that Taz1p does not require a membrane potential to translocate into mitochondria, as most inner membrane proteins do. They found it requires both TOM20 and 70 receptors on the surface, but likely does not require TIM22 or 23. Taz1p seems to interact with Tim10p instead. Although, human TAZ contains the recognition motif (Cx3C) for the MIA complex, it is further away from the identified mitochondrial targeting signals than in other MIA substrate proteins, suggesting it may not be functional.. Recent studies have found that overexpression of mitochondrial translocation proteins can affect mitochondrial protein import, therefore it is possible that overexpression of TOM20-tdTomato in our experiments could interfere with the proper localization of TAZ [29, 30]. Another caveat with our study is that the eGFP moiety may also prevent complete translocation to the IMM. The use of human proteins in a rat cell line may also lead to subtle differences in mitochondrial localization due to variations in protein sequence.
4.5 TAZ-eGFP does not co-localize with peroxisomes or lysosomes
A great deal of evidence has accumulated demonstrating the dynamic interplay between different organelles in the cell. The connection between mitochondria and endoplasmic reticulum (ER) has long been established, but more recently mitochondria derived vesicles (MDVs) have been shown to mediate transport from mitochondria to both peroxisomes and lysosomes [31]. Peroxisomes are arguably closely related to mitochondria; these two organelles not only share biogenesis and division markers, but recent studies have shown that mitochondria are required for de novo synthesis of peroxisomes [31] It has been postulated that MDVs populate newly formed peroxisomes with proteins and lipid products derived from mitochondria, in addition to facilitating transport of fatty acids and oxidized products [32]. CL, which is synthesized in the mitochondria, but has been detected in the peroxisomes of yeast, could be one such candidate [33]. Functionally, both organelles perform β-oxidation of fatty acids and neutralize oxidized products [31].
Our result that TAZ-eGFP was not detected in peroxisomes indicates that it is unlikely to be carried over to the peroxisomes via MDVs. TAZ is not normally detected in peroxisomes, despite MDVs having been shown to bud off regions of both inner and outer mitochondrial membranes for delivery to peroxisomes. It is possible that the domains of mitochondrial membrane targeted for peroxisomes are not those where TAZ tends to be enriched, or that TAZ is endogenously present in such low amounts it cannot be detected.
Lysosomes are responsible for degrading molecules taken up by the cell by endocytosis, as well as cytoplasmic proteins and organelles (such as mitochondria) via auto/mitophagy [34]. In addition, MDVs have been shown to bud off damaged sections of mitochondria and transport them to lysosomes, thereby avoiding the more costly mitophagy. Overexpression of a membrane protein such as TAZ (or parts of it) could lead to the formation of aggregates, which would make our TAZ-eGFP a target for lysosomal degradation. In addition, the over-expression of TAZ-eGFP could affect mitochondrial health, therefore, making these organelles targets for autophagy. However, we did not observe any TAZ-eGFP signal localized in lysosomes.
4.6 Conclusions and Implications
Identification of minimal, non-functional protein domains that promote mitochondrial targeting but do not affect mitochondrial import is useful for localization studies and for development of targeted mitochondrial therapies. In this study, we have demonstrated that a short 12 amino acid fragment of TAZ (84–95) is sufficient to target eGFP to mitochondria and thus can be used for such an application. The IMM in particular is the site of oxidative phosphorylation and location of many important components of the electron transport apparatus, many of which have been implicated in mitochondrial dysfunction in human disease [35]. Development of a peptide reagent that facilitates the delivery of therapeutic molecules to the IMM may lead to development of novel therapies for mitochondrial diseases that result from respiratory dysfunction.
Highlights.
Human tafazzin contains two distinct mitochondrial localization signals that function independently
One of these localization domains is small (12 amino acids), unique to tafazzin and lacks homology to any other mitochondrial localization sequences and may be involved in targeting the inner mitochondrial membrane
These peptides can direct mitochondrial localization of a heterologous protein, eGFP, to mitochondria, in both wild type and tafazzin knockout cells
Selected missense mutations from Barth Syndrome families located within these peptides do not affect mitochondrial localization but may affect submitochondrial localization
These findings are informative about the pathogenesis of Barth Syndrome and provide novel reagents for mitochondrial localization studies and development of mitochondrially targeted therapies
Acknowledgments
Authors thank the Lynn and Mike Garvey Cell Imaging Lab at the Institute of Stem Cell and Regenerative Medicine, University of Washington and the Viral Vector and Transgenic Mouse Core facility for generating the guide RNA targeting plasmid.
This work was supported by grants from the Barth Syndrome Foundation and the NIH (GM116890) to MTC. AAD was supported by NIH training grants HL007312 and GM 007270.
Footnotes
Disclosures
MTC has a significant stock ownership interest in TransCellular Therapeutics, an early stage pharmaceutical company developing an enzyme replacement therapy for Barth Syndrome.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet. 1996;12(4):385–9. doi: 10.1038/ng0496-385. [DOI] [PubMed] [Google Scholar]
- 2.Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, Barth PG. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun. 2000;279(2):378–82. doi: 10.1006/bbrc.2000.3952. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez IL. Barth syndrome: TAZ gene mutations, mRNAs, and evolution. Am J Med Genet A. 2005;134(4):409–14. doi: 10.1002/ajmg.a.30661. [DOI] [PubMed] [Google Scholar]
- 4.Kirwin SM, Manolakos A, Barnett SS, Gonzalez IL. Tafazzin splice variants and mutations in Barth syndrome. Mol Genet Metab. 2014;111(1):26–32. doi: 10.1016/j.ymgme.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Xu Y, Malhotra A, Ren M, Schlame M. The enzymatic function of tafazzin. The Journal of biological chemistry. 2006;281(51):39217–24. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- 6.Houtkooper RH, Turkenburg M, Poll-The BT, Karall D, Perez-Cerda C, Morrone A, Malvagia S, Wanders RJ, Kulik W, Vaz FM. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim Biophys Acta. 2009;1788(10):2003–14. doi: 10.1016/j.bbamem.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Schlame M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J Lipid Res. 2008;49(8):1607–20. doi: 10.1194/jlr.R700018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlame M, Acehan D, Berno B, Xu Y, Valvo S, Ren M, Stokes DL, Epand RM. The physical state of lipid substrates provides transacylation specificity for tafazzin. Nat Chem Biol. 2012;8(10):862–9. doi: 10.1038/nchembio.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mileykovskaya E, Zhang M, Dowhan W. Cardiolipin in energy transducing membranes. Biochemistry (Mosc) 2005;70(2):154–8. doi: 10.1007/s10541-005-0095-2. [DOI] [PubMed] [Google Scholar]
- 10.Schlame M, Towbin JA, Heerdt PM, Jehle R, DiMauro S, Blanck TJ. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol. 2002;51(5):634–7. doi: 10.1002/ana.10176. [DOI] [PubMed] [Google Scholar]
- 11.Friedman JR, Mourier A, Yamada J, McCaffery JM, Nunnari J. MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. Elife. 2015:4. doi: 10.7554/eLife.07739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schagger H. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278(52):52873–80. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- 13.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125(7):1241–52. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 14.Dudek J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front Cell Dev Biol. 2017;5:90. doi: 10.3389/fcell.2017.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herndon JD, Claypool SM, Koehler CM. The Taz1p transacylase is imported and sorted into the outer mitochondrial membrane via a membrane anchor domain. Eukaryot Cell. 2013;12(12):1600–8. doi: 10.1128/EC.00237-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Hattab AW, Scaglia F. Mitochondrial Cardiomyopathies. Front Cardiovasc Med. 2016;3:25. doi: 10.3389/fcvm.2016.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, Wang J, Xie Y, Yan Z, Zhang Y, Chow BY, Surek B, Melkonian M, Jayaraman V, Constantine-Paton M, Wong GK, Boyden ES. Independent optical excitation of distinct neural populations. Nat Methods. 2014;11(3):338–46. doi: 10.1038/nmeth.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, Joung JK. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014;32(6):569–76. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu YW, Galbraith L, Herndon JD, Lu YL, Pras-Raves M, Vervaart M, Van Kampen A, Luyf A, Koehler CM, McCaffery JM, Gottlieb E, Vaz FM, Claypool SM. Defining functional classes of Barth syndrome mutation in humans. Human molecular genetics. 2016;25(9):1754–70. doi: 10.1093/hmg/ddw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Zhang S, Malhotra A, Edelman-Novemsky I, Ma J, Kruppa A, Cernicica C, Blais S, Neubert TA, Ren M, Schlame M. Characterization of tafazzin splice variants from humans and fruit flies. The Journal of biological chemistry. 2009;284(42):29230–9. doi: 10.1074/jbc.M109.016642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hijikata A, Yura K, Ohara O, Go M. Structural and functional analyses of Barth syndrome-causing mutations and alternative splicing in the tafazzin acyltransferase domain. Meta Gene. 2015;4:92–106. doi: 10.1016/j.mgene.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138(4):628–44. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dudek J, Rehling P, van der Laan M. Mitochondrial protein import: common principles and physiological networks. Biochim Biophys Acta. 2013;1833(2):274–85. doi: 10.1016/j.bbamcr.2012.05.028. [DOI] [PubMed] [Google Scholar]
- 24.Stojanovski D, Muller JM, Milenkovic D, Guiard B, Pfanner N, Chacinska A. The MIA system for protein import into the mitochondrial intermembrane space. Biochim Biophys Acta. 2008;1783(4):610–7. doi: 10.1016/j.bbamcr.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Whited K, Baile MG, Currier P, Claypool SM. Seven functional classes of Barth syndrome mutation. Human molecular genetics. 2013;22(3):483–92. doi: 10.1093/hmg/dds447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abe Y, Shodai T, Muto T, Mihara K, Torii H, Nishikawa S, Endo T, Kohda D. Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell. 2000;100(5):551–60. doi: 10.1016/s0092-8674(00)80691-1. [DOI] [PubMed] [Google Scholar]
- 27.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–49. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 28.Acehan D, Malhotra A, Xu Y, Ren M, Stokes DL, Schlame M. Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys J. 2011;100(9):2184–92. doi: 10.1016/j.bpj.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chacinska A, Lind M, Frazier AE, Dudek J, Meisinger C, Geissler A, Sickmann A, Meyer HE, Truscott KN, Guiard B, Pfanner N, Rehling P. Mitochondrial presequence translocase: switching between TOM tethering and motor recruitment involves Tim21 and Tim17. Cell. 2005;120(6):817–29. doi: 10.1016/j.cell.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 30.Harner M, Neupert W, Deponte M. Lateral release of proteins from the TOM complex into the outer membrane of mitochondria. The EMBO journal. 2011;30(16):3232–41. doi: 10.1038/emboj.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. The EMBO journal. 2014;33(19):2142–56. doi: 10.15252/embj.201488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohanty A, McBride HM. Emerging roles of mitochondria in the evolution, biogenesis, and function of peroxisomes. Frontiers in physiology. 2013;4:268. doi: 10.3389/fphys.2013.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wriessnegger T, Gubitz G, Leitner E, Ingolic E, Cregg J, de la Cruz BJ, Daum G. Lipid composition of peroxisomes from the yeast Pichia pastoris grown on different carbon sources. Biochim Biophys Acta. 2007;1771(4):455–61. doi: 10.1016/j.bbalip.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nature reviews. 2013;14(5):283–96. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dhillon VS, Fenech M. Mutations that affect mitochondrial functions and their association with neurodegenerative diseases. Mutat Res Rev Mutat Res. 2014;759:1–13. doi: 10.1016/j.mrrev.2013.09.001. [DOI] [PubMed] [Google Scholar]