

ABSTRACT
Pandemic outbreaks of influenza type A viruses have resulted in numerous fatalities around the globe. Since the conventional influenza vaccines (CIV) provide less than 20% protection for individuals with weak immune system, it has been considered that broadly cross-neutralizing antibodies may provide a better protection. Herein, we showed that a recently generated humanized mouse (DRAGA mouse; HLA-A2. HLA-DR4. Rag1KO. IL-2Rgc KO. NOD) that lacks the murine immune system and expresses a functional human immune system can be used to generate cross-reactive, human anti-influenza monoclonal antibodies (hu-mAb). DRAGA mouse was also found to be suitable for influenza virus infection, as it can clear a sub-lethal infection and sustain a lethal infection with PR8/A/34 influenza virus. The hu-mAbs were designed for targeting a human B-cell epitope (180WGIHHPPNSKEQ QNLY195) of hemagglutinin (HA) envelope protein of PR8/A/34 (H1N1) virus with high homology among seven influenza type A viruses. A single administration of HA180-195 specific hu-mAb in PR8-infected DRAGA mice significantly delayed the lethality by reducing the lung damage. The results demonstrated that DRAGA mouse is a suitable tool to (i) generate heterotype cross-reactive, anti-influenza human monoclonal antibodies, (ii) serve as a humanized mouse model for influenza infection, and (iii) assess the efficacy of anti-influenza antibody-based therapeutics for human use.
KEYWORDS: Humanized mice, Human monoclonal antibodies, Human influenza infection model, Anti-flu antibody therapy
Introduction
Influenza viruses are enveloped orthomyxoviruses with a segmented RNA genome of negative polarity containing eight segments that encode ten proteins in the case of type A and B viruses.1 Hemagglutinin (HA) is the most abundant immunogenic envelope protein of influenza viruses with critical role in viral invasion of the lung cells.2,3 Pandemic outbreaks of influenza type A viruses often rampage the global health resulting in numerous fatalities such as the 1918 Spanish pandemic that killed 20 million people across Europe and 50 million worldwide.4 Influenza infections kill 30,000 people on average in a non-epidemic year in USA alone, particularly those with weak immune system like children, elderly, and immunocompromised individuals. In addition, seasonal mutations in the HA protein like expected for the avian and swain influenza viruses may pose significant challenges to the immune system and design of conventional influenza vaccines (CIV). CIV formulations rely on viral strains detected in previous years that cannot optimally protect against unpredictable seasonal viral mutants. In addition, current CIV preparations cannot be administrated in already infected individuals.7 Attempts to design more versatile vaccines like parental adjuvant-combined vaccines, non-parental vaccine with nasal or epidermal split product of whole virion, inactivated virion, DNA-based vaccines, live vaccine preparations using recombinant live viruses, or virus-vectored vaccines, all showed still a limited protection in humans and animal models.5,6
It has been recently considered that the use of broadly cross-neutralizing human specific Abs against multiple influenza virus strains could be a more efficient vaccine/therapeutic approach to protect against mutated influenza viruses than the current CIV preparations.8–10 The choice of cross-neutralizing antibodies over the CIV is based on experiments in animal models showing that the anti-viral HA antibody response is critical for protection against influenza infection. Thus, IgA and IgM Abs can efficiently clear the virus from the lungs in a primary infection, while the IgG Abs can inhibit viral replication in the lungs.11,12 A number of murine monoclonal cross-neutralizing Abs have been already generated and their cross-protective effect was assessed in animal models of influenza infection.13–25 However, the major caveat of murine Abs in humans is that repeated administrations lead to the induction of human anti-mouse antibodies (HAMA), which can significantly diminish or abolish their therapeutic efficacy,26–31 and in some cases could even lead to serious adverse effects like anaphylactic shock.32 To minimize HAMA effects, genetic engineering approaches were aimed at “humanizing” murine antibodies or designing complementary immunomodulatory strategies. Although the humanized Abs showed some therapeutic effect in some cancer and autoimmune diseases, a complete suppression of HAMA effects has not been achieved yet.33–39 For this reason, “fully” human Abs are being now considered the safest vaccination or therapeutic approach for humans. “Fully” human anti-influenza monoclonal Abs (hu-mAbs) were generated from EBV-immortalized B cells collected from infected or vaccinated individuals and their viral neutralization capacity was successfully tested in mouse models for influenza infections.40,41 However, EBV-generated hu-mAbs lack the known epitope specificities, which confines the design of a broadly cross-protective Ab preparation against influenza infection. In contrast, the use of humanized mice to generate “fully” humanized Abs with known target specificities seems to be a more versatile approach.
Several humanized mouse models engineered on SCID and NOD background allowed different stages of human B-cell development, but inefficient progression of naïve human B cells toward a mature status as required for antibody production and immunoglobulin class switch.42–47 However, depending on the mouse strain and method of reconstitution, several limitations still exist: transfer of T and B memory cells from the donor through PMBC infusion is either inefficient or leads to acute or chronic GVDH, interference of murine innate immunity, lack of immunoglobulin class switch, poor HSC engraftment, inefficient human T and/or B cells expansion and homeostasis.48 To avoid some of these limitations, we developed two new NRG strains of humanized mice (DRAG mouse: HLA-DR*0401+/IL-2gc KO, RAG1 KO, NOD; and DRAGA mouse: HLA-DR*0401+ /HLA-A2.1+/IL-2gc KO, RAG1 KO, NOD)49,50 that can efficiently reconstitute a human immune system upon engraftment with HLA-matched human hematopoietic stem cells (HSC), and produce high number of functional human T cells, B cells, dendritic cells, and FOXP3+ T regulatory cells.49–51 DRAG and DRAGA mice elicit specific, “fully” human Abs upon immunization with foreign antigens.49,51 Herein, we used a conserved HA180-195 epitope with shared homology among several strains of influenza virus (PR8-H1N1, WSN-H1N1, Adachi-H2N2, Aichi-H3N2, Memphis-H3N2, VN1194-H4N1, Hok67-H5N3, W213-H9N2, and Maryland-H13N6)9 to immunize DRAGA mice and to ultimately generate cross-reactive hu-mAbs. By establishing DRAGA mouse as the first humanized mouse model for PR8 influenza infection, we were also able to assess the therapeutic effect of a cross-reactive hu-mAb.
Results
Selecting the immunogenic, human B-cell epitope as target for cross-reactive hu-Abs
Previous data showed HA amino acid homology among several influenza A heterotypes.9 We have thus selected a homologous HA epitope shared by several influenza type A heterotypes (180WGIHHPPNSKEQQ NLY195) as a target for hu-mAbs. The HA180-195 epitope of PR8/A/34-H1N1 influenza virus has high homology among 7 other influenza type A viruses: WSN-H1N1, Adachi-H3N2, Aichi-H3N2, Memphis-H3N2, VN1194-H5N1, Hokkaido 67-H5N3, and W213-H9N2. This epitope is located in the HA head and partially comprised by one of the four HA1 antigenic sites (Sb antigenic site52) (Fig. 1A) as a surface exposed a-helix flanked by two b-sheets (Fig. 1B). De novo modeling of HA180-195 epitope in solution by the PEP-FOLD server53,54 led to the prediction of a similar helicoid folding to that in HA protein of the PR8/A/34 and Hokkaido viruses, but not of Memphis virus (Fig. 1C). The “de novo” software can model peptide structures in solution based on the amino acid composition and peptide bond properties, and it does not use any template or already known structure to generate a homology model. In addition, the I-TASSER software that uses an already solved structure from database and relies on sequence homology,55–57 also predicted a similar 3D folding of HA180-195 epitope in the HA1 protein of PR8/A/34 and Hokkaido viruses, but not of Memphis virus. This is consistent with a 100% homology of HA180-195 epitope at the positions 181–186 in all studied virus strains except the Memphis virus, which suggests that these amino acid residues are critical for the HA180-195 helicoidally and/or interaction with our hu-mAbs.
Figure 1.

HA180-195 epitope homology among different influenza virus heterotypes. (A) Sequence alignment of the HA proteins from several influenza virus strains and homologous residues 180–195 comprising part of the Sb antigenic site. (B) Ribbon representation of the HA protein (PDB ID 1ru7:A, residues 54–270) revealing the Sb antigenic site in red. Shown is in detailed view of the Sb antigenic site, the 180–195 residues side chains in ball and stick representation together with their carbon atoms colored in grey, nitrogen in blue and oxygen in red. The first and last residues of the Sb site are indicated with a black dot and labeled, and the highly exposed residues of the HA180-195 epitope located within the a-helix are labeled. (C) Overlapped conformations of soluble HA180-195 epitope from PR8/A/34 (red ribbon), Memphis (yellow ribbon) and Hokkaido (blue ribbon) viruses according to “de novo” modeling approach and PEP-FOLD server (described in materials and methods section).
We next questioned whether the HA180-195 stretch of amino acids is a human T-cell or B-cell epitope. For this, two DRAGA mice were injected in the foot pads with KLH-HA180-195 conjugate and 2 weeks later boosted with the same conjugate. Two weeks after the boost, the splenic cells were incubated for 2 and 4 days with HA180-195 synthetic peptide and the secretion of T-cell cytokines was measured by Luminex. A lack of IL-2, IL-4, IL-5, IL-10, and IFN-g secretion (not shown) ruled out the possibility that the HA180-195 epitope is a human T-cell epitope.
To find out if the HA180-195 is a B-cell epitope able to induce specific hu-Abs in DRAGA mice, the HA180-195 synthetic peptide was covalently coupled to the KLH protein and the conjugate was used to immunize DRAGA mice (described in materials and methods section). Two weeks after the boost with KLH-HA180-195 conjugate, sera from immunized mice was measured by ELISA for human specific Abs using plates coated with BSA-HA180-195 conjugate in parallel with plates coated with PR8 virus. As illustrated in Figure 2A, immunized DRAGA mice elicited human IgM and IgG antibodies to the HA180-195 epitope, which demonstrated that the HA180-195 peptide consensus is an immunogenic human B-cell epitope able to prime human B cells toward secretion of specific IgM and IgG Abs. These results together with finding of human AID master of immunoglobulin class switch in the spleen of naïve DRAGA mice and its up-regulated expression upon PR8 infection of DRAGA mice (Fig. 2B), demonstrated the ability of B cells to undergo immunoglobulin class switch.
Figure 2.
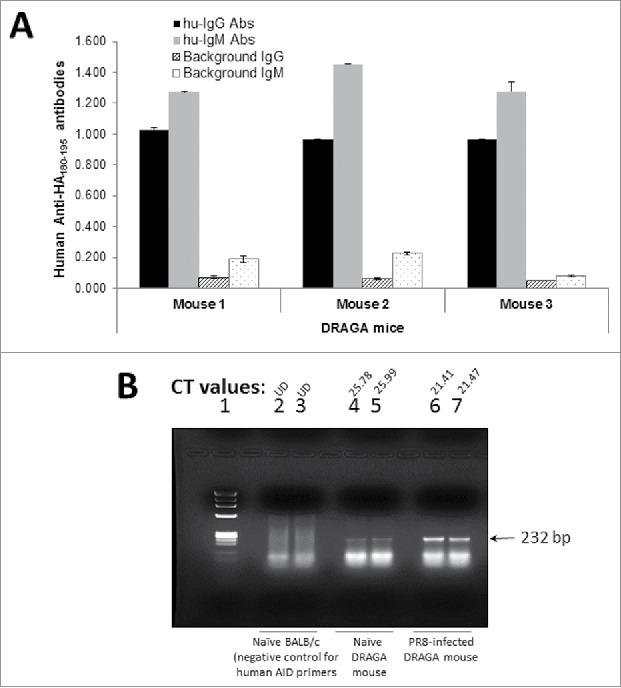
HA180-195 specific hu-Ab responses and human AID expression in DRAGA mice. (A) DRAGA mice were immunized and boosted 2 weeks later with KLH-HA180-195 conjugate. Two weeks after the boost, sera of immunized mice was assessed in ELISA plates coated with rHA of PR8 virus (2 μg/well) for titers of human anti-HA180-195 IgM and IgG antibodies. Shown are the specific antibody titers for 3 representative mice and their corresponding signal-to-noise background of secondary antibody. (B) DRAGA mice (n = 3) were infected or not by the intranasal route with a sub-lethal dose of PR8 /A/34 virus (LD50) and 7 days later the splenic RNA was extracted and analyzed by RT-qPCR using our designed primers for human AID (described in the materials and methods section). One representative DRAGA infected mouse (lanes 6 and 7) and naive DRAGA mouse (lanes 4 and 5) are shown. Control negative for human AID primers specificity was splenic RNA extracted from naive BALB/c mice (lanes 2 and 3). The CT values for duplicate samples are shown for each mouse. Arrow indicates the size of human AID amplicon.
Immunochemical and structural characteristics of HA180-195-specific hu-mAbs
Several hybridoma clones secreting HA180-195 specific hu-mAbs were generated upon fusion of splenic cells from KLH-HA180-195-immunized DRAGA mice with K6/H6 myeloma cells. Hybridoma cells were cloned and sub-cloned, and further selected for stable, highly hu-Ig secreting clones specific for HA180-195 epitope by ELISA-coated plates with PR8 virus or BSA-HA180-195 conjugate. The majority of hybridoma clones (85%) were specific for the KLH protein carrier. Two hybridoma clones secreting HA180-195 specific IgG hu-mAbs were detected during ELISA screening, but did not survive most likely because of loss of chromosomes during re-cloning. Among all specific hu-mAbs, 5 highly secreting, stable HA180-195-specific IgM hybridoma clones (16D11, 6C2 clones #2 and #9, 3E10, and 10B2) were selected for further characterization. IgM hu-mAbs were affinity purified from the cell culture supernatants of hybridoma cells on columns made of goat anti hu-IgM Abs covalently coupled to Sepharose CL-4B gel matrix. The results of ELISA isotyping of HA180-195-specific hu-mAbs was confirmed by Western Blot (WB) as IgM/λ hu-mAbs (Fig. 3). The hybridoma clone 3E10 was found to secrete monomeric, soluble human k-chains (Fig. 3) with no specificity for PR8 virus or HA180-195 epitope according to ELISA and WB analysis (not shown).
Figure 3.
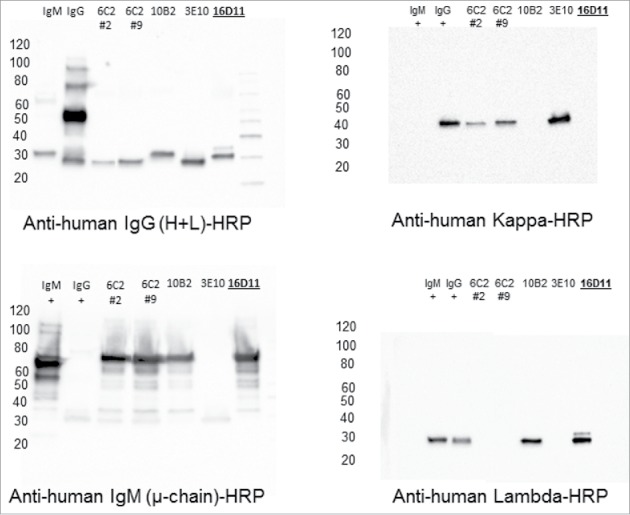
Western Blot isotyping of HA180-195 specific hu-mAbs. Shown are the HA180-195 specific hu-mAbs selected for this study as assessed by Western blot for the presence of IgG and IgM heavy and light chains. Affinity purified hu-mAbs were SDS-denatured and 2ME-reduced, applied at 5 μg/lane in 8–16% gradient gels of polyacrylamide, separated by SDS-PAGE, gels electro-transferred onto PVDF membranes, and the membranes were probed with specific antibodies for the μ, λ, κ, and λ chains followed by incubation with specie-specific secondary Abs-HRP conjugates, as indicated in each panel.
Affinity purified IgM hu-mAbs subjected to SDS-PAGE under denaturing and reducing conditions showed 2 major bands between 70–75 and 27–30 KDa corresponding to the heavy chains and respectively light chains of an IgM molecule (Fig. 4A). Both immunoelectrophoresis (Fig. 4B) and agarose electrophoresis in Titan gels (Fig. 4C) revealed the monoclonality and distinct electrophoretic mobility for each IgM hu-mAb. The molecular assembly of IgM hu-mAbs was determined by FPLC size exclusion chromatography under native conditions, which found that all selected hu-mAbs were secreted by the hybridoma cells as pentamers (Fig. 4D). Together, these assays revealed that the four analyzed anti-HA180-195 hu-mAbs were of IgM/λ monoclonal pentamers with different electrophoretic mobility. The unique electrophoretic mobility for each of IgM hu-mAbs suggested differences in their amino acid composition. To find out if this is the case, we next sequenced all four hu-mAbs. Indeed, sequencing analysis of the heavy and light chain CDRs showed several amino acid differences between 16D11, 8D12, 10B2, and 13C10 hu-mAbs (Fig. 5). Thus, the 8D12 hu-mAb showed a valine substituted for leucine in the signal peptide of heavy chain at the position 89 and in CDR1 of the light chains at position 53, as well as an alanine substituted for glycine in the signal peptide of the light chain at position 111. The 13C10 hu-mAb had a proline substituted for glycine in the frame 3 of light chains at position 97. The 16D11 hu-mAb showed the highest amino acid diversity in the CDR3 of heavy chain. The CDR3 regions of 8A4 and 25-3 isotype controls (IgM/λ hu-mAb) were also quiet diverse when compared with 16D11 hu-mAb.
Figure 4.

Structural analyses of anti-HA180-195 hu-mAbs. (A) Silver stain of 8–16% gradient SDS-PAGE gels ran under denaturing and reducing condition for four affinity purified, HA180-195 specific hu-mAbs at 1 μg/lane. (B) Immunoelectrophoresis of 16D11 hu-mAb showing the monoclonal bands of human μ heavy chain and human l light chain as compared with the human polyclonal m heavy chain and l light chain. (C) Agarose Titan gel analysis showing the monoclonality and difference in the electrophoretic mobility of HA180-195 specific hu-mAbs. (D) Histograms of FPLC analysis showing intact pentameric molecules of 16D11 hu-mAb. Arrows in each histogram indicates the earlier elution time for 16D11 IgM and control human IgM pentameric molecules than for human control IgG monomeric molecules as detected at 280 nm.
Figure 5.

Amino acid sequences of CDR3 VH and VL regions of hu-mAbs and their isotype controls. (A) CDR3 VH sequences of four HA180-195 specific IgM hu-mAbs (16D11, 10B2, 8D12, and 13C10) and two non-specific, isotype control IgM/lambda hu-mAbs (8A4 and 25-3 hu-mAbs). (B) CDR3 VL sequences of the same hu-mAbs like in panel A. Shown are the signal peptides for both the Heavy and Light chains, the flanking frame regions FR1 to FR4, the CDR3s, and 10 amino acids adjacent to constant regions CH1 and CL1. Similar amino acid residues among all hu-mAbs are highlighted in green, those with more than 80% similarity in light green, those between 60 to 80% similarity in yellow, and those with less than 60% similarity are left uncolored. Blue arrows indicate the position where amino acid differences occurred for 10B2, 8D12, and 13C10 hu-mAbs.
Nucleotide sequence of 16D11 VH and VL chains using the IMGT/V-QUEST algorithm58,59 identified the variable (V), diversity (D) and joining (J) gene segments used to assemble its heavy and light chain. The VH chain of 16D11 hu-mAb used the V gene from IGHV3-23*01 or IGHV3-23D*01 germlines, and the J gene from germline IGHJ4*02. Based on the amino acid junction analysis, the germ line IGHD2-21*02 was identified for the D gene to be in the reading frame 3 (Fig. 6A). Same analysis identified the CDR3 VL region to belong to IGLV3-1*01 germline, and the J gene to IGLJ1*01 germline (Fig. 6B). The IMGT/V-QUEST tool also identifies the framework and CDRs in the VL and VH chains by comparison with the closest genes and alleles. The length and sequence corresponding to each region of VH and VL chains are shown according to the IMNGT standardized nomenclature and IMGT unique numbering in a Collier de Perles representation59 (Figs. 6A & 6B). In addition, the homology model of 16D11 Fab containing the VL and VH chains revealed an HA180-195 binding groove built mainly by the CDR3 of both VH and VL regions (Fig. 6C). As depicted in Figure 6 panels 6D and 6E, the surface exposed binding groove to HA180-195 epitope was mainly made of sequestered non-charged and positive charged areas.
Figure 6.

Germline identification and 3D homology model for 16D11 hu-mAb. (A) Sequence alignment of the VH and VL chain of 16D11 hu-mAb and their corresponding germlines. Sequence corresponding to the CDRs are colored, and those corresponding to the V, D and J genes are indicated at the top of alignment. Protein sequence corresponding to the junction is highlighted in gray. Dots indicate sequence conservation, and dashes indicate gaps. (B) Colliers de Perles representation of the VH and VL protein chains. Residues corresponding to CDR loops are colored as in A, and square boxes represent the boundary residues between the framework and the CDR loops. Positions with red and bold letters indicate the five-conserved position of a V domain. (C) Ribbon and surface representation of the homology model of 16D11 Fab showing the VH and VL chains in dark, and respectively light grey. CDR loops are labeled and colored as in B. (D) Coulomb surface coloring of 16D11 homology model. (E) Showed are the areas with no potential charges in red and positive charges in blue.
HA cross-reactivity and binding affinity of 16D11 hu-mAb
The homology of HA180-195 epitope among 7 influenza A strains (shown in fig.1A) led to the question as to what extend 16D11 hu-mAb may cross-react with the HA proteins of these virus strains. ELISA showed that 16D11 hu-mAb bound strongly to the PR8 virus, and cross-reacted with the rHA of PR8-H1N1, WSN-H1N1, Aichi-H3N2, Hokaido-H5N3, and Vietnam-H5N1 viruses, but not of Memphis and Hong Kong viruses (Fig. 7A). The 8D12, 10B2, 13C10 hu-mAbs and the human sera (positive control) also bound to the PR8 virus and cross-reacted in ELISA with the rHA of the same 5 virus strains recognized by 16D11 hu-mAb, but not with the rHA of Memphis and Hong Kong viruses (not shown).
Figure 7.

HA cross-reactivity and binding affinity of 16D11 hu-mAb. (A) Binding of 16D11 hu-mAb to rHA proteins from influenza virus heterotypes in ELISA. Shown are duplicate rHAs-coated wells and+/- SD for 99% confidence. Signal-to-noise background of the anti-human IgM-HRP secondary antibody (0D 450 nm = 0.045 average) has been subtracted from each sample. PR8 virus-coated wells and repository human sera (HRS) were used as controls. (B) SPR comparative binding of 16D11 hu-mAb at 200 nanoMoles to rHA proteins of PR8/a/34 virus (black), Hokkaido virus (grey) and Memphis virus (red). 16D11 injection point and association and dissociation phases are indicated. Representative Sensograms of different concentrations of 16D11 hu-mAbs across biosensor surfaces coupled to rHA protein of PR8 virus (C) and Hokkaido virus (D) at 30 ml/min and 25°C. Sensograms were analyzed using a simultaneous fit algorithm (BIAevaluation 3.1) to calculate the kinetic parameters and binding affinities (as shown in Table 1). SPR sensograms for each response are shown as gray lines whilst fit analyses are shown as black lines.
We next measured the affinity and kinetics of 16D11 interaction with rHA of PR8 and Hokkaido viruses (to which 16D11 and the other hu-mAbs showed the strongest binding in ELISA, Fig. 7A), and the affinity and kinetics of 16D11 interaction with rHA of Memphis (to which none of the hu-mAbs antibody bound in ELISA). Control IgM/λ isotypes for the kinetics and binding affinities were 25-3 and 8A4 hu-mAbs generated in DRAGA mice and lacking specificity for the rHA of all 7 investigated influenza virus strains (not shown). The affinity and kinetics parameters were measured by Surface Plasmon Resonance (SPR) in a Biacore3000 instrument (see materials and methods section). We first compared the binding of 16D11 at 200 nM to the rHA of PR8, Hokkaido, and Memphis viruses. SPR revealed a direct and strong interaction of 16D11 hu-mAb with the rHA of PR8 and Hokkaido viruses, but not of Memphis virus (Fig. 7B). Thus, SPR sensograms confirmed the ELSA results showing the lack of 16D11 cross-reactivity with the rHA of Memphis virus. The kinetics and affinity interactions of 16D11 hu-mAb with the rHA of PR8 (Fig. 7C) and Hokkaido (Fig. 7D) viruses showed similar ka, kd, and KD values. Both interactions were characterized by a fast association constant (ka) of 22.4-25.8 × 10+3 M-1 s-1, and slow dissociation (kd) constant of 4.39-3.32 × 10−3 s-1 with affinity binding in the nano molar range (199 and 130 nM, respectively). Based on the dissociation rate, the half-life time for 16D11 hu-mAb/rHA-PR8 complex was 2.6 minutes and 3.5 minutes for 16D11hu-mAb/rHA-Hokkaido complex (Table 1). These data revealed similar kinetics and binding affinities for the interaction of 16D11 hu-mAb with rHA of Hokkaido and PR8 viruses, and confirmed the ELISA data showing its lack of cross-reactivity with the rHA of Memphis virus, and its cross-reactivity with the rHA of 5 influenza viruses.
Table 1.
Kinetic analysis of 16D11 hu-mAb interaction with rHAs from PR8, Hokkaido and Memphis influenza viruses.
| Ka (mean ± SD) M−1 s−1 | Kd (mean ± SD) s−1 | KD nM | t1/2 min | |
|---|---|---|---|---|
| PR8/A/34 | 199 | 2.62 | ||
| Hokaido | 130 | 3.46 | ||
| Memphis | n.b.d* | n.b.d | n.b.d | N/A |
SPR sensograms for the interaction of 16D11 with rHA proteins as shown in Figure 7. The kinetic data were fit using a 1:1 Langmuir binding model for the estimation of the association (ka) and dissociation (kd) rates and affinity (KD=ka/kd). No binding was detected (*nbd.) for the interaction of 16D11 with rHA of Memphis virus. The complex half-life was calculated as t1/2 = Ln/kd.
Neutralization of PR8/A/34 influenza virus by HA180-195-specific hu-mAbs
We next measured the neutralization capacity of HA180-195-specific hu-mAbs against the PR8 virus by hemagglutination inhibition assay (HIA). The results depicted from Table 2 show that 16D11 hu-mAb has the highest neutralization capacity at 12.5 μg/ml, as compared with 10B2 hu-mAb at 50 μg/ml, and 6C2 (clone #2) and 13C10 hu-mAbs at 100 μg/ml. The 25-3 hu-mAb isotype control show no neutralization activity against PR8A/34 virus up to 300 μg/ml. The positive control for this assay that neutralized PR8/A/4 virus at 1/160 dilution was a human repository sera (HRS) from an individual vaccinated with seasonal CIV.
Table 2.
HIA for PR8 virus neutralization by anti-HA180-195 hu-mAbs.
| anti-HA180-195 hu-mAbs |
|||||
|---|---|---|---|---|---|
| #16D11 | #6C2 (#2) | #13C10 | #10B2 | #25-3 (hu-lgM control) | human sera (dilution factor) |
| 12.5 | 100 | 100 | 50 | >300 | 1/160 |
First row indicates the denomination for hu-mAbs used in HIA; Second row indicates the amount of hu-mAbs required to induce virus neutralization (RBC hemagglutination) (μg/ml) in HIA.
Testing the murine xenogeneic response to 6D11 hu-mAb.
Xenogeneic responses to murine and “partially” humanized antibodies can significantly lower their therapeutic efficacy.26–32 Herein, we questioned whether a xenogeneic system like a wild mouse model for influenza infection would be the appropriate approach to test the potential therapeutic effect of 16D11 hu-mAb. We first measured the murine antibody response to 16D11 hu-mAb vs. its span-life in the blood circulation of BALB/c mice. For this, BALB/c mice were injected intraperitoneally (i.p) once or twice with 600 μg of 16D11 hu-mAb and 9 days later the murine primary antibody response to 16D11 hu-mAb was measured by ELISA. Data depicted in Fig. 1SA revealed a strong and early murine IgM anti-16D11 hu-mAb response, which may explain the 99.7% loss of 16D11 in the BALB/c blood circulation. Some 1.8 μg and respectively 2 μg of remaining 16D11 hu-mAb were detected by ELISA in the sera of BALB/c mice, 9 days post injection (Fig. 1SB). In contrast, the same experiment carried out in DRAGA mice showed that 12% of 16D11 hu-mAb was still present in the blood circulation of DRAGA mice, 9 days after i.p. injection of 600 μg (not shown). This indicated that the span-life of 16D11 hu-mAb in BALB/c mice was 40 times shorter than in DRAGA mice. To find out whether a significant loss of 16D11 hu-mAb in the BABL/c blood circulation may interfere with its potential therapeutic, PR8 lethally-infected BALB/c mice were i.p. injected with 600 μg of 16D11 hu-mAb 2 hours prior infection, and their body-weights was monitored every other 3rd day. Data in Figure 2S show no significant difference in the rate of body-weight loss between infected vs. infected and treated BALB/c mice during a 20 day period of follow up. Together, the results showing a strong murine antibody response against 16D11 hu-mAb vs. its significantly short span-life in BALB/c blood circulation, as well as the lack of therapeutic effect of this antibody in PR8-infected BALB/c mice strongly suggest that a xenogeneic murine response may have interfered with its potential anti-flu therapeutic effect.
Modeling the PR8 virus infection in DRAGA mouse
To avoid a possible murine xenogeneic interference with the in vivo effect of 16D11 hu-mAb, we thought that DRAGA mouse having the murine immune system replaced with a functional human immune system would be an appropriate model for testing its anti-flu therapeutic effect. For this, we first investigated whether DRAGA mice are suitable for PR8 virus infection. The results in Figure 8A show that DRAGA mice (n = 6) inoculated intranasally (i.n.) with a sub-lethal dose (LD25) of PR8 virus did not lose weight post-infection except for one mouse with a 5% temporary loss while the HA viral RNA was detected in the lungs. The HA viral RNA was totally cleared between day 7 and 14 post-infection according to RT-qPCR (Fig. 8B), and afterwards mice were considered free of infection. Furthermore, just like the BALB/c mice, DRAGA mice were able to sustain a PR8 i.n. lethal infection for 2 to 3 weeks (Fig. 8C & 8D). These experiments clearly demonstrated that DRAGA mouse is a suitable model for influenza infection, and thereby appropriate to assess the in vivo anti-flu effect of 16D11 hu-mAb.
Figure 8.

Body-weights of PR8-infected DRAGA mice vs. titers of viral HA expression in the lungs. (A) Sub-lethal infection of DRAGA mice with PR8 virus. Body-weights of PR8 sub-lethally infected DRAGA mice (n = 6) were monitored every other 3rd or 4th day post-infection (dpi). (B) Agarose gel electrophoresis of the HA amplicons obtained by RT-qPCR and corresponding CT values in the lungs of DRAGA mice from panel A (sub-lethal infection), as measured at 7, 14, and 21 dpi. Shown are duplicate samples from a representative naïve (not infected) DRAGA mouse (lanes 2 and 3), and sublethally-infected 7 dpi (4–5) and 21 dpi (lanes 7–8 and 9–10). Arrow indicates the size (160 bp) amplicon of HA of PR8 virus (positive control, lane 11). Lane 1, molecular markers; Lane 12 distilled water (primers control). (C) Body-weights of PR8 lethally-infected (LD100) BALB/c mice and naïve BALB/c mice (n = 4 mice/group) monitored every other 3rd or 4th day post-infection (dpi). Lower panel, the agarose gel electrophoresis of HA amplicons in the lungs measured by RT-qPCR, and CT corresponding values in duplicate samples of lethally-infected BALB/c mice (lanes 2 to 9) at 18 dpi; Lane 1, molecular markers; Lane 10, the 160 bp amplicon of HA of PR8 virus (positive control); Lane 11, a representative naïve (non-infected) BALB/c mouse. (D) Body-weights of PR8 lethally-infected (LD100) DRAGA mice. Mice were monitored every other 3rd or 4th day. Lower panel, agarose gel electrophoresis of HA amplicons in the lungs measured by RT-qPCR, and CT corresponding values in duplicate samples. Lane 1, molecular markers; lanes 2-3, naïve DRAGA mouse; Lanes 4–5 and 6–7, two representative DRAGA mice lethally infected, as measured at 14 dpi and 18 dpi; Lanes 8–9, 160 bp amplicon of HA of PR8 virus (positive control).
Testing the anti-flu effect of 16D11 hu-mAb in DRAGA mice.
Since 16D11 hu-mAb showed the highest in vitro neutralization capacity against PR8A/34 virus among all four anti-HA180-195 hu-mAbs, we tested its in vivo effect against lethal infection with PR8 virus. Having set the DRAGA mouse model for PR8/A/34 virus infection, we injected a single i.p. dose of 16D11 hu-mAb (600 μg/mouse) in these mice at the time of i.n. lethal infection. Control groups were: (i) non-infected/untreated mice (control naive group), (ii) lethally-infected/untreated mice (control infection group), and (iii) lethally-infected and treated with isotype control 25-3 hu-mAb. All DRAGA mice showed 15.1 to 35.6% CD19+ B cells and 14.2 to 22.9% CD3+ T cells in blood circulation on day 0 of this experiment. DRAGA mice in the control infection group started to rapidly lose weight by day 2 post-infection and survived 2 to 3 weeks post-lethal infection (Fig. 9A & 9B). Lung analysis at the time of death in mice from control infection group showed massive damage to the lungs with heavy lymphocyte and blood infiltration, and distorted alveolar architecture (Fig. 9C lower panels). In contrast, all treated DRAGA mice started to lose weight 10 days post-infection and came close to the highest body-weight loss 2 weeks later than those in the control infection group. Interesting enough, mice with the lowest number of B-cells (15.1% to 17.5% CD19+ cells) accounted for the shortest survival, whereas those with higher number of B cells (28.4-35.6% CD19+ cells) showed longer survival in both the infection control group and infection/treated group. In contrast, no correlation was found between the survival rates and number of CD3+ T-cells in either group of mice. The most resilient DRAGA mouse to the infection in the infected/treated group showed diminished damage to the lungs, and fewer areas with distorted alveolar architecture and inter-alveolar infiltration with lymphocytes and blood, when compared with the control infection group (Fig. 9C, middle panels vs. lower panels). The isotype control group (n = 3) that was lethally-infected with PR8 virus and treated i.p. with 600 μg/mouse of 25-3 isotope control hu-mAb at the time of infection showed a similar pattern of body-weight loss and rate of survival to that of control infection group (not shown).
Figure 9.

The effect of 16D11 hu-mAb in PR8 lethally-infected DRAGA mice. (A) Body-weights of naïve (non-infected) DRAGA mice and PR8 lethally-infected (LD100) DRAGA mice with and without 16D11 hu-mAb treatment. A single i.p. injection of 600 μg per mouse (n = 3-4 mice /group) was administered at the time of infection. Mice were monitored every other 3rd or 4th dpi. Brown star indicates the most resilient mouse in the treatment group. (B) Survival rates for groups of DRAGA mice described in panel A. Of note, the most resilient DRAGA mouse in the PR8-infected/treated group showed 40% less loss in body-weight at day 43 post-infection when was sacrificed for analysis, which is a significantly when compared with the average loss in body-weight for the control infection group (p = 0.026 according to Mantel-Cox test, and p = 0.016 according to pairwise curves comparisons of Gehan-Breslow-Wilcoxon test). (C) Lung analysis of DRAGA mice described in panel A. Upper panels, lungs and Hematoxilin-Eosin (HE) staining of lung sections from a representative naïve DRAGA mouse; Middle panels, lungs and HE staining of lung sections from the most resilient DRAGA mouse to PR8 lethal infection upon 16D11 hu-mAb treatment as analyzed 40 days post-infection. Shown is a mild lung damage in the lower lobe of the right lung (diffuse grayish area) with slightly distorted alveolar architecture and scattered lymphocyte infiltrates (yellow arrow); Lower panels, lungs and HE staining of lung sections from a representative PR8-lethally DRAGA mouse left untreated, and analyzed 20 days post-infection. Shown is massive pneumonia in both lungs (dark reddish areas) with heavily distorted alveolar architecture, and interstitial and intra-alveolar lymphocyte infiltration.
Together, these in vivo experiments demonstrated that DRAGA mouse is the first humanized mouse model for PR8/A/34 influenza virus infection, and that a single dose of 16D11 hu-mAb targeting a single HA epitope significantly delayed (by more than 2 weeks) the lethality of PR8/A/34 influenza virus infection.
Discussion
This study addressed several questions about the bio applicability of a new humanized mouse strain (DRAGA: HLA-A2. HLA-DR4. Rag1 KO. IL-2Rgc KO. NOD). We first questioned whether DRAGA mouse can be used to generate “fully” human monoclonal antibodies (hu-mAbs) targeting a homologous HA epitope among different influenza type A virus heterotypes (WSN-H1N1, Adachi-H3N2, Aichi-H3N2, Memphic-H3N2, VN1194-H5N1, Hokkaido 67-H5N3, and W213-H9N2). The amino acid sequence (180WGIHHPPNSKEQQ NLY195) of the human B-cell epitope used to raise cross-reactive hu-mAbs in DRAGA mice was chosen from the HA of PR8/A/34 virus and is part of the immunogenic Sb site of HA1 chain.52
Immunization of DRAGA mice with a KLH-HA180-195 conjugate induced IgG and IgM hu-mAbs specific for PR8/A/34 influenza virus, and the hybridization process using K6/H6 human IgM myeloma cells rendered 4 stable, highly IgM/λ-secreting hybriboma cells with specificity for HA180-195 epitope. Human anti-flu IgM antibodies can protect against influenza infection, as recent work demonstrated that human IgM+ memory B cells induced upon influenza vaccination secreted cross-protective antibodies to human and avian viruses type A (H5N1 and H1N1 heterotypes)13,40 and to the Sb antigenic site of HA protein.14 Our IgM hu-mAbs specific for HA180-195 epitope showed structural integrity and were secreted by the hybriboma cells as pentameric molecules.
The antigen binding site of antibodies resides mostly in the CDR3 pocket of the heavy chain, and the nature of antigens can dictate unique CDR3 amino acid compositions aimed at establishing optimal interaction with the antigen.60,61 Interestingly enough, although the same HA180-195 epitope was used in this study for immunization, all selected hu-mAbs recognized this epitope with 16D11 hu-mAb showing the highest amino acid diversity in the CDR3 VH region. The high CDR3 structural diversity of 16D11 hu-mAb was attributed to immunization-induced B-cell hypermutations in DRAGA mice. Similar observations were reported by Tsibane et al.41,62,63 showing that two different human monoclonal antibodies (1F1 and HC63) isolated from influenza-infected individuals and recognizing the same HA epitope shared by 1918, 1943, and 1977 influenza A-H1N1 heterotypes, displayed diversity in amino acid diversity with respect with the antigen binding site in CDR3 VH region. The authors showed that 1F1 and HC63 hu-mAbs used similar germline V, D, and J regions to assemble the CDR3 region of the heavy chain. On the other hand, two human monoclonal antibodies using similar V, D, and J gene segments for the heavy chain but having different light chains, displayed different virus neutralization capacity, as one of them cross-neutralized three different H1N1 influenza heterotypes and the other one did not.41,64 These data, together with our SPR sensograms strongly suggest that amino acid composition in the HA receptor binding pocket of CDR3 VH region may affect not only the binding strength, but also the unique 3D folding of the antibody-HA binding site.
Among hu-mAbs specific for HA180-195 epitope generated in this study, the 16D11 hu-mAb showed the highest neutralization capacity in vitro against PR8/A/34 influenza virus, and cross-reacted with the HA viral protein from PR8, WSN, Aichi, Hokkaido and Vietnam influenza heterotypes. SPR sensograms revealed that 16D11 hu-mAb has relative high binding affinity to the HA protein of PR8 and Hokkaido viruses, and it forms stable and long half-life 16D11-rHA complexes in solution. In contrast, the failure of 16D11 hu-mAb to recognize the HA proteins from Memphis and Hong Kong viruses was attributed to low binding affinity constants. A 100% homology of HA180-195 epitope at the positions 181–186 in all studied virus strains except the Memphis virus, suggested that these residues are critical for the HA180-195 helicoidally and/or interaction with our HA180-195-specific hu-mAbs. In addition, the HA180-195 epitope of Memphis and Hong Kong viruses lacks a conserved histidine at position 183 and a glutamic acid at position 191, and these substitutions could also be critical for maintaining the epitope helicoid structure required for the interaction with our HA180-195-specific hu-mAbs.
Xenogeneic antibodies can significantly lower and even abolish an antibody-based therapy.26–31 This was likely the case of our 16D11 hu-mAb, which showed a very short span-life and anti-flu effect in BALB/c mice. In contrast, a considerable 40 times longer span-life of 16D11 hu-mAb was detected in DRAGA mice. Based on previous reports and our data, one may consider that “fully’ human antibodies lacking xenogeneic reactions would be more efficient therapeutics when administered repeatedly in humans, though a number of “partially” humanized antibodies were quite successful in clinical trials for infectious disease, cancer, and autoimmune diseases. To rule out any possible murine xenogeneic interference with the in vivo effect of our 16D11 hu-mAb, and considering that DRAGA mice (lacking the murine immune system and expressing a human immune system) are suitable for sub-lethal and lethal infections with PR8A/34 virus, we used these mice to assess the in vivo anti-flu effect of 16D11 hu-mAb. PR8 lethally-infected DRAGA mice and treated with a single dose of 16D11 hu-mAb showed a significant 2-week delay in body-weight loss and survival as compared with those infected and left untreated. Treated mice also showed less lung damage, which suggests a delayed viral replication in the lungs. It remains to be further investigated whether repeated injections of 16D11 hu-mAb with different time spacing between treatments, or a cocktail of broadly cross-neutralizing hu-mAbs targeting several conserved HA epitopes may provide better therapeutic efficacy than a single injection of a hu-mAb targeting a single conserved HA epitope. Cross-neutralization provided by a cocktail of antibodies targeting multiple HA viral epitopes may overcome the seasonal mutations.
In summary, this work demonstrated first, that DRAGA mouse is a reliable tool to generate heterotype cross-reactive, human anti-influenza monoclonal antibodies. Secondly, our results showed that DRAGA mouse is the first humanized animal model for influenza infection. Since DRAGA mouse can sustain the malaria cycle of Plasmodium falciparum,51 as well as HIV65 and Dengue infections (manuscripts in preparation), one may consider that this mouse can be used to establish humanized mouse models for various infectious diseases including those of potential biothreat or resistant to antibiotic therapy. Thirdly, our results demonstrated that DRAGA mice can be used to assess the therapeutic effect of heterotype cross-reactive, human anti-influenza monoclonal antibodies.
Materials and methods
Mice
BALB/c female mice of 4 months of age were purchased from Jackson Labs. Humanized mice like DRAGA mice (HLA-A2.1. HLA-DR-0401. Rag1 KO. IL-2Rgc KO. NOD) are generated and reconstituted with human immune system in our laboratories.50 The humanized mice (DRAGA) were monitored twice a month by FACS for human T and B cell reconstitution and by ELISA kits for the amount of antibodies in sera, and used in experiments at the age of 5–6 months when full reconstitution was achieved. All animal procedures were conducted under IACUC protocols approved by USUHS (ID#MED-14-902) and WRAIR/ NMRC (ID#16-IDD-43) in compliance with the Animal Welfare Act and in accordance with the principles set forth in the “Guide for the Care and Use of Laboratory Animals,” Institute of Laboratory Animals Resources, National Research Council, National Academy Press, 1996.
Generation of HA180-195-specific hu-mAbs
We have recently reported the generation of a new humanized mouse strain (DRAGA mice, NOD/Rag1 KO/IL-2Rgc KO/HLA-DR*0401+, HLA-A2.1+) that can reconstitute functional human T and B cells upon infusion of CD34+ human hematopoietic stem cells (HSC).42 Briefly, HLA-DRB1*0401+ umbilical cord bloods (UCB) purchased from AllCells and Promocell were enriched for CD34+ stem cells to more than 60% when using EasySep, Human Progenitor Cell Enrichment kit (StemCell Technologies, Vancouver, BC). Three month-old female/male, irradiated DRAGA mice at 350 rads were injected intravenously with 8 × 105 CD34+-enriched stem cells, and the extent of human T and B cells reconstitution was monitored weekly by FACS using human CD3 Ab and respectively, human CD19 Ab. In parallel, mice were monitored for the levels of human IgM and IgG in sera by ELISA kits (Bethyl Laboratories) according to the manufacturer's instructions. Three HSC- reconstituted DRAGA mice (six month of age) showing between 1.0 and 1.5 mg/ml of human serum IgM and IgG were then immunized intraperitoneally (i.p.) with 100 μg/mouse of KLH-HA180-195 conjugate in complete Freund's adjuvant CFA) and boosted two weeks later with 100 μg/mouse of the same conjugate in incomplete Freund's adjuvant. The levels of HA180-195-specific and PR8-specific IgG and IgM hu-mAbs in sera were monitored weekly by ELISA using BSA-HA180-195 coated plates (100 μg/ml) and PR8 virus (Charles Rivers) coated plates (50 μg/ml), and bound antibodies were revealed with goat anti-human IgM-HRP (Thermo Fisher Scientific) and respectively, goat anti-human IgG-HRP (Bethyl Laboratories) on a TMB (3,3', 5,5' tetramethylbenzidine, BD Biosciences) according to the manufacturer's instructions. To generate human hybridoma cells, spleen cells from immunized DRAGA mice were fused with K6H6/B5 myeloma cell partner (ATCC® CRL-1823™) using ClonaCell-HY Hybridoma kit (cat #03800, StemCell Technologies, Inc.) according to the manufacturer's instructions. Stable hypoxanthine-aminopterin-thymidine (HAT, Sigma Aldrich) hybrids were then cloned, and positive clones were re-cloned at 0.1 cell/well in 96-well plates in HAT-supplemented DMEM with 20% FCS. High and stable producers were confirmed by ELISA in plates coated with BSA-HA180-195 conjugate or PR8 virus. Secreted IgM hu-mAbs specific for HA180-195 were purified from the cell culture supernatants by affinity chromatography on Sepharose CL-4B (GE Healthcare Life Sciences) coupled to goat anti-human IgM Abs (Helena Labs), and dialyzed/concentrated on Amicon filter units (#UFC710008, Millipore) with 100 kDa molecular weight cut-off.
SDS-Page
Samples containing 1 μg of affinity-purified hu-mAbs were denatured at 100°C for 5 min in Laemmli sample buffer (Bio-Rad) containing 5% 2-mercaptoethanol, and subjected 1 h to 100 Volts electrophoresis in 4–20% Mini-Protean TGX gels (Bio-Rad). Gels were silver stained and analyzed for hu-mAbs purity and the molecular weight of the hu-mAbs heavy and light chains.
Size exclusion chromatography
To determine whether the native hu-mAb molecules were properly assembled, we measured their molecular weight by a FPLC Superose 12 column coupled to an AKTA prime plus instrument (Amersham Biosciences, GE, Co). The samples, molecular markers, and column were equilibrated in PBS and samples were run at 1 ml/min flow-rate.
Immunoelectrophoresis
Isotyping and monoclonality of anti-HA hu-mAbs was carried out by visualization of hu-mAbs by immunoelectrophoresis (IEP kits, Helena Labs.). Some 5 μg of purified hu-mAbs in 5 μL barbital buffer were electrophoresed on pre-casted 1% agarose gels, and stained according to the manufacturer's instructions.
Titan-gel electrophoresis
Monoclonality of the anti-influenza hu-mAbs was also confirmed by protein electrophoresis in Titan agarose gels (Protein Titan gels, Helena Labs) according to the manufacturer's instructions. Briefly, 15 μg of purified hu-mAbs in 5 μL barbital buffer were applied on the gel and electrophoresed for 60 min at 150 Volts. Gels were stained with Amido Black for 30 min at room temperature, dried under hot air blower, and de-stained as per manufacturer's instructions.
Enzyme-Linked Immunosorbent Assays (ELISA)
ELISA was used to select positive hybridoma clones secreting anti-HA hu-mAbs and to isotype these antibodies, to determine the cross-reactivity of hu-mAbs to rHAs from various influenza virus heterotypes, to measure the BALB/c mouse Ab response to anti-HA hu-mAb treatment and to PR8 virus infection, and to determine the span-life of 16D11 anti-HA hu-mAb in the blood circulation of BALB/c mice; (i) Selection of stable hybridoma clones secreting anti-HA hu-mAbs in the cell culture supernatant and isotyping the affinity-purified hu-mAbs by these clones was carried out by semi-quantitative ELISA kits (Bethyl Laboratories) according to the manufacturer's instructions; (ii) Cross-reactivity of hu-mAbs to rHAs from various influenza virus heterotypes was measured in 96-well plates were coated overnight at 4°C with 20 μg/ml of recombinant HA proteins from A/Puerto Rico/8/1934 (Cat# 11684-V08H), A/WSN/1933 (Cat# 11692-V08H), A/Aichi/2/1968 (Cat# 11707-V08H), A/Memphis/1/1968 (Cat# 40101-V08H1), A/Vietnam/1194/2004 (Cat# 11062-V08H1), A/Hokkaido/167/2007 (Cat# 11696-V08H), and A/Hong Kong/2009 (Cat# 40174-V08H1) (Sino Biological) in 0.05M Bicarbonate buffer at pH 9.6. Coated plates were blocked with 5% BSA in 1X PBS overnight at 4°C, and hu-mAbs (5 μg /ml) were added to the plate and incubated for 2 h at room temperature. Plates were washed 3 times with 1X PBS containing 0.05% Tween, and secondary goat anti-human m-chain-HRP conjugate (ThermoFisher Scientific) was added at 1:2,000 dilution for 1 h at room temperature. Plates were washed 3 times with 1X PBST, TMB substrate (BD Biosciences) was added to the wells for 10 minutes followed by stop solution (0.18M H2SO4), and the optical density (OD) was read at 450 nm in an ELISA reader (Molecular Devices); (iii) To measure the BALB/c mouse xenogeneic response to anti-HA hu-mAbs, mice were injected i.p. with 250 μg of 16D11 hu-mAb on days 1 and 2, and 7 days later harvested sera at 1:100 dilution was incubated overnight at 4° C in 96-well plates coated with 16D11 hu-mAb (5 μg/well). Plate was blocked with 5% BSA in 1X PBS for 2 h at room temperature, washed 3 times with 1X PBS containing 0.05% Tween, and a secondary goat anti-IgM-HRP conjugate (Jackson Labs.) was added at 1:20,000 dilution for 2 h at room temperature. Plate was washed 3 times with 1X PBST, and TMB substrate (BD Biosciences) was added to the wells for 10 minutes followed by stop solution (0.18M H2SO4). Optical density (OD) was read at 450 nm in an ELISA reader (Molecular Devices); (iv) To determine the span-life of anti-HA hu-mAbs in the blood circulation of BALB/c mice, sera from the same BALB/c mice treated with 16D11 hu-mAb described above was harvested and 7 days post-injection and the amount of remaining 16D11 hu-mAb was measured by semi-quantitative ELISA kits (Bethyl Laboratories) according to the manufacturer's instructions.
Western Blot analysis
Isotype confirmation of hu-mAbs by ELISA kits (Bethyl Laboratories) was carried out by Western blot analyses. Briefly, hu-mAbs separated in SDS-PAGE gels were transferred to nitrocellulose (NC) membranes in semidry conditions using an semi-dry iBlot electro-transfer equipment (ThermoFisher Scientific), and blocked overnight in 5% powdered milk plus 3% BSA in 1X PBST. Triplicate NC membranes were then probed for 2 h at room temperature with goat anti-human m-chain-HRP conjugate (ThermoFisher Scientific), or goat anti-human l-chain-HRP conjugate (Southern Biotech.), or goat anti-human k-chain-HRP conjugate (Southern Biotech.) followed by 3 washes in 1X PBST and exposed 10 min to SuperSignal West Pico Chemiluminescent Substrate (ThermoFisher Scientific).
Rapid Amplification of cDNA Ends (RACE) for hu-mAbs sequencing
RACE procedure was used for amplification of heavy and light chain genes. Total RNA was extracted from hybridoma cells using RNeasy Mini Kit (Qiagen), and cDNA synthesized using our designed primers, IgM-Mu-R primer for heavy chain (5’-AACGGCCACGCTGCT CGTATC 3’), and Lambda-new-R primer for light chain (5’-TATGAACATTCTGTAGGGGC-3’). After the first strand of cDNA synthesis, the original mRNA template was removed by treatment with the RNAse mix. Unincorporated dNTPs, primers, and other proteins were separated from cDNA using the S.N.A.P. column. Terminal deoxynucleotidyl transferase (TdT) was used to add homopolymeric tails to the 3’ ends of cDNA, and tailed cDNA was then amplified by PCR using 5’ Abridged Anchor Primer (5’-GGCCACGCGTCGACTAGTACGGG IIGGGIIGGGIIG-3’) and a primer specific to IgM heavy chain (5’GGAGACGAGGGGGAA AAG-3’) or light chain primers (5’-TGGCTTGAAGCTCCTCAGAG-3’). PCR amplified products were run in 1% LMP Agarose gel (ThermoFisher Scientific) using cyan/yellow tracker dye (ThermoFisher Scientific). Bands corresponding to the molecular weights of expected cDNA products were excised from the gel and extracted using QIAquick gel extraction kit (Qiagen). Briefly, gel slices were weighed and three volumes of buffer QG were added to one volume of gel, and then incubated at 50°C for 5 min. Samples were vortexed every 2–3 min to help dissolve the gel. One gel volume of isopropanol was added to the sample and mixed. Sample was applied to QIAquick column and centrifuged for 1 min at 13,000 rpm. Flow through was discarded, 500ul of Buffer QG was added to the QIAquick column and then centrifuged for 1 min at 13000 rpm. To wash column, 750ul of buffer PE was added to the QIAquick column, and then centrifuged for 1 minute. Flow through was discarded, and QIAquick column placed into a clean 1.5 ml microcentrifuge tube. To elute DNA, 50ul of DEPC-treated water was added to the column, and allow to incubate for 4 minutes, and then centrifuged. DNA sample for IgM heavy chain was sequenced at USUHS's Biomedical Instrumentation Center facility using primer (5’-GGAGACGAGGGGGAAAAG-3’), and for light lambda chain using primer (5’-TGGCTTGAAGCTCCTCAGAG-3’). Sequencing results were verified using forward primers for the IgM heavy chain (5’-ATGGAGTTTGGGCTGAGCTGG-3’), and light chain (5’-TGG CATGGATCCCTCTCTTC-3’).
Reverse Transcription-quantitative Polymerase Chain Reaction (RT-qPCR)
As a positive control for the expression of PR8 HA protein in the lungs, we used RNA extracted from live A/PR/8/34 (H1N1) (Charles River). Briefly, 200ug PR/8 virus was homogenized in RLT buffer using a syringe and a needle. Homogenized sample was used to purify total RNA using RNEasy Mini Kit (Qiagen) following the manufacture's protocol, and then quantitated by a NanoDrop-8000 instrument (ThermoFisher). Serial dilution of viral RNA from influenza virus was used as positive controls to establish limit of detection in gel in RT-qPCR.
To estimate the rate of virus clearance in PR8-infected and treated BALB/c with 16D11 hu-mAb, lungs from individual mice were sectioned, snap frozen, and stored in liquid nitrogen at −196°C till used. Some 30 mg snap-frozen lung tissue was lysed using lysing matrix D and FastPrep-24 instrument (MP Biomedicals) in RLT buffer as follows: 5 cycles at speed of 6 m/s with 5 min rest period in between cycles. Homogenized samples were spun at 14,000 rpm for 10 min at 4°C. Total lung RNA of individual mice was extracted using RNeasy Mini Kit (Qiagen) following the manufacture's protocol, and then quantitated by a NanoDrop-8000 instrument (Thermo Fisher). 2.5 ug total RNA of each mouse sample was used for reverse transcription with SuperScript™ II Reverse Transcriptase (Fisher; Cat: 18064014). Quantitative PCR (Q-PCR) was performed using an ABI 7500 Real-Time PCR System and software SDS v1.4. Amplification of hemagglutinin (HA) cDNA of influenza virus Puerto Rico/8/A/34 (PR8) was carried out using our designed forward primer 5'- GACACTGTTGACACAGTACTC-3' and reverse primer 5'-AGAGCCATCCGGCGATGTTAC-3. Thermocycling parameters used were: 10 min at 95°C, 30 sec at 95°C followed by 1 min at 60°C. The threshold cycle (CT) is proportional to the number of target copies present in the sample, and is defined as the PCR cycle in which the gain in fluorescence due to accumulating amplicon products exceeds 10 standard deviations of the mean baseline fluorescence. We have used CT data taken from cycles 10 to 28 with CT values higher than 28 considered below the sensitivity limit.
GAPDH was included as an internal control to make sure that a similar amount of starting PCR products were equally loaded in each agarose well. Amplification of cDNA GAPDH internal control from lung of individual corresponding mice was carried out using commercially available forward and reverse primers (Qiagen, Cat: QT01658692). Q-PCR reactions were performed in a total volume of 20 ul in SYBR Green Master Mix (ABI & Thermo Fisher; REF: 4309155) containing forward and reverse primers at 1.25 uM, respectively. The normalized signal level was calculated based on the ratio to the corresponding GAPDH signal. All RT-qPCR samples were electrophoresed at the same starting RNA concentration (before retro-transcription) on a 2% agarose gel containing 0.2% ethydium bromide.
To measure the level of AID expression in naïve and PR8-infected DRAGA mice, 5 ug total splenic RNA from individual mice was used for reverse transcription with SuperScript™ II Reverse Transcriptase using the same RT conditions described above for the HA of PR8 virus, and 45 cycles of PCR amplification. Our designed primers for human AID (forward: GGTTATCTTCGCAATAAGAAC, and reverse: TCGGGCTCAGCCTTGCGGTCC) revealed a 232 base pairs (bp) amplicon in 2% agarose gel electrophoresis.
3D Structural model of 16D11 hu-mAb
A structural model of 16D11 hu-mAb was generated by using the Rosetta Antibody 3.0 software.66 The best templates for the VL and VH scaffold identified by Rosetta based of sequence homology were PDB ID 3mlr for the VL chain and PDB ID 3kdm for the VH chain. “De novo” modeling was requested for modeling the hyper variable CDRH3 loop. The model with the best score was selected from the ten models generated by Rosetta antibody 3.0 software. N-terminal and one b-strand of the VL model generated by Rosseta Antibody software was miss folded and iTasser63 was used to model the VL chain using the structure PDB ID 4aiz as a template. The VL model generated was superposed over the Fab model originally generated by Rosetta Antibody.
PROCHECK, a program to check the stereochemical quality of protein structures67 was use to assess the quality of the 16D11 model. From the 189 non-glycine and non-proline residues contained in the VL-VH model, 81% are in the most favorable region of the Ramachandran plot. PDB files were visualized with UCSF Chimera software.68 Chimera was also used for Coulomb surface coloring representation and to generate the 16D11 structure figures. Arrows to the encircled brown areas indicate the architecture of antibody combining site made between the CDR3 regions of VH and VL chains.
Surface Plasmon Resonance (SPR) analysis
SPR was used to measure the binding kinetics of hu-mAbs to HA protein of PR8/A/34 virus using a Biacore 3000 instrument (GE Healthcare). Briefly, rHA protein from several influenza A viral strains were coupled via amine-coupling procedure to the flow cells of CM5 sensor chip until levels of 2000, 1500 and 1000 resonance units were reached. Hu-mAbs at concentrations ranging between 200 to 1.25 nM diluted in 0.01 M HEPES buffer, pH 7.4, 0.15 M NaCl, 3mM EDTA, and 0.005% (v/v) Surfactant P20) (HBS-EP) were injected separately into the flow cells at 30 mL/min and 25°C during the 2 min association phase using HBS-EP as running buffer. The dissociation phase, initiated by passage of HBS-EP buffer was carried out over a period of 4 min. Collected sensograms were aligned and fitted to a 1:1 Langmuir binding model using BIAevaluation software v4.1.1 to calculate the affinity and kinetics (KD, ka and kd) constants between the 16D11 hu-mAb and rHAs from several influenza virus heterotypes. The kinetic constants were determined from fitting containing 6 concentration sensograms. Rmax values for the 16D11-Hokkaido and 16D11-PR8 interaction were 38.4± 0.1 and 44.7±0.4, respectively. All binding curves were corrected for background and bulk refractive index contribution by subtraction of the reference flow cells. To further validate the kinetics and affinity calculations, we tried to perform the SPR experiments by immobilizing the 16D11 hu-mAb on a CM5 chip surface and subsequently inject the rHA proteins, but immobilization of 16D11 by amine coupling was not possible, probably due to the low isoelectric point of the hu-mAb.
Hemagglutination inhibition assay (HIA)
Virus neutralization capacity of hu-mAbs was measured by hemagglutinin inhibition assay (HIA), as we previously described.69 Briefly, 5 ml of fresh chicken red blood cells (RBC, Lampire Biologicals Labs.) was first washed with 1X PBS at 1,200 rpm at 4°C till the supernatant was clear of hem lysate. To establish the hemagglutination titer of the virus, mixtures of 2 fold dilutions of PR8 virus (Charles River) in 1% chicken red blood cells (RBCs) were suspended 60 min at room temperature in 1X PBS in round-bottom 96 well plates. To determine the hemagglutination inhibition titer of hu-mAbs, the hu-mAbs at 200 μg/ml were serially diluted with 1X PBS in 96 well round bottom plates, and a standardized influenza virus concentration was added to the wells in the presence of 1% chicken RBCs for 2 h at room temperature. The HI titer was defined as the minimum concentration of antibody that inhibit hemagglutination of 1% chicken RBCs.
Hematoxilin-Eosin (HE) staining of lung sections
Lungs from naive and infected mice with PR8 virus were fixed in formalin 10%, and 5 μ sections were stained with HE according to a standard technique to analyze the alveolar architecture and lymphocyte infiltration.
Biostatistics
Body-weight curves were compared by the Mantel-Cox log rank test, and for statistical significance by pairwise curves comparison by Gehan-Breslow-Wilcoxon test with Bonferroni's corrected threshold of significance for multiple groups. Statistical comparisons were made between PR8-infected vs. PR8-infected and treated groups of mice. ELISA and HIA virus titer analyses were analyzed by one-way analysis of variance (ANOVA).
Supplementary Material
Funding Statement
This work was supported by USUHS grants (RO83193816 and G287252016) to TDB.
Disclosure of potential conflicts of interest
The authors declare no potential conflicts of interests.
Acknowledgments
We thank Xinyue Qiu for assistance with ELISA, RT-qPCR, and WB assays, and Mike Flora for assistance with FPLC assays. TDB, and SC are US Government employees. The work of these individuals was prepared as part of official government duties. Title 17 U.S.C. §105 provides that ‘Copyright protection under this title is not available for any work of the United States Government.’ Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person's official duties. The views expressed in this paper are those of the authors and do not necessarily represent the views of Uniformed Services University, Department of Defense, or other Federal Agencies.
References
- 1.Kilbourne ED. Influenza. New York, NY: Plenum Medical Book Co.; 1987 [Google Scholar]
- 2.Li OT, Poon LL. One step closer to universal influenza epitopes. Expert Rev Anti Infect Ther. 2009;7(6):687–690 doi: 10.1586/eri.09.48. PMID:19681695 [DOI] [PubMed] [Google Scholar]
- 3.Velkov T, Ong C, Baker MA, Kim H, Li J, Nation RL, Huang JX, Cooper MA, Rockman S. The antigenic architecture of the hemagglutinin of influenza H5N1 viruses. Mol Immunol. 2013;56(4):705–719 doi: 10.1016/j.molimm.2013.07.010. PMID:23933511 [DOI] [PubMed] [Google Scholar]
- 4.Osterholm MT. Preparing for the next pandemic. N Engl J Med. 2005;352:1839–1842 doi: 10.1056/NEJMp058068. PMID:15872196 [DOI] [PubMed] [Google Scholar]
- 5.McMurry JA, Johanson BE, De Groot AS. A call to cellular and humoral arms. Human Vaccines. 2008;4(2):148–157 doi: 10.4161/hv.4.2.5169. PMID:18382131 [DOI] [PubMed] [Google Scholar]
- 6.Epstein SL. Control of influenza virus infection by immunity to conserved viral features. Expert Rev Anti Infect Ther. 2003;1:627–638 doi: 10.1586/14787210.1.4.627. PMID:15482160 [DOI] [PubMed] [Google Scholar]
- 7.Cassetti MC, Couch R, Wood J, Pervikov Y. Report of meeting on the development of influenza vaccines with broad spectrum and long-lasting immune responses. WHO, Geneva, Switzerland, 26–27 February 2004, Vaccine. 2005;23:1529–1533 doi: 10.1016/j.vaccine.2004.09.004. PMID:15754468 [DOI] [PubMed] [Google Scholar]
- 8.Nilvebrant J, Dunlop DC, Sircar A, Wurch T, Falkowska E, Reichert JM, Helquera G, Piccione EC, Brack S, et al. . IBC's 22nd annual antibody engineering and 9th annual antibody therapeutics international conferences and the 2011 annual meeting of the Antibody Society. December 5–8, 2011, San Diego, CA J MAbs. 2012;4(2):153–181 doi: 10.4161/mabs.4.2.19495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshida R, Igarashi M, Ozaki H, Kishida N, Tomabechi D, Kida H, Ito K, Takada A. Cross-protective potential of a novel monoclonal antibody directed against antigenic site B of the Hemagglutinin of influenza viruses. Plos Pathogens. 2009;5:e1000350 doi: 10.1371/journal.ppat.1000350. PMID:19300497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lambkin R, McLain L, Jones SE, Aldridge SL, Dimmock NJ. Neutralization escape mutants of type a influenza virus are readily selected by antisera from mice immunized with whole virus: a possible mechanism for antigen drift. J General Virology. 1994;75:3493–3502 doi: 10.1099/0022-1317-75-12-3493. [DOI] [PubMed] [Google Scholar]
- 11.Murphy BR, Nelson DL, Wright PE, Tierney EL, Phelan MA, Chanock RM. Secretory and systemic immunological response in children infected with live attenuated influenza A virus vaccines. Infect Immun. 1982;36:1102–1108 PMID:7095844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burlington DB, Clemens ML, Meiklejohn G, Phelan M, Chanok RM. Hemagglutinin specific antibody response in immunoglobulin G, A, and M isotype as measured by ELISA after primary or secondary infection of human with influenza A virus. Infect Immunity. 1983;41:540–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Throby M, van den Brink E, Jongeneelen M, Poon LL, Alard P, Cornelissen L, Bakker A, Cox F, van Deventer E, et al. . Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. Plos ONE. 2008;3(12):e3942 doi: 10.1371/journal.pone.0003942. PMID:19079604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshida R, Igarashi M, Ozaki H, Kishida N, Tomabechi D, Kida H, Ito K, Takada A. Cross-protective potential of a novel monoclonal antibody directed against antigenic site B of the Hemagglutinin of influenza viruses. Plos Pathogens. 2009;5(3):e1000350 doi: 10.1371/journal.ppat.1000350. PMID:19300497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janice Oh H-L, Akerstrom S, Shen S, Bereczky S, Karlberg H, Klingström J, Lal SK, Mirazimi A, Tan YJ. An antibody against a novel and conserved epitope in the Hemagglutinin 1 subunit neutralizes numerous H5N1 influenza viruses. J Virology. 2010;84:8275–8286 doi: 10.1128/JVI.02593-09. PMID:20519402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y, Luo W, Wu WL, Fang Z, Xia L, Gui X, Chen Y, Chen H, Shih JW, Xia N. Humanized antibodies with broad-spectrum neutralization to avian influenza virus H5N1. Antiviral Res. 2010;87:81–84 doi: 10.1016/j.antiviral.2010.04.012. PMID:20450935 [DOI] [PubMed] [Google Scholar]
- 17.Zheng Q, Xia L, Wu WL, Zheng Z, Huo Y, Wu J, Liu Y, Yu H, Chen Y, Lau SY, Chen H, Luo W, Xia N. Properties and therapeutic efficacy of broadly reactive chimeric and humanized H5-specific monoclonal antibodies against H5N1 influenza viruses. Antimicrob Agents Chemother. 2011;55(4):1349–1357 doi: 10.1128/AAC.01436-10. PMID:21245446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang TT, Tan GS, Hai R, Pica N, Petersen E, Moran TM, Palese P. Broadly protective monoclonal antibodies against H3 Influenza viruses following sequential immunization with different hemagglutinins. Plos Pathogens. 2010;6(2):e1000796 doi: 10.1371/journal.ppat.1000796. PMID:20195520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanson BJ, Boon AC, Lim AP, Webb A, Ooi EE, Webby RL. Passive immunoprophylaxis and therapy with humanized monoclonal antibody specific for influenza A H5 hemagglutinin in mice. Resp Res. 2006;7:126–136 doi: 10.1186/1465-9921-7-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du L, Jin L, Zhao G, Sun S, Li J, et al. . Identification and structural characterization of a broadly neutralizing antibody targeting a novel conserved epitope on the influenza virus H5N1 hemagglutinin. J. Virol. 2013;87(4):2215–2225 doi: 10.1128/JVI.02344-12. PMID:23221567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujimoto Y, Tomioka Y, Takakuwa H, Uechi G, et al. . Cross-protective potential of anti-nucleoprotein human monoclonal antibodies against lethal influenza A virus infection. J Gen Virol. 2016;97(2):2104–2116 PMID:27260213 [DOI] [PubMed] [Google Scholar]
- 22.Wrammert J, Koutsananos D, Li G-M, Edupuganti S, Sui J, et al. . Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J Exp Med. 2011;208(1):181–193 doi: 10.1084/jem.20101352. PMID:21220454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klausberger M, Tschelliesing R, Neft S, Nachbagauer R, Wohlbold TJ, Wilde M, Palmberger D, Krammer F, Jungbauer A, Grabherr R . Globular head-displayed conserved influenza H1 hemagglutinin stalk epitopesconfer protection against heterologous H1N1 virus. PlosONE. 2016;11(4):e0153579 doi: 10.1371/journal.pone.0153579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clementi M, Criscuolo E, Castelli M, Mancini N, Clementi M, Burioni R. Influenza B-cells Protective epitope characterization: a passkey for the rational design of new broad-range anti-influenza vaccines. Viruses. 2012;4(11):3090–3108 doi: 10.3390/v4113090. PMID:23202517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen HH, Tumpey TM, Park H-J, Byun Y-Ho, Tran LD, Nguyen VD, Kilgore PE, Czerkinsky C, Katz JM, et al. . Prophylactic and therapeutic efficacy of avian antibodies against influenza virus H5N1 and H1N1 in mice. PloS One. 2010;5(4);e10152 doi: 10.1371/journal.pone.0010152. PMID:20405007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tjandra JJ, Ramadi L, McKenzie IF. Development of human anti-murine antibody (HAMA) response in patients. Immunol Cell Biol. 1990;68:367–376 doi: 10.1038/icb.1990.50. PMID:1711007 [DOI] [PubMed] [Google Scholar]
- 27.Collins AM, Roberton DM, Hosking CS, Flannery GR. Oral immunization with xenogeneic antibodies stimulates the production of systemic and mucosal anti-idiotypic antibodies. Immunology. 1991;73(4):388–393 PMID:1916890 [PMC free article] [PubMed] [Google Scholar]
- 28.Clark JI, Alpaugh RK, von Mehren M, Schultz J, Gralow JR, Cheever MA, Ring DB, Weiner LM. Induction of multiple anti-c-erbB-2 specificities accompanies a classical idiotypic cascade following 2B1 bispecific monoclonal antibody treatment. Cancer Immunol Immunother. 1997;44(5):265–272 doi: 10.1007/s002620050382. PMID:9247561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimball JA, Norman DJ, Shield CF, Schroeder TJ, Lisi P, Garovoy M, O'Connell JB, Stuart F, McDiarmid SV, et al. . The OKT3 antibody response study: a multicenter study of human anti-mouse antibody (HAMA) production following OKT3 use in solid organ transplantation. Transpl Immunol. 1995;3(3):212–221 doi: 10.1016/0966-3274(95)80027-1. PMID:8581409 [DOI] [PubMed] [Google Scholar]
- 30.Scrhroff RW, Foon KA, Beatty SM, Oldham RK, Morgan AC. Human anti-murine immunoglobulin responses in patients receiving monoclonal antibody therapy. Cancer Res. 1985;45(2):879–885 PMID:3871353 [PubMed] [Google Scholar]
- 31.Reynolds JC, Del Vecchio S, Sakahara H, Lora ME, Carrasquillo JA, Neumann RD, Larson SM. Anti-murine antibody response to mouse monoclonal antibodies: clinical findings and implications. Int J Rad Appl Instrum B. 1989;16(2):1221–125 doi: 10.1016/0883-2897(89)90182-7. [DOI] [PubMed] [Google Scholar]
- 32.Baudouin V, Crusiaux A, Haddad E, Schandene L, Goldman M, Loirat C, Abramowicz D. Anaphylactic shock caused by immunoglobulin E sensitization after treatment with the chimeric interleukin-2 receptor monoclonal antibody basiliximab. Transplantation. 2003;76(3):459–463 doi: 10.1097/01.TP.0000073809.65502.8F. PMID:12923429 [DOI] [PubMed] [Google Scholar]
- 33.Gonzales NR, Padlan EA, De Pascalis R, Schuck P, Scholm J, Kashmiri SV. Minimizing immunogenicity of the SDR-grafted humanized antibody CC49 by genetic manipulation of the framework residues. Mol Immunol. 2003;40(6):337–349 doi: 10.1016/S0161-5890(03)00166-4. PMID:14522015 [DOI] [PubMed] [Google Scholar]
- 34.Hosono M, Endo K, Sakahara H, Watanabe Y, Saga T, Nakai T, Kawai C, Matsumori A, Yamada T, et al. . Human/mouse chimeric antibodies show low reactivity with human anti-murine antibodies (HAMA). Brit J Cancer. 1992;65(2):197–200 doi: 10.1038/bjc.1992.41. PMID:1739617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luiten RM, Warnaar SO, Sanborn D, Lamers CH, Bolhuis RL, Litvinov SV, Zurawski VR, Coney LR. Chimeric bispecific OC/TR monoclonal antibody mediates lysis of tumor cells expressing the folate-binding protein (MOv18) and displays decrease immunogenicity in patients. J Immunother. 1997;20(6):496–504 doi: 10.1097/00002371-199711000-00010. PMID:9409456 [DOI] [PubMed] [Google Scholar]
- 36.Chan KT, Cheng SC, Xie H, Xie Y. A humanized monoclonal antibody constructed from intronless expression vectors targets human hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2001;284(1):157–167 doi: 10.1006/bbrc.2001.4837. PMID:11374885 [DOI] [PubMed] [Google Scholar]
- 37.Slavin-Chiorini DC, Kashmiri SV, Lee HS, Milenic DE, Poole DJ, Bernon E, Schlom J, Hand PH. A CDR-grafted (humanized) domain-deleted antitumor antibody. Cancer Biother Radiopharm. 1997;12(5):305–316 doi: 10.1089/cbr.1997.12.305. PMID:10851481 [DOI] [PubMed] [Google Scholar]
- 38.Bitoh S, Lang GM, Kierek-Jaszczuk D, Fujimoto S, Sehon AH. Specific immunosuppression of human anti-murine antibody responses in hu-PBL-SCID mice. Hum Antibodies Hybridomas. 1993;4(3):134–143 PMID:8357957 [PubMed] [Google Scholar]
- 39.Sivolapenko GB, Kanariou M, Edwards RJ, Epenetos AA, Ritter MA. Immunosuppression by immunoglobulin deaggregation is not effective in reducing the anti-xenogeneic immunoglobulin response: experimental and clinical studies. Br J Cancer. 1990;61(2):347–358 doi: 10.1038/bjc.1990.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simmons CP, Bernasconi NL, Suguitan AL, Mills K, Ward JM, Chau NV, Hien TT, Sallusto F, Hado Q, et al. . Prophylactic and therapeutic efficacy of human monoclonal antibodies against H5N1 influenza. Plos Med. 2007;4(5):e178 doi: 10.1371/journal.pmed.0040178. PMID:17535101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du T, Eckiert DC, Krause JC, Martinez O, Crow Jr JE, Wilson IA, Basler CF. Influenza human monoclonal antibody 1F1 interacts with three major antigenic sites and residues mediating human receptor specificity in H1N1 viruses. Plos Pathogens. 2012;8(12):e1003067 doi: 10.1371/journal.ppat.1003067. PMID:23236279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, Manz MG. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. 2004;304:104–107 doi: 10.1126/science.1093933. PMID:15064419 [DOI] [PubMed] [Google Scholar]
- 43.Watanabe Y, Takahashi T, Okajima A, Shiokawa M, Ishii N, Katano I, Ito R, Ito M, Minegishi M, et al. . The analysis of the functions of human B and T cells in humanized NOD/shi-scid/gcnull (NOG) mice (hu-HSC NOG mice). Int Immunol. 2009;21:843–858 doi: 10.1093/intimm/dxp050. PMID:19515798 [DOI] [PubMed] [Google Scholar]
- 44.Matsumura T, Kametani Y, Ando K, Hirano Y, Katano I, Ito R, Shiina M, Tsukamoto H, Saito Y, et al. . Functional CD5+ B cells develop predominantly in the spleen of NOD/SCID/ gammac(null) (NOG) mice transplanted either with human umbilical cord blood, bone marrow, or mobilized peripheral blood CD34+ cells. Exp Hematol. 2003;31:789–797 doi: 10.1016/S0301-472X(03)00193-0. PMID:12962725 [DOI] [PubMed] [Google Scholar]
- 45.Baenziger S, Tussiwand R, Schlaepfer E, Mazzucchelli L, Heikenwalder M, Kurrer MO, Behnke S, Frey J, Oxenius A, et al. . Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2-/-gamma c-/- mice. Proc Natl Acad Sci USA. 2006;103:15951–15956 doi: 10.1073/pnas.0604493103. PMID:17038503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajesh D, Zhou Y, Jankowska-Gan E, Roenneburg DA, Dart ML, Torrealba J, Burlingham WJ. Th1 and Th17 immunocompetence in humanized NOD/SCID/_C-KO mice. Human Immunol. 2010;71:551–559 doi: 10.1016/j.humimm.2010.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jaiswal S, Pearson T, Friberg H, Shultz LD, Greiner DL, Rothman AL, Mathew A. Dengue virus infection and Virus-Specific HLA-A2 restricted immune responses in humanized NOD-scid IL2rcnull Mice. PLoS One. 2009;4:e7251 doi: 10.1371/journal.pone.0007251. PMID:19802382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akkina R. New generation humanized mice for virus research: comparative aspects and future prospects. Virology. 2013;435(1):14–28 doi: 10.1016/j.virol.2012.10.007. PMID:23217612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danner R, Chaudhari SN, Rosenberger J, Surls J, Richie TL, Brumeanu T-D, Casares S. Expression of HLA class II molecules in humanized NOD.Rag1KO.IL2Rg KO mice is critical for the development and function of human T and B cells. PlosONE. 2011;6:e19826 doi: 10.1371/journal.pone.0019826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Majji S, Wijayalath W, Shashikumar S, Pow-Sang L, Villasante EF, Brumeanu TD, Casares S. Differential effect of HLA class-I versus class-II transgenes on human T and B cell reconstitution and function in NRG mice. Sci Rep. 2016;6:28093 doi: 10.1038/srep28093. PMID:27323875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wijayalath W, Majji S, Villasante EF, Brumeanu T-D, Richie TL, Casares S. Humanized HLA-DR4.RagKO.IL2RγcKO.NOD (DRAG) mice sustain the complex vertebrate life cycle of Plasmodium falciparum malaria. Malaria J. 2014;13:386 (1–14) doi: 10.1186/1475-2875-13-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soundararajan V, Zheng S, Patel N, Warnock K, Raman R, Wilson IA, Raguram S, Sasisekharan V, Sasisekharan R. Networks link antigenic and receptor-binding sites of influenza hemagglutinin: Mechanistic insight into fitter strain propagation. Sci Rep. 2011;1:200 doi: 10.1038/srep00200. PMID:22355715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen Y, Maupetit J, Derreumaux P, Tufféry P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J Chem Theor Comput. 2014;10:4745–4758 doi: 10.1021/ct500592m. [DOI] [PubMed] [Google Scholar]
- 54.Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012;40:288–293 doi: 10.1093/nar/gks419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: Protein structure and function prediction. Nature Methods. 2015;12:7–8 doi: 10.1038/nmeth.3213. PMID:25549265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature Protocols. 2010;5:725–738 doi: 10.1038/nprot.2010.5. PMID:20360767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40 doi: 10.1186/1471-2105-9-40. PMID:18215316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brochet X, Lefranc MP, Giudicelli V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008;36:W503–8 doi: 10.1093/nar/gkn316. PMID:18503082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruiz M, Lefranc MP. IMGT gene identification and Colliers de Perles of human immunoglobulins with known 3D structures. Immunogenetics. 2002;53 (10-11):857–83 doi: 10.1007/s00251-001-0408-6. PMID:11862387 [DOI] [PubMed] [Google Scholar]
- 60.Xu JL, Davis MM. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity. 2000;13(1):37–45 doi: 10.1016/S1074-7613(00)00006-6. PMID:10933393 [DOI] [PubMed] [Google Scholar]
- 61.Barrios Y, Jirholt P, Ohlin M. Length of the antibody heavy chain complementarity determining region 3 as a specificity-determining factor. J Mol Recognit. 2004;17(4):332–338 doi: 10.1002/jmr.679. PMID:15227640 [DOI] [PubMed] [Google Scholar]
- 62.Daniels PS, Jeffries S, Yates P, Schild GC, Rogers GN, Paulson JC, Wharton SA, Douglas AR, Skehel JJ, et al. . The receptor-binding and membrane-fusion properties of influenza virus variants selected using anti-haemagglutinin monoclonal antibodies. The EMBO Journal. 1987;6:1459–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barbey-Martin C, Gigant B, Bizebard T, Calder I, Wharton SA, Douglas AR, Skehel JJ, Wiley DC. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology. 2002;294:70–74 doi: 10.1006/viro.2001.1320. [DOI] [PubMed] [Google Scholar]
- 64.Yu X, Tsibane T, McGraw PA, House FS, Keefer C, Hicar MD, Tumpey TM, Pappas C, Perrone LA, Martinez O, et al. . Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature. 2008;455:532–536 doi: 10.1038/nature07231. PMID:18716625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim J, Peachman KK, Jobe O, Morrison EB, Allam J L, Casares S, Rao M. Tracking Human Immunodeficiency Virus-1 infection in the humanized DRAGA mouse model. Frontiers in Immunology. 2017;8:1405. Epub 2017/11/23. doi: 10.3389/fimmu.2017.01405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weitzner BD, Kuroda D, Marze N, Xu J, Gray JJ. Blind prediction performance of RosettaAntibody 3.0: grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins. 2014;82(8):1611–1623 doi: 10.1002/prot.24534. PMID:24519881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231(4):1049–1067 doi: 10.1006/jmbi.1993.1351. PMID:8515464 [DOI] [PubMed] [Google Scholar]
- 68.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;13:1605–1612 doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 69.Brumeanu T-D, Casares S, Bot A, Bona CA. Immunogenicity of a contiguous T-B synthetic epitope of the PR8/A/34 influenza virus. J Virol. 1997;10:129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.