

Abstract
Oscillatory activity occurs in cortical and hippocampal networks with specific frequency ranges thought to be critical to working memory, attention, differentiation of neuronal precursors, and memory trace replay. Synchronized activity within relatively large neuronal populations is influenced by firing and bursting frequency within individual cells, and the latter is modulated by changes in intrinsic membrane excitability and synaptic transmission. Published work suggests that dopamine (DA), a potent modulator of learning and memory, acts on dopamine receptor 1-like dopamine receptors (D1Rs) to influence the phosphorylation and trafficking of glutamate receptor subunits, along with long-term potentiation (LTP) of excitatory synaptic transmission in striatum and prefrontal cortex. Prior studies also suggest that dopamine can influence voltage gated ion channel function and membrane excitability in these regions. Fewer studies have examined dopamine’s effect on related endpoints in hippocampus, or potential consequences in terms of network burst dynamics. In the present study, we record action potential activity using a micro-electrode array system to examine the ability of dopamine to modulate baseline and glutamate-stimulated bursting activity in an in vitro network of cultured murine hippocampal neurons. We show that dopamine stimulates a D1R-dependent increase in number of overall bursts within minutes of its application. Notably, however, at the concentration used herein, dopamine did not increase the overall synchrony of bursts between electrodes. Although the number of bursts normalizes by 40 minutes, bursting in response to a subsequent glutamate challenge is enhanced by dopamine pretreatment. Dopamine-dependent potentiation of glutamate-stimulated bursting was not observed when the two modulators were administered concurrently. In parallel, pretreatment of murine hippocampal cultures with dopamine stimulated lasting increases in the phosphorylation of the glutamate receptor subunit GluA1 at serine 845. This effect is consistent with the possibility that enhanced membrane insertion of GluAs may contribute to a more slowly evolving dopamine-dependent potentiation of glutamate-stimulated bursting. Together, these results are consistent with the possibility that dopamine can influence hippocampal bursting by at least two temporally distinct mechanisms, contributing to an emerging appreciation of dopamine-dependent effects on network activity in the hippocampus.
Keywords: hippocampus, neuronal networks, bursting, dopamine, glutamate
Summary Schematic
In this issue we show that dopamine can stimulate a short-term increase in neuronal network activity. Dopamine can also potentiate increased network activity in response to a later glutamate challenge. These findings may be of relevance to enhanced memory that is associated with reward.
Introduction
Dopamine (DA) is a catecholamine modulator that acts on specific G protein coupled receptor subtypes to impact learning, working memory, and attention (reviewed in (Tritsch & Sabatini 2012, Surmeier et al. 2010, Gerfen & Surmeier 2011)). Receptor subtypes fall into two broad families, D1 and D2 like, with opposing effects on adenylate cyclase activity. D1 like receptors (D1R) are linked to increased adenylate cyclase and protein kinase A (PKA) activity, and represent the most strongly expressed subtype in brain regions including prefrontal cortex and hippocampus. D1R positive cells in hippocampus include glutamatergic projection neurons of the dentate gyrus as well as CA1 pyramidal neurons and CA1/CA3 interneurons (Gangarossa et al. 2012).
Published studies have linked dopamine and D1 like receptor activation to enhancement of glutamate mediated synaptic transmission (Andre et al. 2010). For example, PKA can increase the surface expression (Sun et al. 2005, Snyder et al. 2000) and open probability of GluAs (Shepherd & Huganir 2007). Related work has shown that methylphenidate, used in the treatment of attention deficit hyperactivity disorder (ADHD) and narcolepsy, can amplify LTP in hippocampal CA1 and surface GluA1 expression through an adrenergic and D1 like receptor dependent pathway (Rozas et al. 2015).
Previous work has also linked dopamine to changes in membrane excitability. Although overall effects of dopamine and D1R agonists on voltage gated ion channel function and intrinsic membrane excitability are complex, a large body of evidence supports models in which D1R activation promote intrinsic excitability in D1R bearing medium spiny neurons of the striatum (Gerfen & Surmeier 2011, Tritsch & Sabatini 2012). Studies in pyramidal neurons, in which synaptic transmission was pharmacologically blocked, also suggest that dopamine can enhance intrinsic membrane excitability (Ceci et al. 1999, Moore et al. 2011).
Overall, previously described synaptic and extrasynaptic effects of dopamine on PKA would be expected to enhance action potential probability and possibly the number of network level population bursts in specific subpopulations of hippocampal neurons. Bursting is a critical physiological event that may contribute to the differentiation of neuronal progenitors and the transfer of hippocampal engrams to neocortex for long-term memory storage (Babu et al. 2009, Ego-Stengel & Wilson 2010, Buzsaki 1989).
Consistent with its ability to influence action potential probability and bursting, recent studies suggest that elevations in dopamine may increase the probability of a specific form of physiological population burst that is relevant to memory, the sharp wave ripple (SWR). These events, at 135–250Hz, occur during quiet rest and slow wave sleep and are thought to allow for rapid replay of memory traces. In one study, which examined SWRs using obliquely cut hippocampal slices, a one-minute bath application of 1μM dopamine stimulated a lasting D1R dependent increase in the frequency and magnitude of events (Miyawaki et al. 2014). In a related and very elegant study, an optogenetic approach was used to stimulate dopaminergic projections with subsequent recording of pyramidal cell assembly reactivation during sleep and rest. Data supported dopamine and D1R dependent reactivation of spatial memory (McNamara et al. 2014).
Dopamine and amphetamine have also been linked to increased synchrony in the gamma frequency band (Komek et al. 2012). These effects may, however, be concentration dependent with moderate levels enhancing synchrony and high or low levels reducing the same.
While intact anatomical interactions between specific brain regions may be important to the generation or consequences of specific in vivo network activity that involves large populations of neurons, dissociated hippocampal networks allow for the study of events including increased burst duration or frequency that may in turn influence the probability of widespread synchronous activity. Moreover, recent work suggests that oscillatory activity may develop in dissociated hippocampal networks (Leondopulos et al. 2012). It has thus been suggested that oscillations can occur in the absence of input from cortex, thalamus or laminar brain architecture. Importantly, cultured hippocampal networks presents a reduced platform that can be easily manipulated to identify molecular mechanisms that might contribute to rhythmic bursting in more complex systems.
In the present study, we examine the potential for dopamine to increase basal and glutamate stimulated bursting in murine hippocampal cultures. We examine bursting within seconds of dopamine application. Since glutamate can increase GluA membrane insertion on a time scale that peaks at 10 minutes and endures for at least 30 minutes (Song et al. 2013), we also examine baseline and glutamate stimulated bursting at 40 minutes following dopamine exposure.
Materials and Methods
Chemicals
We used commercially available chemicals as follows: Dopamine (Sigma Aldrich, H8502), SKF 83566, a dopamine D1-like receptor (D1R) antagonist, (Tocris Bioscience, Cat#1586), glutamate (Sigma Aldrich, G1626). Dopamine was freshly prepared at a concentration of 5mM just prior to use. SKF83566 was prepared in water at a stock concentration of 1mM. Glutamate was diluted in water to a stock concentration of 5mM. Stock solutions were stored at −20 °C.
Cell culture
Ethics Statement
All experimental procedures were carried out in accordance with the Georgetown University Animal Care and Use Committee (GUACUC). For hippocampal neuronal cell cultures, we applied previously described techniques modified from (Beaudoin III et al. 2012). Briefly, hippocampal tissues were harvested from male and female postnatal 1 day C57BL/6 wild type mice, obtained through in house breeding of mice from Jackson laboratories. One litter of approximately 6 pups were used for each cell culture preparation. Tissues were finely chopped and digested with both 0.1% trypsin and mechanical trituration. Cells were diluted at a final concentration of 3×105 cells/ml and were plated onto micro-electrode arrays (MEA, Multi Channel Systems MCS GmbH, Reutlingen, Germany) plates which were used for electrophysiological recordings. For western blot studies, cells at a concentration of 3×105 cells/ml were plated onto 12 well plates. All plates used in this study were previously treated with poly-d-lysine and laminin (Sigma, St. Louis, MO). Cultures were maintained in Neurobasal A medium with B27 (Invitrogen, Carlsbad, CA), with bi-weekly changes, and stored in a humidified 5% CO2 and 95% O2 incubator at 37°C. Experiments were performed on cultures at 14 days in vitro (DIV14).
MEA recordings
All electrical activity on cultured hippocampal neurons was recorded using a micro-electrode array (MEA) as previously described (Niedringhaus et al. 2012b). Each MEA consists of 59 titanium nitride electrodes, one reference electrode and four auxiliary analog channels, all arranged on an 8×8 square array. Each electrode is 30 μm in diameter and the inter-electrode spacing is 200 μm. Spiking network activity was recorded from cultured neurons at DIV14, as this is a time point at which network connectivity is well-established (Wagenaar et al. 2006). We use the MEA1060 preamplifier and sampled electrical activity at a 10kHz acquisition rate to allow for the detection of multi-unit spikes. The data is digitized and stored on a Dell personal computer (Round Rock, TX). Possible exposure to contaminants and fluctuations in osmolality and pH were significantly reduced during the data acquisition period by the use of an MEA cover made of a hydrophobic membrane and short recording durations (5 minutes). This membrane provides a seal that is semi-permeable to CO2 and O2 and is largely impermeable to water vapor. To ensure reproducibility of results across animals, experimental groups were derived from multiple experimental preparations. Results obtained from cultures within and across different preparations were not significantly different.
Application of dopamine, glutamate, and D1R antagonist for MEA recordings and/or Western blot
10 μl of vehicle alone or vehicle containing specified chemicals was added to 1 ml of media within the MEA culture dish. Application was on a sterile bench adjacent to the recording set up and care was taken to minimize mechanical disruption. Recordings were subsequently performed as follows. All MEAs underwent 5 minutes of baseline recordings. Subsequently, for cells treated with vehicle or 50 μM glutamate, recordings were performed immediately after treatment. For cells treated with dopamine, recordings were obtained either immediately after treatment or 40 minutes after treatment as indicated. To examine the dopamine-dependent potentiation of the glutamate response, cells were pretreated with dopamine for 40 minutes and were subsequently exposed to glutamate with recordings obtained immediately thereafter. Lastly, for experiments treated with the D1R antagonist, SKF83566 (10 μM), the compound was applied 15 minutes prior to dopamine and subsequent recordings. In all experiments, pretreatments with either dopamine or the D1R antagonist were not washed out prior to the addition of glutamate. All recordings lasted for 5 minutes and were performed in a chamber maintained at 37° C. For Western blot experiments, cells were pretreated with dopamine for 5 to 40 minutes as indicated below and, without washing out, subsequently treated with glutamate for 5–20 minutes as indicated. Cell lysates were harvested at the end of each treatment.
Western blot
Western blot assays were performed on lysates as previously described (Conant et al. 2011). In brief, cells were washed with 1X PBS twice. The lysates were prepared via the addition of lysis buffer containing: 50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1% NP-40, 0.5% sodium deoxycholate, 0.2 mM phenylmethylsulfonyl fluoride, 0.5 mM dithiothreitol, 1Xprotease and phosphatase inhibitor cocktail (Thermo Scientific, 78440). The mixture was placed into a microfuge tube, sonicated for 10 s, kept on ice for 20 min, and then spun at 13,000 rpm for 15 min at 4°C in a microcentrifuge. Protein concentrations in the lysates were quantified with the bicinchoninic acid assay (BCA assay). An equal quantity of protein for each lane line was mixed with loading buffer containing β-mercaptoethanol and then heated for 5 min at 95°C. After resolution through gel electrophoresis, using 4–20% precast gels (Bio-Rad, Cat#456-1094), and transfer to nitrocellulose, blots were probed with a commercially available antibodies to phospho-GluA1 (S845) (R&D Systems, PPS008, 1:1000 dilution) or loading control (Actin or GAPDH as indicated) as previously described (Lonskaya et al. 2013). Molecular weights were inferred by comparison to prestained markers (BioRad). Following exposure of membranes to western-lighting plus-ECL (PerkinElmer. lnc, 203-73391) and film, bands were quantified with Image J software.
MEA Analysis
For determination of the numbers of spikes and bursts, we removed low frequency fluctuations by high-pass filtering all voltage traces at 200 Hz in order to obtain a stable baseline for spike extraction. Extracellularly recorded spikes were detected using a threshold algorithm from Offline Sorter (Plexon Inc., Dallas TX). The threshold is calculated as a multiple of the standard deviation (3σ) of the biological noise. For this study, we did not discriminate and sort spikes within each electrode as we focused on overall network dynamics and the signal from each electrode suitably reflects these dynamics. This spike identification process results in an M x N matrix where M corresponds to the electrode number and the N’s are the time stamps of the spikes.
We analyzed network dynamics that were recorded from the cultured hippocampal networks using custom-written software written in MATLAB (The MathWorks, Natick, MA). We chose a common temporal feature, the burst, found in cultured networks and in vivo (Buzsaki et al. 2002) to assess changes in network activity due to the dopamine-dependent potentiation of the glutamate response. The burst represents a collective neural response, similar to rhythmic oscillations with its manifestation is due to the occurrence of high frequency action potential firing (Paladini & Roeper 2014). In our experiments, we analyze bursts from each individual electrode and we define a burst from each electrode to consist of no less than 3 spikes with a maximum interspike interval of 20 ms. We generated log histograms of burst durations from each MEA as well as bar graphs of burst durations for all treatments.
To investigate the time evolution of the dynamics within the networks, we used a short time, Fourier transform based time-frequency analysis (STFT - The Mathworks, Natick, MA). We used the unfiltered data sets and down-sampled at 5 KHz. This STFT analysis will result in a power spectrum as a function of time in which the power at a particular frequency will be displayed as a color map. We then convolved the down-sampled data with a Gaussian filter (5 ms width) in order to extract the slowly-varying envelope of the burst and use this in a correlation-based method that calculates the average of the similarity of burst and spike trains between pairs of active electrodes (Schreiber et al. 2003). This results in a measure of the reliability of activity and is calculated as follows:
where M is the number of active electrodes and is the low-pass filtered burst envelope from electrode i; 0 < Rcorr <1.
Statistics
Statistical analyses were performed with GraphPad Prism 5 (GraphPad Software, Inc.). Data are shown as mean ± SEM. Student’s t-test was used for two group comparisons. For comparison of more than two groups, one-way ANOVA followed by Tukey post hoc test was used to determine significance (*p< 0.05, ** p<0.01, ***p<0.001).
Results
I. Dopamine stimulates a D1 receptor dependent increase in the number of bursts
Previously published work has demonstrated that dopamine may enhance overall membrane excitability (Tritsch & Sabatini 2012). Multiple conductances may be affected, including that mediated by voltage gated L-type calcium channels (Surmeier et al. 1995, Gray & Johnston 1987). Increased membrane excitability may in turn increase action potential probability to enhance bursting within hippocampal networks (Wojtowicz & Mozrzymas 2014). As shown in figure 1, dopamine stimulates a qualitative change in the spiking activity pattern (1a) as well as an increase in the number of spikes per burst (figures 1b and 1d). Burst duration is also increased shortly after application as seen in the increasing bin counts and the tail in the histogram of figure 1c2 as compared to that of figure 1c1. This effect is not long-lasting as activity returns to baseline levels 40 minutes post dopamine treatment (figure 1c3). The number of spikes within a burst and number of bursts are increased shortly after dopamine application (figures 1d and 1e) whereas a D1 like receptor antagonists inhibits this effect (1f). Importantly, vehicle control did not increase spike or burst number (0.96 +/− S.D. =0.030 baseline and 0.97 +/− 0.027 S.D. vehicle spike number; 0.91 +/− 0.39 S.D. baseline and 0.93 +/− 0.51 S.D. vehicle burst number in n=4 MEAs, p>0.05).
Figure 1. 1. Dopamine stimulates a D1 receptor dependent increase in the number of bursts.

As shown in figure 1, dopamine stimulates a qualitative change in the spiking activity pattern (1a) as well as an increase in the number of spikes per burst (figures 1b and 1d). Burst duration is also increased shortly after application as seen in the increasing bin counts and the tail in the histogram of figure 1c2 as compared to that of figure 1c1. This effect is not long-lasting as activity returns to baseline levels 40 minutes post dopamine treatment (figure 1c3). The number of spikes within a burst and number of bursts are increased shortly after dopamine application (figures 1d and 1e) whereas a D1 like receptor antagonists inhibits this effect (1f). Data in 1d and 1e represent the mean and standard error from n=8 MEAs and the data in 1f from n=5 MEAs.
II. Concurrent administration of dopamine does not enhance glutamate stimulated increases in number of bursts
Since dopamine could increase the number of bursts in a manner that was inhibited by a D1 like dopamine receptor antagonist (1f), and since D1 like dopamine receptors are expressed on glutamate responsive hippocampal neurons, we next examined dopamine for its potential to increase glutamate stimulated activity. As shown in figure 2, glutamate treatment elicited a significant increase in number of bursts. Interestingly, however, concomitant glutamate treatment with dopamine did not potentiate the effect.
Figure 2. 2. Concurrent administration of dopamine does not enhance glutamate stimulated increases in number of bursts.
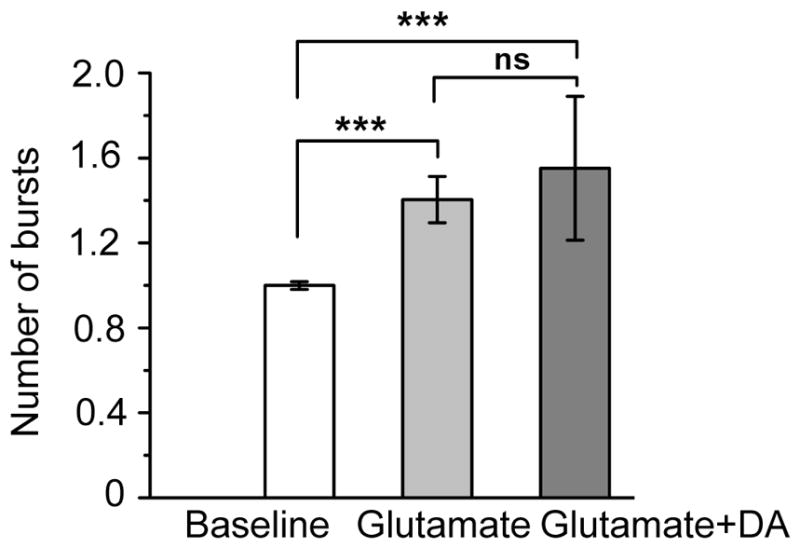
As shown in figure 2, glutamate treatment elicited a significant increase in number of bursts. Interestingly, however, concomitant glutamate treatment with dopamine did not potentiate the effect. Data represent the mean and standard error from n=7 MEAs.
III. Pretreatment with dopamine enhances subsequent glutamate-stimulated bursting
Despite the fact that dopamine had only a transient positive increase on network dynamics (figure 1), we asked whether pretreatment with dopamine would potentiate a subsequent glutamate response. We incubated cultured hippocampal neurons in dopamine or vehicle for 40 minutes and then applied glutamate. Representative raster plot data is shown in figure 3a, number of bursts in 3b and burst duration data in 3c. There was a 50% increase in the number of bursts in the dopamine pretreatment group as compared with glutamate alone (3b). In addition, there is a lengthening of the tail in the histogram of burst durations showing that not only are there more bursts taking place with dopamine pretreatment, but many of these bursts are sustained in duration. This is highlighted in the representative raster plot in figure 3a.
Figure 3. 3. Pretreatment with dopamine enhances subsequent glutamate-stimulated bursting.

Cultured hippocampal neurons were pre-incubated in dopamine or vehicle for 40 minutes and then glutamate was applied. Representative raster plot data is shown in figure 3a, number of bursts in 3b and burst duration data in 3c. There was an increase in the number of bursts in the dopamine pretreatment group as compared with glutamate alone (3b). In addition, there is a lengthening of the tail in the histogram of burst durations (3c). This is highlighted in the representative raster plot data (3a). Data represent the mean and standard error from n=7 MEAs.
IV. Glutamate and dopamine differentially modulate rhythmic activity
We present representative distributions of inter-burst intervals (IBIs) in figure 4a. Panel 1 is the distribution for baseline and panel 2 is the distribution after the treatment with glutamate. IBIs for the networks that have been pre-treated with dopamine before glutamate stimulation is displayed in panel 3. The maximum interval for the baseline is nearly 20 seconds and we note that all three distributions are exponential in nature. This maximum decreases after treatment with glutamate to approximately 16 seconds. Most notable is the case where the cells were pre-treated with dopamine before glutamate stimulation. The distribution is truncated by nearly a factor of two from baseline with a maximum interval of about 12 seconds. These IBI distributions also inform how the bursting activity changes with each treatment. The counts within each bin of the IBI distribution are displayed on a log scale. The bursting activity moderately increases after glutamate but when the networks are first treated with dopamine before glutamate is applied, there is nearly an order of magnitude increase from baseline.
Figure 4. 4. Glutamate and dopamine modulate synchronization parameters.

(a) Inter-burst intervals (IBIs) (1) Baseline activity displays an exponential distribution with a broad range of intervals. (2) After application of glutamate, the IBI distribution remains exponential with the tail shortened. (3) With pre-treatment of dopamine followed by a glutamate application, the tail of the exponential decreases considerably. (b) Short time, Fourier transform based time-frequency spectrum (STFT) (1) Baseline activity shows short, low-power bursts of activity with a narrow range of frequency components. The peak frequency is around 40 Hz. (2) After treatment with glutamate, the power in the bursts of activity increases and the spectrum spans the range to 100 Hz. (3) When the networks are first pre-treated with dopamine and followed with a glutamate application, the burst of activity smear to nearly uniform pattern with both low power. (c) Reliability coefficient (1) Baseline activity shows a normal-like distribution that peaks around 0.15. (2) The reliability coefficient increases after application of glutamate with a uniform-like distribution around larger values. (3) The average coefficient for baseline activity is significantly smaller than that for networks treated with glutamate (mean +/− S.D.; n=4 per group, Student’s t test). However, the coefficient for networks that were pre-treated with dopamine before glutamate was nearly zero (data not shown).
This change in the tail of the IBIs is further characterized in the frequency-time distributions depicted in Figure 4b. In the baseline activity of panel b1, network activity is present in discrete bursts with peak frequencies near 40 Hz and low power in each epoch. Visible gaps in the spatial maps correspond to the range of IBIs presented in panel a1. When glutamate is added to the networks, activity becomes more rhythmic with higher power in the single hertz range (panel b2). However, there is spectral power present in the networks within the entire range up to 100 Hz. In contrast, while the overall activity clearly increases when glutamate is added to networks previously treated with dopamine, there is no longer an established periodicity to the activity as seen in panel b3. Lastly, we calculated the reliability coefficient in panel c. Baseline activity (panel c1) shows a nearly normal distribution skewed towards smaller coefficients whereas the distribution after glutamate is applied (panel c2) is more uniform and skewed towards larger coefficients. Panel c3 summarizes the coefficients with the glutamate-treated networks having a significantly larger average coefficient than baseline. We note that when the application of glutamate followed a pre-treatment with dopamine, the coefficient was reduced (data not shown).
V. Dopamine stimulates increased phosphorylation of GluA at serine 845
To address potential mechanisms that might contribute to dopamine’s ability to potentiate the effect of subsequent glutamate administration, we examined neurotransmitter dependent phosphorylation of GluA1, an AMPA receptor subunit that is important to long-term potentiation of synaptic activity in the hippocampus (Makino & Malinow 2009, Lee et al. 2010). Enhanced dendritic membrane insertion of GluA1 is thought to precede its lateral diffusion to post synaptic contacts, and membrane insertion is facilitated by phosphorylation of the subunit at serine 845. Since D1R activation has been associated with substantial increases in PKA activity, which targets serine 845, we examined dopamine-stimulated phosphorylation of GluA1 at this site. As previously described (Song et al. 2013), an increase can be observed by 5 min (figure 5a and b). This increase in phosphorylation is sustained at 40 min (figure 5c and d). Importantly, dopamine also prevents or partially reverses a glutamate associated decrease in serine-845 phosphorylation of this subunit (figures 5a and b).
Figure 5. 5. Dopamine stimulates increased phosphorylation of GluA at serine 845.

Lysates from cultured hippocampal neurons were examined by Western blot for phospho-GluA1 serine 845). A dopamine associated increase in signal can be observed by 5 min (figure 5a and b). The increase in phosphorylation is also observed at 40 min (figure 5c and d). Importantly, dopamine also prevents or partially reverses a glutamate associated decrease in serine-845 phosphorylation of this subunit. Representative Western blot images are shown in a and c. Respective densitometric analysis from n=4 data points is shown with mean and standard error in c and d (* denotes p < 0.05).
Discussion
In the present study, we examine a potential molecular effector of bursting in cultures of murine hippocampal neurons. We examine dopamine dependent effects as a function of select time points, as well as the D1R dependence of observed effects. Our focus on dopamine derives from its role as an important modulator of attention and memory consolidation, as well as its potential to influence endpoints such as synaptic and membrane excitability that have the potential to enhance bursting (Tritsch & Sabatini 2012). Though we focus on bursts detected at single electrodes and that are thus representative of small neuronal subpopulations, we note that an increase in burst frequency within small subpopulations could influence the probability of synchronous bursting on a larger spatial scale.
We show that within seconds of exposure to dopamine, overall burst frequency is increased. This effect is dependent on D1 like dopamine receptor signaling in that it is abrogated by pretreatment of cultures with the D1 receptor antagonist SKF 83566.
The D1 dependence of our results is consistent with a predominance of D1 as opposed to D2 receptor expression in hippocampus, as well as previously described D1 dependent intracellular signaling cascades (Tritsch & Sabatini 2012, Gangarossa et al. 2012). For example, D1 receptor activation can enhance PKA dependent increases in GluN and GluA mediated excitatory post-synaptic currents (Gonzalez-Islas & Hablitz 2003). And while D1 receptor coupled Gαs can enhance PKA activity, associated βγ subunits may enhance PKC activity to increase pyramidal cell excitability through modulation of ion flux through sodium channels (Franceschetti et al. 2000). In prefrontal cortex, dopamine and PKA also enhance the excitability of select interneuron populations (Tritsch & Sabatini 2012). For example, PKA can suppress leak, inward rectifying and depolarization-activated potassium channels (Gorelova et al. 2002).
We also observe that the ability of dopamine to enhance burst frequency on its own does not persist in that by 40 minutes, dopamine-treated cultures show a burst frequency that is similar to control. One possibility is that changes in ion channel function are relatively transient. In comparison to vehicle, the 40-minute dopamine pretreatment enhanced burst frequency in response to a subsequent glutamate challenge. A potential explanation is that while dopamine could stimulate a relatively rapid and transient change in ion channel function, later potentiation of the glutamate response may be at least in part dependent on lasting changes at the level of GluA phosphorylation and membrane insertion. This is supported by prior studies in which a D1 agonist or PKA can modulate L type calcium channels within a matter of seconds to potentially enhance excitability (Surmeier et al. 1995, Gray & Johnston 1987), while dopamine can also stimulate a more slowly emerging and lasting (detectable at approximately 5 minutes and still elevated at 30 minutes) increase in GluA1 membrane insertion (Song et al. 2013). Our Western blot results with hippocampal lysates are also consistent with a lasting increase in GluA1 membrane insertion in that dopamine stimulated relatively long lasting GluA phosphorylation of serine 845, a change linked to enhanced membrane incorporation of the subunit (Lee et al. 2010). Although future studies will be necessary to evaluate the kinetics of membrane and synaptic contributions to bursting, we propose a schematic summarizing a hypothetical model in figure 5.
Temporally coincident activity in a large population of neurons has varied physiological and pathological consequences. The former includes potential “binding” of multiple features of a complex experience such as a visual scene (Buzsaki & Silva 2012, Singer & Gray 1995). Memory consolidation may also be favored by specific bursting patterns (Ego-Stengel & Wilson 2010), through mechanisms such as spike timing dependent plasticity (STDP) (Pawlak et al. 2010). The pathological consequences of bursting, in which strict bounds of burst duration and synchrony are exceeded, include epilepsy (Buzsaki & Silva 2012).
In vivo and in vitro slice data suggest that specific brain regions and/or their chemical and anatomical interactions may be critical to synchronous bursting. In addition, rhythmic burst occurrence, such as the sharp wave ripple, is facilitated when slice preparations are at least 350 μm and ideally 400 μm (Schlingloff et al. 2014). Interestingly, however, are studies suggesting that rhythmic oscillatory activity can still be observed in hippocampal cultures that are devoid of intact anatomy (Leondopulos et al. 2012). These cultures include pyramidal neurons, interneurons, and potential gap junction coupling subnetwork components that may contribute to population rhythms (Traub & Bibbig 2000). Moreover, the study of bursting and oscillatory activity within dissociated hippocampal networks allows for ease of pharmacological manipulation as well as potential identification of emergent mechanisms which may occur in a manner that is independent of intact anatomy.
We acknowledge that our burst analysis focused on spikes from small subpopulations of neurons that are detected at the single electrode level. We propose that increased bursting at this level could enhance the probability of synchronous bursting within larger subpopulations. To assess this, we also performed analyses focused on measures of synchrony. Of interest, while these analyses supported a glutamate stimulated increase in correlated activity, dopamine pretreatment reduced the same. This does not rule out the possibility that the absolute number of synchronous and asynchronous events are both increased by dopamine pretreatment; it may be that the relative increase in asynchronous activity is of importance. We used a relatively high concentration of dopamine. Of relevance, is work by the Cho group suggesting that at relatively low or high concentrations, dopamine can reduce synchrony in the gamma range (Komek et al. 2012). It has been suggested that dopamine thus has an inverted-U shaped effect on synchrony which, based on modeling work, may follow from its effects on fast spiking interneurons (Komek et al. 2012). Though beyond scope of the present study, it will be of interest to explore dopamine dependent effects on glutamate-stimulated synchrony as a function of dopamine exposure time and concentration, and to examine effects on specific cellular subpopulations. While we suggest that dopamine dependent increases in spike and burst number at the individual electrode level may result from changes in pyramidal cell excitability – since this cell type is relatively numerous in the hippocampus (Trevino 2016) – we acknowledge that network synchrony relevant effects are likely to be more complex.
An additional caveat of our study is that burst frequency is an endpoint to which multiple changes both at the molecular and cellular level contribute. Though our discussion has focused on changes in ligand and non-ligand gated ion channels that may influence burst frequency, it is important to note that changes in gap junction coupling between constitutive network neurons can influence large scale bursts and oscillations (Traub & Bibbig 2000). Our manuscript also highlights molecular mechanisms that have been linked to direct effects of DA signaling. We recognize, however, that at later time points following DA administration, additional neuromodulators could be upregulated. For example, DA stimulates rapid changes in the activity of matrix metalloproteinases (Li et al. 2016), and these enzymes have been shown to increase synaptic and intrinsic excitability in addition to bursting activity (Lonskaya et al. 2013, Wojtowicz & Mozrzymas 2014, Niedringhaus et al. 2012a, Dityatev et al. 2010, Nagy et al. 2006).
Lastly, with respect to the relatively high concentration of dopamine (micromolar levels) in our network and biochemical studies, micromolar levels are observed in the central nervous system in the setting of psychostimulant exposure (Bradberry et al. 2000, Jones et al. 1998, Schmitz et al. 2001). Moreover, as compared to nanomolar concentrations, micromolar concentrations may best stimulate sustained cAMP production by D1Rs in the early endosome (Kotowski et al. 2011).
In summary, we show that dopamine significantly modulates basal and glutamate- stimulated spike and burst frequency in cultured hippocampal networks. These studies contribute to an emerging appreciation of this modulator as important to both memory encoding and burst-dependent consolidation. Future studies using genetically encoded calcium indicators in specific hippocampal subpopulations could better explore underlying mechanisms important to this effect, and additional slice or in vitro experiments could validate the same.
Acknowledgments
We would like to acknowledge funding from the National Institutes of Health (NS083410), the Von Matsch professorship for neurological diseases, and the National Science Foundation (PHY-1205919). We would also like to thank Chau Tran and P. Lorenzo Bozzelli for assistance with graphics.
Abbreviations
- DA
dopamine
- D1R
dopamine type-1 receptor
- LTP
long term potentiation
- MEA
multielectrode array
- STFT
fourier transform based time-frequency analysis
- PKA
protein kinase A
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- Andre VM, Cepeda C, Cummings DM, Jocoy EL, Fisher YE, William Yang X, Levine MS. Dopamine modulation of excitatory currents in the striatum is dictated by the expression of D1 or D2 receptors and modified by endocannabinoids. Eur J Neurosci. 2010;31:14–28. doi: 10.1111/j.1460-9568.2009.07047.x. [DOI] [PubMed] [Google Scholar]
- Babu H, Ramirez-Rodriguez G, Fabel K, Bischofberger J, Kempermann G. Synaptic Network Activity Induces Neuronal Differentiation of Adult Hippocampal Precursor Cells through BDNF Signaling. Front Neurosci. 2009;3:49. doi: 10.3389/neuro.22.001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoin GM, III, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nature protocols. 2012;7:1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- Bradberry CW, Barrett-Larimore RL, Jatlow P, Rubino SR. Impact of self-administered cocaine and cocaine cues on extracellular dopamine in mesolimbic and sensorimotor striatum in rhesus monkeys. J Neurosci. 2000;20:3874–3883. doi: 10.1523/JNEUROSCI.20-10-03874.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G. Two-stage model of memory trace formation: a role for “noisy” brain states. Neuroscience. 1989;31:551–570. doi: 10.1016/0306-4522(89)90423-5. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Csicsvari J, Dragoi G, Harris K, Henze D, Hirase H. Homeostatic maintenance of neuronal excitability by burst discharges in vivo. Cereb Cortex. 2002;12:893–899. doi: 10.1093/cercor/12.9.893. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Silva FL. High frequency oscillations in the intact brain. Prog Neurobiol. 2012;98:241–249. doi: 10.1016/j.pneurobio.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceci A, Brambilla A, Duranti P, Grauert M, Grippa N, Borsini F. Effect of antipsychotic drugs and selective dopaminergic antagonists on dopamine-induced facilitatory activity in prelimbic cortical pyramidal neurons. An in vitro study. Neuroscience. 1999;93:107–115. doi: 10.1016/s0306-4522(99)00123-2. [DOI] [PubMed] [Google Scholar]
- Conant K, Lonskaya I, Szklarczyk A, Krall C, Steiner J, Maguire-Zeiss K, Lim ST. Methamphetamine-associated cleavage of the synaptic adhesion molecule intercellular adhesion molecule-5. Journal of neurochemistry. 2011;118:521–532. doi: 10.1111/j.1471-4159.2010.07153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dityatev A, Schachner M, Sonderegger P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat Rev Neurosci. 2010;11:735–746. doi: 10.1038/nrn2898. [DOI] [PubMed] [Google Scholar]
- Ego-Stengel V, Wilson MA. Disruption of ripple-associated hippocampal activity during rest impairs spatial learning in the rat. Hippocampus. 2010;20:1–10. doi: 10.1002/hipo.20707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschetti S, Taverna S, Sancini G, Panzica F, Lombardi R, Avanzini G. Protein kinase C-dependent modulation of Na+ currents increases the excitability of rat neocortical pyramidal neurones. J Physiol. 2000;528(Pt 2):291–304. doi: 10.1111/j.1469-7793.2000.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangarossa G, Longueville S, De Bundel D, Perroy J, Herve D, Girault JA, Valjent E. Characterization of dopamine D1 and D2 receptor-expressing neurons in the mouse hippocampus. Hippocampus. 2012;22:2199–2207. doi: 10.1002/hipo.22044. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Hablitz JJ. Dopamine enhances EPSCs in layer II–III pyramidal neurons in rat prefrontal cortex. J Neurosci. 2003;23:867–875. doi: 10.1523/JNEUROSCI.23-03-00867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelova N, Seamans JK, Yang CR. Mechanisms of dopamine activation of fast-spiking interneurons that exert inhibition in rat prefrontal cortex. J Neurophysiol. 2002;88:3150–3166. doi: 10.1152/jn.00335.2002. [DOI] [PubMed] [Google Scholar]
- Gray R, Johnston D. Noradrenaline and beta-adrenoceptor agonists increase activity of voltage-dependent calcium channels in hippocampal neurons. Nature. 1987;327:620–622. doi: 10.1038/327620a0. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komek K, Bard Ermentrout G, Walker CP, Cho RY. Dopamine and gamma band synchrony in schizophrenia--insights from computational and empirical studies. Eur J Neurosci. 2012;36:2146–2155. doi: 10.1111/j.1460-9568.2012.08071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotowski SJ, Hopf FW, Seif T, Bonci A, von Zastrow M. Endocytosis promotes rapid dopaminergic signaling. Neuron. 2011;71:278–290. doi: 10.1016/j.neuron.2011.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103:479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leondopulos SS, Boehler MD, Wheeler BC, Brewer GJ. Chronic stimulation of cultured neuronal networks boosts low-frequency oscillatory activity at theta and gamma with spikes phase-locked to gamma frequencies. J Neural Eng. 2012;9:026015. doi: 10.1088/1741-2560/9/2/026015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Partridge J, Berger C, Sepulveda-Rodriguez A, Vicini S, Conant K. Dopamine increases NMDA-stimulated calcium flux in striatopallidal neurons through a matrix metalloproteinase-dependent mechanism. Eur J Neurosci. 2016;43:194–203. doi: 10.1111/ejn.13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonskaya I, Partridge J, Lalchandani RR, Chung A, Lee T, Vicini S, Hoe HS, Lim ST, Conant K. Soluble ICAM-5, a product of activity dependent proteolysis, increases mEPSC frequency and dendritic expression of GluA1. PLoS One. 2013;8:e69136. doi: 10.1371/journal.pone.0069136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara CG, Tejero-Cantero A, Trouche S, Campo-Urriza N, Dupret D. Dopaminergic neurons promote hippocampal reactivation and spatial memory persistence. Nat Neurosci. 2014;17:1658–1660. doi: 10.1038/nn.3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki T, Norimoto H, Ishikawa T, Watanabe Y, Matsuki N, Ikegaya Y. Dopamine receptor activation reorganizes neuronal ensembles during hippocampal sharp waves in vitro. PLoS One. 2014;9:e104438. doi: 10.1371/journal.pone.0104438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AR, Zhou WL, Potapenko ES, Kim EJ, Antic SD. Brief dopaminergic stimulations produce transient physiological changes in prefrontal pyramidal neurons. Brain Res. 2011;1370:1–15. doi: 10.1016/j.brainres.2010.10.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy V, Bozdagi O, Matynia A, et al. Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J Neurosci. 2006;26:1923–1934. doi: 10.1523/JNEUROSCI.4359-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedringhaus M, Chen X, Dzakpasu R, Conant K. MMPs and soluble ICAM-5 increase neuronal excitability within in vitro networks of hippocampal neurons. PLoS One. 2012a;7:e42631. doi: 10.1371/journal.pone.0042631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedringhaus M, Chen X, Dzakpasu R, Conant K. MMPs and soluble ICAM-5 increase neuronal excitability within in vitro networks of hippocampal neurons. PLoS One. 2012b:7. doi: 10.1371/journal.pone.0042631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini CA, Roeper J. Generating bursts (and pauses) in the dopamine midbrain neurons. Neuroscience. 2014;282C:109–121. doi: 10.1016/j.neuroscience.2014.07.032. [DOI] [PubMed] [Google Scholar]
- Pawlak V, Wickens JR, Kirkwood A, Kerr JN. Timing is not Everything: Neuromodulation Opens the STDP Gate. Front Synaptic Neurosci. 2010;2:146. doi: 10.3389/fnsyn.2010.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas C, Carvallo C, Contreras D, Carreno M, Ugarte G, Delgado R, Zeise ML, Morales B. Methylphenidate amplifies long-term potentiation in rat hippocampus CA1 area involving the insertion of AMPA receptors by activation of beta-adrenergic and D1/D5 receptors. Neuropharmacology. 2015;99:15–27. doi: 10.1016/j.neuropharm.2015.07.003. [DOI] [PubMed] [Google Scholar]
- Schlingloff D, Kali S, Freund TF, Hajos N, Gulyas AI. Mechanisms of sharp wave initiation and ripple generation. J Neurosci. 2014;34:11385–11398. doi: 10.1523/JNEUROSCI.0867-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S, Fellous JM, Whitmer D, Tiesinga P, Sejnowski TJ. A new correlation-based measure of spike timing reliability. Neurocomputing. 2003;52–54:925–931. doi: 10.1016/S0925-2312(02)00838-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Singer W, Gray CM. Visual feature integration and the temporal correlation hypothesis. Annu Rev Neurosci. 1995;18:555–586. doi: 10.1146/annurev.ne.18.030195.003011. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Allen PB, Fienberg AA, Valle CG, Huganir RL, Nairn AC, Greengard P. Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J Neurosci. 2000;20:4480–4488. doi: 10.1523/JNEUROSCI.20-12-04480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song RS, Massenburg B, Wenderski W, Jayaraman V, Thompson L, Neves SR. ERK regulation of phosphodiesterase 4 enhances dopamine-stimulated AMPA receptor membrane insertion. Proc Natl Acad Sci U S A. 2013;110:15437–15442. doi: 10.1073/pnas.1311783110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Shen W, Day M, Gertler T, Chan S, Tian X, Plotkin JL. The role of dopamine in modulating the structure and function of striatal circuits. Prog Brain Res. 2010;183:149–167. doi: 10.1016/S0079-6123(10)83008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Bibbig A. A model of high-frequency ripples in the hippocampus based on synaptic coupling plus axon-axon gap junctions between pyramidal neurons. J Neurosci. 2000;20:2086–2093. doi: 10.1523/JNEUROSCI.20-06-02086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevino M. Inhibition Controls Asynchronous States of Neuronal Networks. Front Synaptic Neurosci. 2016;8:11. doi: 10.3389/fnsyn.2016.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Sabatini BL. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron. 2012;76:33–50. doi: 10.1016/j.neuron.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar DA, Pine J, Potter SM. An extremely rich repertoire of bursting patterns during the development of cortical cultures. BMC neuroscience. 2006;7:11. doi: 10.1186/1471-2202-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtowicz T, Mozrzymas JW. Matrix metalloprotease activity shapes the magnitude of EPSPs and spike plasticity within the hippocampal CA3 network. Hippocampus. 2014;24:135–153. doi: 10.1002/hipo.22205. [DOI] [PubMed] [Google Scholar]