

Abstract
Panic disorder (PD) affects about four million Europeans, with women affected twice as likely as men, causing substantial suffering and high economic costs. The etiopathogenesis of PD remains largely unknown, but both genetic and environmental factors contribute to risk. An epigenome-wide association study (EWAS) was conducted to compare medication-free PD patients (n = 89) with healthy controls (n = 76) stratified by gender. Replication was sought in an independent sample (131 cases, 169 controls) and functional analyses were conducted in a third sample (N = 71). DNA methylation was assessed in whole blood using the Infinium HumanMethylation450 BeadChip. One genome-wide association surviving FDR of 5% (cg07308824, P = 1.094 × 10-7, P-adj = 0.046) was identified in female PD patients (N = 49) compared to controls (N = 48). The same locus, located in an enhancer region of the HECA gene, was also hypermethylated in female PD patients in the replication sample (P = 0.035) and the significance of the association improved in the meta-analysis (P-adj = 0.004). Methylation at this CpG site was associated with HECA mRNA expression in another independent female sample (N = 71) both at baseline (P = 0.046) and after induction by dexamethasone (P = 0.029). Of 15 candidates, 5 previously reported as associated with PD or anxiety traits also showed differences in DNA methylation after gene-wise correction and included SGK1, FHIT, ADCYAP1, HTR1A, HTR2A. Our study examines epigenome-wide differences in peripheral blood for PD patients. Our results point to possible sex-specific methylation changes in the HECA gene for PD but overall highlight that this disorder is not associated with extensive changes in DNA methylation in peripheral blood.
Introduction
Panic disorder (PD) is the most disabling anxiety disorder, causing substantial suffering, and high economic and social costs. It affects about four million Europeans (12-month prevalence estimate is about 2%) with women being twice as likely to be affected as men1. PD is characterized by sudden episodes of acute anxiety (panic attacks) occurring without any apparent reason. It can be accompanied by a persistent concern of having additional attacks or worry about the possible consequences of the attacks (e.g., suffering of a heart attack, dying, losing control) and significant behavioral changes to avoid future panic attacks2. First onset for PD is in adolescence and early adulthood2 and it is highly co-morbid with other mental disorders, especially agoraphobia3.
Despite the substantial long-term disability, PD appears to be underdiagnosed and undertreated in mental health settings. The overall heritability of PD is substantial with heritability estimates up to 48%4. Genetic studies (genome-wide as well as candidate gene approaches) have identified some loci possibly contributing to disease risk such as variants in the TMEM132D locus5, 6, COMT 7, CRHR1 8, SLC6A4, MAOA, HTR1A 9, but overall genetic linkage or association signals are usually not consistently replicated.
Exposure to negative life events are the complementing risk factors to genetics so that both genetic and environmental components are likely important mechanisms influencing disease risk10. The differential contribution of environmental and genetic factors in risk for anxiety disorders, personality disorders, alcohol use disorders has been previously supported by epidemiologic and twin studies11–13. Adverse life events have been shown to associate with specific epigenetic modifications, such as DNA methylation, which may mediate the lasting cellular consequences of these exposures in psychiatric disorders14, also in the context of gene–environment interactions15. Exploring DNA methylation may thus be able to give an integrated view of both environmental and genetic risk factors. While epigenetic changes including DNA methylation are mainly tissue specific, some sites show cross tissue relevance16, 17 and furthermore changes in peripheral tissues such as blood could serve as potential biomarker for disease risk.
To date, a few studies have investigated differences in DNA methylation in candidate genes in PD5, 18–21. Recently, one epigenome-wide study of DNA methylation in a relatively small Japanese PD case-control sample (48 patients vs. 48 controls) detected significant associations at 40 sites with overall small methylation differences22. So far, studies in the European population are lacking. To perform such a study in two independent samples of patients with PD vs. control was the aim of our study. To reduce confounding due to effects of drug treatment, both patients and controls were free of psychotropic medication. Given that both the prevalence of PD as well as DNA methylation pattern show large gender differences23, a gender-stratified analysis was undertaken and complemented by a meta-analysis.
Previous studies report that hits identified in genome-wide association studies (GWAS) show changes in DNA methylation in peripheral blood, e.g., in schizophrenia24 or bipolar disorder25. For this reason, in addition to an unbiased approach, we also investigated DNA methylation changes in candidate genes that have emerged from genome-wide or candidate gene studies for PD, anxiety disorders or anxiety related phenotypes either in humans or animals5,26, 27.
PD has a high comorbidity rate not only with other psychiatric disorders like agoraphobia and depression, but also with other medical conditions, e.g., cardiovascular disorders, asthma and epilepsy1. DNA methylation age has been previously correlated with morbidity and mortality28–31. Therefore, we also investigated whether age acceleration was occurring in PD patients compared to controls.
Materials and Methods
Max Planck Institute of Psychiatry PD cohort
PD patients included in the discovery and replication sample were recruited in the anxiety disorders outpatient unit at the MPIP in Munich5. PD was the primary diagnosis; mild secondary depression was allowed (Table 1). The diagnosis was ascertained by trained psychiatrists according to the Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV criteria. All patients underwent the Structured Clinical Interviews for DSM-IV (SCID I and II). PD due to a medical or neurological condition or the presence of a comorbid Axis II disorder was an exclusion criterion. All patients underwent a thorough medical examination including EEG, ECG and detailed hormone laboratory assessment.
Table 1.
Characteristics of the participants included in the study
| Variable | Controls | Cases | Total |
|---|---|---|---|
| Discovery | |||
| Participants, N (%) | 76 (46%) | 89 (54%) | 165 |
| Male, N (%) | 28 | 40 | 68 (41%) |
| Female, N (%) | 48 | 49 | 97 (59%) |
| Age, years (SD) | 37 (7.5) | 36 (10.4) | |
| Diagnosis | None | PDA 72% | |
| PD 28% | |||
| Comorbidity:MDD 13.5% | |||
| Replication | |||
| Participants, N (%) | 169 (56%) | 131 (44%) | 300 |
| Male, N (%) | 48 | 48 | 96 (32%) |
| Female, N (%) | 121 | 83 | 204 (68%) |
| Age, years (SD) | 38 (7.2) | 38 (11.6) | |
| Diagnosis | None | PDA 61% | |
| PD 39% | |||
| Comorbidity:MDD 13% | |||
| Meta-analysis | |||
| Participants, N (%) | 245 (55%) | 220 (45%) | 465 |
| Male, N (%) | 76 | 88 | 164 (34%) |
| Female, N (%) | 169 | 132 | 301 (66%) |
| Age, years (SD) | 38 (7.3) | 38 (11) | |
| Diagnosis | None | PDA 65% | |
| PD 35% | |||
| Comorbidity:MDD 13% | |||
| Dex-treatment study | |||
| Participants, N (%) | 29 (41%) | 42 (59%) | 71 |
| Male, N (%) | 0 | 0 | 0 |
| Female, N (%) | 29 | 42 | 71 |
| Age, years (SD) | 44 (11.4) | 44 (13.7) | |
| Diagnosis | None | MDD | |
PDA panic disorder with agoraphobia, PD panic disorder without agoraphobia, MDD major depressive disorder
Control subjects were recruited from a Munich-based community sample and screened for the absence of axis I psychiatric disorders using the Munich version of the Composite International Diagnostic Interview32. Controls were age-matched and sex-matched with patients.
To reduce confounding due to effects of drug treatment, both patients and controls were free of psychotropic medication for at least 4 weeks before the blood draw. All subjects were Caucasian and provided written informed consent. The Ethics Committee of the Ludwig Maximilians University, Munich, Germany, in accordance with the Declaration of Helsinki approved all procedures.
Microarray processing and quality control in the MPIP PD cohort
Genomic DNA was extracted from peripheral blood using the Gentra Puregene Blood Kit (Qiagen). DNA quality and quantity was assessed using NanoDrop 2000 Spectrophotometer (Thermo Scientific) and Quant-iT Picogreen (Invitrogen). To minimize batch effects, samples were randomized with respect to case-control status, sex and age.
Genomic DNA was bisulfite converted using the Zymo EZ-96 DNA Methylation Kit (Zymo Research) and DNA methylation levels were assessed for >480,000 CpG sites using the Illumina HumanMethylation450 BeadChip array. Hybridization and processing were performed according to the instructions of the manufacturer.
The Bioconductor R package minfi (version 1.10.2) was used for the quality control of methylation data including intensity read outs, normalization, cell type composition estimation, β-value and M-value calculation. Outliers, i.e., samples whose behavior deviated from that of others in terms of median intensity, were excluded from the analysis (N = 3 in the discovery sample, N = 5 in the replication sample) as well as samples with a discordant methylation-predicted vs. reported sex (N = 1 in the replication sample).
Failed probes were excluded based on a detection P-value larger than 0.01 in >50% of the samples. X and Y chromosome were removed to avoid a possible gender effect and also non-specific binding probes33. We also excluded probes if single nucleotide polymorphisms (SNPs) were documented in the interval for which the Illumina probe is designed to hybridize. Probes located close (10 bp from query site) to a SNP which had a minor allele frequency of ≥0.05, as reported in the 1000 Genomes Project, were also removed. This yielded a total of around 425,000 CpG sites in the discovery and replication sample for further analysis.
The data were then normalized with functional normalization (FunNorm)34, an extension of quantile normalization included in the R package minfi.
Batch effects were identified by inspecting the association of principal components of the methylation levels with possible technical batches using linear regressions and visual inspection of principal component analysis plots using the Bioconductor R package shinyMethyl (version 0.99.3). Identified batch effects (i.e., bisulfite conversion plate and plate position) were removed using the Empirical Bayes’ method ComBat 35. Batch corrected M-values after ComBat were used for all further statistical analyses.
Epigenome-wide association analysis
Linear regression models were fit for each probe to test for a case vs. control difference within the R package MatrixEQTL (version 2.1.1)36. Sex, age and imputed white blood cell distribution from the Houseman projection37 were included as covariates. Population stratification was investigated using multidimensional scaling and could not be observed (Supplementary Fig. S1). Significance after multiple testing was adjusted using false discovery rate (FDR) of 5%. As a first step all the samples were analyzed together (Table 1) but, given the higher prevalence of PD in females, we performed a sex-stratified analysis, first in the discovery and then in the replication sample. A fixed-effect meta-analysis across both samples was performed in Plink v1.938 following identification of hits in the individual analyses.
In order to investigate whether we can find clusters of association in the epigenome-wide analysis, we performed the differentially methylated region (DMR) analysis on the combined results from both samples based on the input of individual P-values of at least 5e-05 and within 500 bp using Comb-P39.
Targeted gene analysis
A high number of studies showed mostly single SNP associations in different genes with PD, however, the replicability of these findings was low. Therefore, we used three lines of approaches to select candidate genes for the targeted methylome analysis: (1) candidate genes from human genetic studies confirmed in the recent meta-analysis of different international PD cohorts (TMEM132D, COMT, NPSR1 and HTR2A)7, (2) and/or having additional evidence from translational studies for anxiety and stress related-phenotypes (CRH, CRHR1, ADCYAP1, ADCYAP1R1, FKBP5, SGK1, BDNF, HTR1A)8, 40–46 and lastly, (3) genes containing loci with previous evidence for differential methylation in PD and anxiety disorders (GAD1, OXTR)20, 47. All the genes examined (N = 15) showed previous evidence of association with stress-related phenotypes not only in clinical (human) studies but also in preclinical (animal) studies5,26,27, 48–52.
The CpGs lying within the target genes were selected from the meta-analysis results of the epigenome-wide association study (EWAS) and FDR correction of 5% was applied for the number of CpGs included in the gene.
Disease association analysis
To investigate a possible enrichment for specific pathways, we conducted a disease association analysis using Web Gestalt53, 54, DAVID55, 56 and the R-package DOSE57.
Tested genes for a disease enrichment were annotated from CpG sites with P-value < 0.001 in the meta-analysis results of the cases vs. controls EWAS in the whole sample (N genes = 312), in the female subset (N genes = 428) and in the male subset (N genes = 379). The analysis was background corrected for the Illumina HumanMethylation450 BeadChip array annotated genes.
DNA Methylation age calculation
DNA methylation age was calculated from peripheral blood of patients and controls included in the discovery (N = 165) and replication sample (N = 300). DNA methylation-based age prediction was performed using the R code and statistical pipeline developed by Horvath58. This predictor was developed using 82 Illumina DNA methylation array datasets (n = 7,844) involving 51 healthy tissues and cell types58. The raw data were normalized using BMIQ normalization method59 implemented in the Horvath DNA methylation-based age predictor R script58. We then tested whether epigenetic age acceleration (∆-age), calculated by subtracting the actual chronological age from DNA methylation age58, was associated with case-control status. Since DNA methylation age is calculated from raw beta values, technical batches identified for discovery and replication sample (96-well plate) were included as covariates in the linear regression model together with age, sex and cell counts (Houseman and Horvath cell counts, specifically: PlasmaBlast, CD8pCD28nCD45Ran, CD8.naive, CD4T, NK, Mono, Gran).
MPIP dexamethasone treatment study
Glucocorticoid-induced methylation and gene expression changes were examined in an independent sample of 71 Caucasian female subjects (29 healthy probands and 42 depressed) recruited at the MPIP. Recruitment strategies and characterization of participants have been previously described60. Baseline whole blood samples were obtained at 6 pm after 2 h of fasting and abstention from coffee and physical activity (baseline). Subjects then received 1.5 mg oral dexamethasone (DEX) and a second blood draw was performed at 9 pm 3 h after DEX ingestion (post-DEX). The study was approved by the local ethics committee and all individuals gave written informed consent.
DNA methylation and gene expression arrays in the MPIP dexamethasone treatment study
Genomic DNA was extracted from whole blood using the Gentra Puregene Blood Kit (QIAGEN) and processed as for the MPIP PD cohort. DNA methylation levels were assessed for >480,000 CpG sites using the Illumina HumanMethylation450 BeadChip arrays. Whole blood RNA was collected using PAXgene Blood RNA Tubes (PreAnalytiX), processed as described previously60. Blood RNA was hybridized to Illumina HumanHT-12 v3 and v4 Expression BeadChips arrays. All methylation and gene expression array probes have been subjected to an extensive quality control including filtering by low p-detection value, normalization (FunNorm for methylation and VSN for gene expression data) and batch correction with ComBat as previously described in ref. 61. Cellular composition was estimated by using CellCode62.
Statistical analysis in the MPIP dexamethasone treatment study
Methylation levels of cg07308824 were tested for association with gene expression levels of the HECA mRNA (ILMN_1770667) using a linear mixed effects model within the lme4 package63.
Results
Genome-wide methylation differences in discovery and replication samples
Genome-wide associations were performed in the discovery sample, combined as well as stratified by gender. While no association survived correction for multiple testing in the overall samples and the male subset, one genome-wide association, cg07308824, surviving FDR of 5% (P = 1.094 × 10-7, P-adj = 0.046) was observed in the female-only discovery sample. QQ plots for each of the analyses are presented in the Supplementary Figs. S2–S3. cg07308824 is located in the promoter of the HECA gene and was hypermethylated in female PD patients (N = 49) compared to controls (N = 48). The association and the direction of the association could be replicated in the second sample (P = 0.035) and yielded a combined P-value of 1.651e-08 in the meta-analysis, that would again survive correction for multiple testing (P-adj = 0.004) (Figs. 1, 2).
Fig. 1. Manhattan plot of the Panic Disorder EWAS in females (meta-analysis results): the x-axis shows chromosomal position and the y-axis shows −log10 (P).

The red line represents the multiple test threshold (P < 1.09 × 10−7)
Fig. 2.
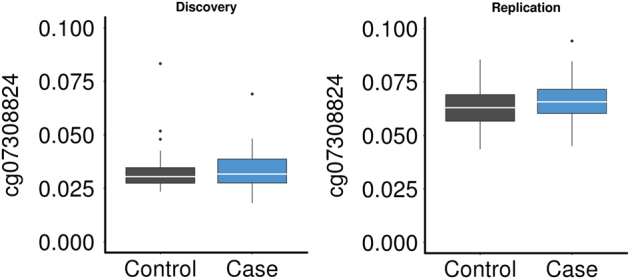
Box plot of DNA methylation levels for the genome-wide significant CpG in the discovery (P-adj = 0.046) and replication sample (P = 0.035)
The DMR analysis using Comb-P revealed no significantly associated regions.
Targeted gene analysis
The targeted gene analysis (see Supplementary Tables S1–S15 for the complete list of genes tested) using the meta-analysis results, yielded in females one significant CpG each (surviving 5% FDR correction over the CpGs in the gene) in ADCYAP1 (P-adj = 0.010) and HTR1A (P-adj = 0.041) (Supplementary Fig. S4). The same analysis yielded one significant CpG in SGK1 (P-adj = 0.035) (Supplementary Fig. S5) in the whole sample and in males in fragile histidine triad (FHIT, P-adj = 0.010) and two significant CpGs in HTR2A (P-adj = 0.015 and P-adj = 0.029) (Supplementary Fig. S6) (Table 2). Single nominal associations have been found in the genes ADCY1P1R1, BDNF, COMT, CRH, CRHR1, GAD1, OXTR and TMEM132D. No differential methylation was detected for NPSR1 between cases and controls.
Table 2.
Targeted gene analysis results for the significant CpGs
| Sample | Gene | CpG | P-adj |
|---|---|---|---|
| Meta-analysis | |||
| Whole | SGK1 | cg00959636 | 0.035 |
| Males | FHIT | cg07351758 | 0.010 |
| HTR2A | cg09361691 | 0.015 | |
| cg06476131 | 0.029 | ||
| Females | ADCYAP1(PACAP) | cg13940693 | 0.010 |
| HTR1A | cg16280141 | 0.041 | |
Disease association analysis
An enrichment for psychiatric disorders could be found in the whole sample (bipolar disorder, P = 1.9e-2; mental disorders, P = 2.5e-2) and in females (response to antipsychotic treatment, P = 5.3e-2; ADHD, P = 9.5e-2) using DAVID55, 56.
Functional characterization of significant results
To assess the functionality of the significant CpG methylation site, association of methylation levels with gene expression of the HECA mRNA was tested. Methylation at this CpG site was associated with mRNA expression of HECA (ILMN_1770667) both at baseline (P = 0.046) and after induction by dexamethasone (P = 0.029) (Fig. 3). Gene expression was significantly altered in the sample following dexamethasone induction (P = 8.78e-05) but not DNA methylation (P = 0.796), indicating that the significant association between gene expression and DNA methylation is specific and not due to dexamethasone.
Fig. 3.
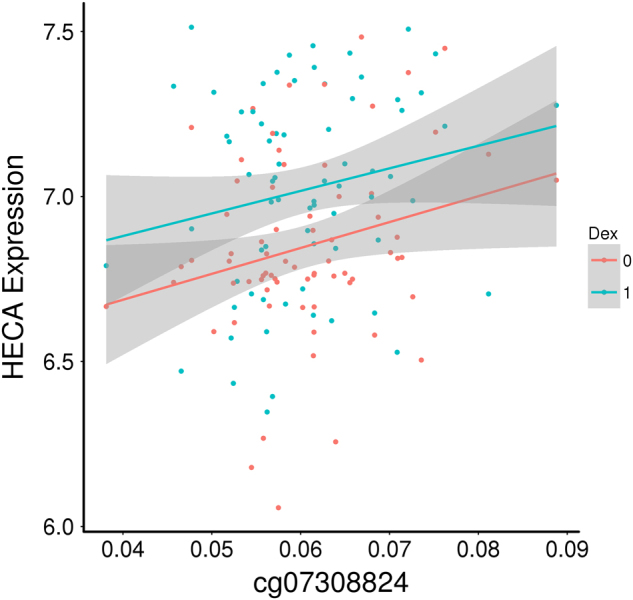
Scatterplot showing the association between DNA methylation (x-axis, beta values) and gene-expression (y-axis, VSN normalized array probe intensity) in an independent female sample at baseline (P = 0.046) and after induction by dexamethasone (P = 0.029)
DNA Methylation age and case-control status
PD is a strong stressor for the people affected and so far no studies have been carried out to determine whether patients affected by PD also develop age acceleration. To answer this question, we compared the ∆-age of PD patients with healthy controls in the whole discovery (N = 165, P = 0.980) and replication sample (N = 300, P = 0.282) and found no significant differences. We then stratified for gender and found no significant results in males (discovery: N = 68, P = 0.835; replication: N = 95, P = 0.467) as well as in females (discovery: N = 97, P = 0.964; replication: N = 204, P = 0.402) (Fig. 4, Supplementary Fig. S7).
Fig. 4. Violin plots of ∆-age by case-control status in the discovery sample.

From the left: whole sample (P = 0.980), males only (P = 0.835), and females only (P = 0.964)
Discussion
Genome-wide association of whole blood DNA methylation with PD cases and matched controls identified a locus (cg07308824), which was hypermethylated in female PD patients compared to healthy controls. This locus was also associated with case-control status in females in another independent sample and results were further confirmed with a meta-analysis (N = 301). No methylation differences were identified at genome-wide level taking both genders together. This is the first and biggest EWAS for PD in a population with European background.
The methylation locus that we identified in females is located in the intragenic and enhancer region64 of the Homo sapiens headcase homolog (Drosophila) (HECA) gene on Chromosome 6. The HECA gene is a cell cycle regulator and may play an important role in human cancers, e.g., hepatocellular carcinoma65; however, only a few publications about this gene are available to date. The potential functional relevance of cg07308824 was further investigated in the UCSC Genome Browser64. An overlap was observed between the location of cg07308824 probe and histone 3 lysine 27 acetylation (H3K27Ac) on seven cell lines from ENCODE66 (Fig. 5). H3K27Ac was previously found near to active regulatory elements suggesting that the sequence where the probe is located is functional67. The HECA gene is expressed in brain at lower levels compared to blood (Supplementary Fig. S8) but we could not see any correlation between the methylation levels of the significant CpG found in blood and brain, which could indicate that the relevance of these results might be limited to blood. No significant correlations were found between cg07308824 methylation levels and four different brain regions (i.e., prefrontal cortex, superior temporal gyrus, entorhinal cortex and cerebellum) in a linear regression model using a publicly available data set16(Supplementary Fig. S9).
Fig. 5. Annotation of the genome-wide significant CpG located in the HECA gene.

The top panel contains the HECA gene model, located on Chr 6. The other two panels show the genome-wide significant CpG and the CpG island where the CpG is located. The bottom panel shows the levels of enrichment of the H3K27Ac mark in the HECA gene. Data were obtained from UCSC Genome Browser and plotted using the R package Gviz87
It is noteworthy considering that methylation levels of the identified locus showed a significant correlation with gene expression levels of the HECA gene in another independent female sample, which points to the functional relevance of the observed methylation change. The fact that the direction of the association is positive (higher gene expression correlated with higher methylation) may be explained by the intragenic location of the significant locus. It has indeed previously been shown68 that a positive correlation with gene expression is expected for CpG probes located in the body of the gene and a negative correlation is expected for CpG probes located close to a gene’s TSS. The authors also report that however this is only partially verified, with one-third of the latter type showing a positive correlation and nearly half of the former type showing a negative correlation.
Notably, the significant changes in DNA methylation presented here are small with less than 1% difference (0.08%) but replicable. While the functional relevance of such small changes is debatable, these effect sizes are in line with other EWAS in psychiatry, including the recent small Japanese EWAS in PD22, where 40 significant CpGs with overall low methylation (mostly under 0.05%) differences have been detected. Sex-specific associations were not reported, most likely because of the small sample size. Furthermore, similar effect sizes were observed in other methylome studies of psychiatric disorders (schizophrenia24) and other complex diseases (rheumatoid arthritis69, multiple sclerosis70 and Alzheimer’s disease71). The EWAS in schizophrenia is largest EWAS in the psychiatric field to date, with 689 schizophrenia patients and 645 controls included in the analysis and shows methylation differences lower than 0.02% that replicate in an independent cohort. More work needs to be performed to understand what contributes to these small differences (e.g., slight differences in cell composition, genotype differences or differences in environmental exposures) and if they are informative beyond serving as biomarker.
Female-specific DNA methylation changes in PD have been previously shown both in mice72 and in humans. A female-specific association and/or correlation of negative life events with decreased overall methylation levels has been shown for GAD1 and MAOA 20, 73. In contrast, female-specific effects in terms of increased methylation levels of promoter region were observed in the FOXP3 gene for PD18. Sex-specific findings regarding the methylation pattern have been also detected in depression, which is highly comorbid with PD, and psychosis74, 75.
Interestingly, the disease association analysis shows an enrichment for psychiatric disorders in the whole sample and in females, but not in males.
In our analysis with Comb-P, no evidence for clusters of differential methylation per gene could be detected, indicating that associated CpGs do not have adjacent signals. The genome-wide findings are thus not supported by DMR analysis. However, the structure of the 450k Illumina array has many loci that are only interrogated with single CpGs, especially in enhancer regions, so that single significant CpGs could reflect a differentially methylated region, but since no adjacent CpGs are probed, this cannot be assessed using only array data. Here bisulfite sequencing approaches are needed.
One of the limitation of the study is that we could not correct for the smoking status of the subjects, due to lack of information. We verified though that our significant genome-wide hit was not one of the top-associated CpGs in the biggest EWAS for cigarette smoking76. Another limitation is the lower number of males compared to females, which is due to the higher prevalence of the disease in the latter. This might also explain why no significant results were found in the male subset but only in females. For this reason a bigger study with a higher number of subjects, with possibly the same ratio between genders, is necessary in order to confirm the sex-specificity of our findings.
We have to point out that we observe increased lambdas in the QQ plots of the replication sample despite correction for population stratification and methodological issues, such as DNA extraction and batch effects. It is noteworthy that female subjects of the replication sample seem to contribute to the higher inflation in our study. Other previously published EWAS in psychiatric disorders have shown similar or even higher inflation24,25, 77–79. The different sample size of discovery and replication sample as well as between female and male subjects might be one reason for these effects. The sample size of female subjects is doubled in the replication sample (97 vs. 204). If we extrapolate the inflation factor from discovery to replication sample80, we see an increase of lambda to 1.11. In our study, we saw the association in the HECA gene first in the discovery sample which showed only moderate inflation and could then confirm the finding in the second sample and meta-analysis, suggesting that this finding cannot be attributed just to the inflation effects in the replication sample. Additionally, if we correct for population stratification using the inflation factor qualitatively results are not altered.
A targeted gene approach was subsequently applied, with the aim of investigating if genes previously associated with PD or anxiety-related phenotypes are affected at the methylation level. Five of the genes we analyzed, i.e., HTR1A, HTR2A, ADCYAP1 (pituitary adenylate cyclase-activating polypeptide (PACAP)), FHIT and SGK1 showed different methylation patterns in PD patients compared to controls. For these genes, the evidence for a correlation with anxiety disorders at the genetic level could be confirmed in our data at the epigenetic level. SGK1 (serum/glucocorticoid regulated kinase 1) is one of the key player in the mediation of fast and chronic stress response and, therefore, could be implicated in the transition of the environmental stress influences via methylation43. It seems to play a role in the expression of conditioned fear in the animal model27 and is one target of miRNAs in the glucocorticoid pathway affecting neurogenesis and leading to anxiogenic and depressiogenic behavior in mice81. Additional evidence from human studies point to the implication of this gene in the pathophysiology of traumatic stress, e.g., PTSD82 and our results point to a possible involvement in PD as well. The second gene which is implicated in the stress response regulation is PACAP. Ressler et al. could demonstrate a female-specific significant correlation of the PACAP38 peptide concentration in blood with PTSD symptoms and diagnosis83. In line with these previous findings, we could also demonstrate a female-specific significant methylation difference in one locus between cases and controls, suggesting that this gene may play an important role in long-lasting stress dependent pathophysiology in PD or other anxiety disorders. Similarly, a female-specific methylation difference could be shown for the gene HTR1A. This serotonin receptor is the most abundant of all serotonin receptors in the brain and HTR1A variants have been shown to be associated with depression and defensive behavior in PD patients41. So far, there is no evidence for gender-specific implication of HTR1A gene in PD or other mental disorders. There are instead already previous studies showing an association of HTR2A and PD7, 84, and supporting evidence comes from our results in males. Similarly, variants in the gene FHIT (fragile histidine triad) were nominally associated in GWAS studies with anxiety85 and PD5, and were not gender-specific. In a recent huge meta-analysis for broad depression phenotype, several variants in FHIT were among the most significant hits86. However, there was no difference in the burden of depressive symptoms or depression diagnosis between the male and female group in our study, therefore, we cannot refer our finding to depression as bias. For HTR1A, HTR2A and FHIT, gender-specific effects presented here need to be replicated and elucidated in further studies.
In the targeted gene analysis we observe five significant results, which is more than expected by chance (2–3 expected by chance). We corrected gene-wide because each gene was regarded as separate analysis driven by the positive evidence for association with anxiety phenotypes. The results might be biased by the selection of the candidate gene itself as one limitation of the gene-targeted approach. Therefore, these findings, while supporting previous results, have to be taken with caution and replicated elsewhere.
Another aim of the study was to determine the effect of PD on epigenetic aging, as measured with the epigenetic clock58. We found no differences between PD patients and healthy controls in the discovery and replication sample, also when separated by gender. This might be explained by the heterogeneity of the samples in terms of age. It was showed by Zannas et al.61 that the effect of personal life stress on ∆-age is stronger in older as compared to younger people. This suggests that the effects in our study, if presents, might be diluted by the age-range. Additionally, the effect sizes of PD on epigenetic age might be low and higher sample sizes are needed to detect significant changes. As cumulative lifetime stress has been shown to contribute to faster epigenetic aging61, analysis of other phenotypes, such as number of panic attacks or duration of the disorder, could be more effective in uncovering epigenetic aging effects of PD. In summary, our study examines epigenome-wide differences in peripheral blood for PD patients. Our results point to possible sex-specific methylation changes in the HECA gene for PD but overall highlight that this disorder is not associated with extensive changes in DNA methylation pattern in peripheral blood.
Electronic supplementary material
Acknowledgements
We thank Maik Ködel and Susann Sauer for the DNA extraction, Torsten Klengel for his assistance in performing the experiments and Monika Rex-Haffner for her technical assistance.
This study was financed by ERA-NET NEURON.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Stella Iurato, Email: stellaiurato@outlook.com.
Angelika Erhardt, Email: erhardt@psych.mpg.de.
Supplementary Information
The online version of this article (10.1038/s41398-017-0026-1) contains supplementary material.
References
- 1.American Psychiatric Association, Diagnostic and Statistical Manual of Mental Disorders (DSM-5®) (American Psychiatric Publishing, Arlington, VA, 2013).
- 2.Goodwin RD, Rosi S, et al. The epidemiology of panic disorder and agoraphobia in Europe. Eur. Neuropsychopharmacol. 2005;15:435–443. doi: 10.1016/j.euroneuro.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Noyes R, Jr, et al. Relationship between panic disorder and agoraphobia. A family study. Arch. Gen. Psychiatry. 1986;43:227–232. doi: 10.1001/archpsyc.1986.01800030037004. [DOI] [PubMed] [Google Scholar]
- 4.Hettema JM, Neale MC, Kendler KSA. Review and meta-analysis of the genetic epidemiology of anxiety disorders. Am. J. Psychiatry. 2001;158:1568–1578. doi: 10.1176/appi.ajp.158.10.1568. [DOI] [PubMed] [Google Scholar]
- 5.Erhardt A, et al. TMEM132D, a new candidate for anxiety phenotypes: evidence from human and mouse studies. Mol. Psychiatry. 2011;16:647–663. doi: 10.1038/mp.2010.41. [DOI] [PubMed] [Google Scholar]
- 6.Erhardt A, et al. Replication and meta-analysis of TMEM132D gene variants in panic disorder. Transl. Psychiatry. 2012;2:e156. doi: 10.1038/tp.2012.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howe AS, et al. Candidate genes in panic disorder: meta-analyses of 23 common variants in major anxiogenic pathways. Mol. Psychiatry. 2016;21:665–679. doi: 10.1038/mp.2015.138. [DOI] [PubMed] [Google Scholar]
- 8.Weber H, et al. Allelic variation in CRHR1 predisposes to panic disorder: evidence for biased fear processing. Mol. Psychiatry. 2016;21:813–822. doi: 10.1038/mp.2015.125. [DOI] [PubMed] [Google Scholar]
- 9.Gottschalk MG, Domschke K. Novel developments in genetic and epigenetic mechanisms of anxiety. Curr. Opin. Psychiatry. 2016;29:32–38. doi: 10.1097/YCO.0000000000000219. [DOI] [PubMed] [Google Scholar]
- 10.Teh AL, et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res. 2014;24:1064–1074. doi: 10.1101/gr.171439.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torvik FA, et al. Longitudinal associations between social anxiety disorder and avoidant personality disorder: a twin study. J. Abnorm. Psychol. 2016;125:114–124. doi: 10.1037/abn0000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.South SC, et al. A population based twin study of DSM-5 maladaptive personality domains. Personal Disord. 2017;8:366–375. doi: 10.1037/per0000220. [DOI] [PubMed] [Google Scholar]
- 13.Kendler KS, et al. A National Swedish Twin-Sibling Study of alcohol use disorders. Twin Res. Hum. Genet. 2016;19:430–437. doi: 10.1017/thg.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slatkin M. Epigenetic inheritance and the missing heritability problem. Genetics. 2009;182:845–850. doi: 10.1534/genetics.109.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klengel T, Binder EB. Epigenetics of stress-related psychiatric disorders and gene × environment interactions. Neuron. 2015;86:1343–1357. doi: 10.1016/j.neuron.2015.05.036. [DOI] [PubMed] [Google Scholar]
- 16.Hannon E, Lunnon K, Schalkwyk L, Mill J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics. 2015;10:1024–1032. doi: 10.1080/15592294.2015.1100786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farre P, Jones MJ, Meaney MJ, Emberly E, Turecki G, Kobor MS. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin. 2015;8:19. doi: 10.1186/s13072-015-0011-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prelog M, et al. Hypermethylation of FOXP3 promoter and premature aging of the immune system in female patients with panic disorder? PLoS. One. 2016;11:e0157930. doi: 10.1371/journal.pone.0157930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ziegler C, et al. MAOA gene hypomethylation in panic disorder-reversibility of an epigenetic risk pattern by psychotherapy. Transl. Psychiatry. 2016;6:e773. doi: 10.1038/tp.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Domschke K, et al. Epigenetic signature of panic disorder: a role of glutamate decarboxylase 1 (GAD1) DNA hypomethylation? Prog. Neuropsychopharmacol. Biol. Psychiatry. 2013;46:189–196. doi: 10.1016/j.pnpbp.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Bayles R, et al. Methylation of the SLC6a2 gene promoter in major depression and panic disorder. PLoS. One. 2013;8:e83223. doi: 10.1371/journal.pone.0083223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimada-Sugimoto M, et al. Epigenome-wide association study of DNA methylation in panic disorder. Clin. Epigenetics. 2017;9:6. doi: 10.1186/s13148-016-0307-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yousefi P, et al. Sex differences in DNA methylation assessed by 450 K BeadChip in newborns. BMC Genom. 2015;16:911. doi: 10.1186/s12864-015-2034-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montano C, et al. Association of DNA methylation differences with schizophrenia in an Epigenome-Wide Association Study. JAMA Psychiatry. 2016;73:506–514. doi: 10.1001/jamapsychiatry.2016.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houtepen LC, van Bergen AH, Vinkers CH, Boks MP. DNA methylation signatures of mood stabilizers and antipsychotics in bipolar disorder. Epigenomics. 2016;8:197–208. doi: 10.2217/epi.15.98. [DOI] [PubMed] [Google Scholar]
- 26.Nieto SJ, Patriquin MA, Nielsen DA, Kosten TA. Don’t worry; be informed about the epigenetics of anxiety. Pharmacol. Biochem. Behav. 2016;146-147:60–72. doi: 10.1016/j.pbb.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knoll AT, Halladay LR, Holmes AJ, Levitt P. Quantitative trait loci and a novel genetic candidate for fear learning. J. Neurosci. 2016;36:6258–6268. doi: 10.1523/JNEUROSCI.0177-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horvath S, et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY) 2015;7:1159–1170. doi: 10.18632/aging.100861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marioni RE, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015;16:25. doi: 10.1186/s13059-015-0584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen BH, et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY) 2016;8:1844–1859. doi: 10.18632/aging.101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christiansen L, et al. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell. 2016;15:149–154. doi: 10.1111/acel.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wittchen, H. U. & Pfister, H. DIA-X-Interviews: Manual für Screening-Verfahren und Interview (Swets & Zeitlinger, Frankfurt, 1997).
- 33.Chen YA, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fortin JP, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:503. doi: 10.1186/s13059-014-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 36.Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Houseman EA, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinforma. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pedersen BS, Schwartz DA, Yang KechrisKJ., IV Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28:2986–2988. doi: 10.1093/bioinformatics/bts545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blaya C, et al. Panic disorder and serotonergic genes (SLC6A4, HTR1A and HTR2A): association and interaction with childhood trauma and parenting. Neurosci. Lett. 2010;485:11–15. doi: 10.1016/j.neulet.2010.08.042. [DOI] [PubMed] [Google Scholar]
- 41.Straube B, et al. The functional −1019C/G HTR1A polymorphism and mechanisms of fear. Transl. Psychiatry. 2014;4:e490. doi: 10.1038/tp.2014.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ressler KJ, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470:492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cattaneo A, Riva MA. Stress-induced mechanisms in mental illness: a role for glucocorticoid signalling. J. Steroid Biochem. Mol. Biol. 2016;160:169–174. doi: 10.1016/j.jsbmb.2015.07.021. [DOI] [PubMed] [Google Scholar]
- 44.Zannas AS, Wiechmann T, Gassen NC, Binder EB. Gene-stress-epigenetic regulation of fkbp5: clinical and translational implications. Neuropsychopharmacology. 2016;41:261–274. doi: 10.1038/npp.2015.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han EJ, et al. Evidence for association between the brain-derived neurotrophic factor gene and panic disorder: a novel haplotype analysis. Psychiatry Investig. 2015;12:112–117. doi: 10.4306/pi.2015.12.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konishi Y, et al. Genexgenexgender interaction of BDNF and COMT genotypes associated with panic disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2014;51:119–125. doi: 10.1016/j.pnpbp.2014.01.020. [DOI] [PubMed] [Google Scholar]
- 47.Ziegler C, et al. Oxytocin receptor gene methylation: converging multilevel evidence for a role in social anxiety. Neuropsychopharmacology. 2015;40:1528–1538. doi: 10.1038/npp.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Desbonnet L, et al. Physiological and behavioural responsivity to stress and anxiogenic stimuli in COMT-deficient mice. Behav. Brain Res. 2012;228:351–358. doi: 10.1016/j.bbr.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 49.Leonard SK, et al. Pharmacology of neuropeptide S in mice: therapeutic relevance to anxiety disorders. Psychopharmacol. (Berl.). 2008;197:601–611. doi: 10.1007/s00213-008-1080-4. [DOI] [PubMed] [Google Scholar]
- 50.Benekareddy M, Vadodaria KC, Nair AR, Vaidya VA. Postnatal serotonin type 2 receptor blockade prevents the emergence of anxiety behavior, dysregulated stress-induced immediate early gene responses, and specific transcriptional changes that arise following early life stress. Biol. Psychiatry. 2011;70:1024–1032. doi: 10.1016/j.biopsych.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mustafa T, Jiang SZ, Eiden AM, Weihe E, Thistlethwaite I, Eiden LE. Impact of PACAP and PAC1 receptor deficiency on the neurochemical and behavioral effects of acute and chronic restraint stress in male C57BL/6 mice. Stress. 2015;18:408–418. doi: 10.3109/10253890.2015.1025044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bahi A, Al Mansouri S, Al Maamari E. Nucleus accumbens lentiviral-mediated gain of function of the oxytocin receptor regulates anxiety- and ethanol-related behaviors in adult mice. Physiol. Behav. 2016;164:249–258. doi: 10.1016/j.physbeh.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 53.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) Nucleic Acids Res. 2013;41:W77–W83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 57.Yu G, Wang LG, Yan GR, He QY. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 2015;31:608–609. doi: 10.1093/bioinformatics/btu684. [DOI] [PubMed] [Google Scholar]
- 58.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teschendorff AE, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29:189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arloth J, et al. Genetic differences in the immediate transcriptome response to stress predict risk-related brain function and psychiatric disorders. Neuron. 2015;86:1189–1202. doi: 10.1016/j.neuron.2015.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zannas AS, et al. Lifetime stress accelerates epigenetic aging in an urban, African American cohort: relevance of glucocorticoid signaling. Genome Biol. 2015;16:266. doi: 10.1186/s13059-015-0828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chikina M, Zaslavsky E, Sealfon SC. CellCODE: a robust latent variable approach to differential expression analysis for heterogeneous cell populations. Bioinformatics. 2015;31:1584–1591. doi: 10.1093/bioinformatics/btv015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw.67, 10.18637/jss.v067.i01 (2015).
- 64.Kent W, Sugnet C, Furey T, Roskin K, Pringle T, Zahler A, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang J, et al. The human homolog of drosophila headcase acts as a tumor suppressor through its blocking effect on the cell cycle in hepatocellular carcinoma. PLoS. One. 2015;10:e0137579. doi: 10.1371/journal.pone.0137579. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Rosenbloom KR, et al. ENCODE Data in the UCGC Genome Browser. Nucleic Acids Res. 2013;41:D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wagner JR, Busche S, Ge B, Kwan T, Pastinen T, Blanchette M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014;15:R37. doi: 10.1186/gb-2014-15-2-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu Y, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 2013;31:142–147. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huynh JL, et al. Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat. Neurosci. 2014;17:121–130. doi: 10.1038/nn.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lunnon K, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 2014;17:1164–1170. doi: 10.1038/nn.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Papale LA, et al. Sex-specific hippocampal 5-hydroxymethylcytosine is disrupted in response to acute stress. Neurobiol. Dis. 2016;96:54–66. doi: 10.1016/j.nbd.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Domschke K, et al. Monoamine oxidase A gene DNA hypomethylation—a risk factor for panic disorder? Int. J. Neuropsychopharmacol. 2012;15:1217–1228. doi: 10.1017/S146114571200020X. [DOI] [PubMed] [Google Scholar]
- 74.Byrne EM, et al. Monozygotic twins affected with major depressive disorder have greater variance in methylation than their unaffected co-twin. Transl. Psychiatry. 2013;3:e269. doi: 10.1038/tp.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Melas PA, et al. Genetic and epigenetic associations of MAOA and NR3C1 with depression and childhood adversities. Int. J. Neuropsychopharmacol. 2013;16:1513–1528. doi: 10.1017/S1461145713000102. [DOI] [PubMed] [Google Scholar]
- 76.Joehanes R, et al. Epigenetic signatures of cigarette smoking. Circ. Cardiovasc. Genet. 2016;9:436–447. doi: 10.1161/CIRCGENETICS.116.001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cecil CA, et al. Epigenetic signatures of childhood abuse and neglect: implications for psychiatric vulnerability. J. Psychiatr. Res. 2016;83:184–194. doi: 10.1016/j.jpsychires.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 78.Houtepen LC, et al. Genome-wide DNA methylation levels and altered cortisol stress reactivity following childhood trauma in humans. Nat. Commun. 2016;7:10967. doi: 10.1038/ncomms10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Viana J, et al. Schizophrenia-associated methylomic variation: molecular signatures of disease and polygenic risk burden across multiple brain regions. Hum. Mol. Genet. 2017;26:210–225. doi: 10.1093/hmg/ddw373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Freedman ML, et al. Assessing the impact of population stratification on genetic association studies. Nat. Genet. 2004;36:388–393. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- 81.Jin J, et al. miR-17-92 cluster regulates adult hippocampal neurogenesis, anxiety, and depression. Cell. Rep. 2016;16:1653–1663. doi: 10.1016/j.celrep.2016.06.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Licznerski P, et al. Decreased SGK1 expression and function contributes to behavioral deficits induced by traumatic stress. PLoS. Biol. 2015;13:e1002282. doi: 10.1371/journal.pbio.1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dias BG, Ressler KJ. PACAP and the PAC1 receptor in post-traumatic stress disorder. Neuropsychopharmacology. 2013;38:245–246. doi: 10.1038/npp.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Unschuld PG, et al. Polymorphisms in the serotonin receptor gene HTR2A are associated with quantitative traits in panic disorder. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2007;144b:424–429. doi: 10.1002/ajmg.b.30412. [DOI] [PubMed] [Google Scholar]
- 85.Luciano M, et al. Genome-wide association uncovers shared genetic effects among personality traits and mood states. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2012;159b:684–695. doi: 10.1002/ajmg.b.32072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Direk N, et al. An analysis of two genome-wide association meta-analyses identifies a new locus for broad depression phenotype. Biol. Psychiatry. 2017;82:322–329. doi: 10.1016/j.biopsych.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hahne F, Ivanek R. Visualizing genomic data using Gviz and bioconductor. Methods Mol. Biol. 2016;1418:335–351. doi: 10.1007/978-1-4939-3578-9_16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.