

Abstract
Despite promising therapeutic utilities for treatment of hematological malignancies, histone deacetylase inhibitor (HDACi) drugs have not proven as effective in the treatment of solid tumors. To expand the clinical indications of HDACi drugs, we developed novel boron-containing prodrugs of belinostat (2), one of which efficiently releases active 2 through a cascade of reactions in cell culture and demonstrates activities comparable to 2 against a panel of cancer cell lines. Importantly, prodrug 7 is more efficacious than belinostat in vivo, not only inhibiting the growth of tumor but also reducing tumor volumes in an MCF-7 xenograft tumor model owing to its superior biocompatibility, which suggests its clinical potential in the treatment of solid tumors.
Keywords: histone deacetylase inhibitors, belinostat, MCF-7 xenograft, boron-containing prodrugs, biocompatibility
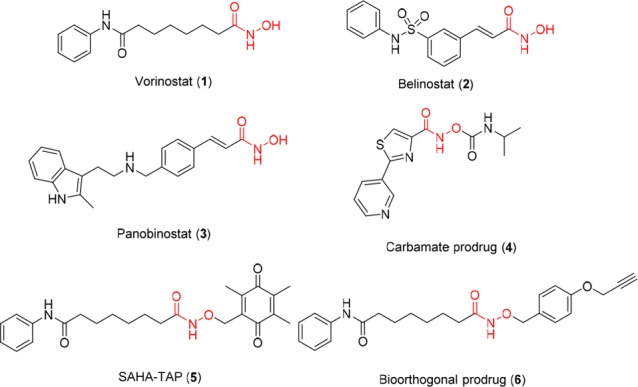
Histone deacetylases (HDACs) play a major role in the epigenetic regulation of gene expression through their effects on the compact chromatin structure. The aberrant epigenetic states (alterations in acetylation levels and overexpression) of HDACs are associated with a variety of pathologies, most notably cancer.1−4 Thus, HDACs have become one of the most promising therapeutic targets, and histone deacetylase inhibitors (HDACi) have been developed as epigenetic therapeutic agents for cancer treatment.5−8 HDACi can be subdivided into several structural classes including hydroxamic acids, cyclic peptides, aliphatic acids, and benzamides.6,9 Three of the four FDA-approved HDAC inhibitors belong to hydroxamate-based molecules, including vorinostat (SAHA, 1), belinostat (2), and panobinostat (3) (Figure 1) that are indicated for cutaneous T-cell lymphoma (CTCL), relapsed/refractory peripheral T-cell lymphoma, and multiple myeloma, respectively.
Figure 1.
Structures of hydroxamate-based HDAC inhibitors and their prodrugs.
Despite promising outcome from treatment of hematological malignancies with these hydroxamate-based HDACi agents, so far clinical trials have not shown that HDACi drugs are as effective in treating solid tumors. The exact reasons are not well understood, but some evidence suggests that the lack of activity may be due to their chemical instability and rapid metabolic elimination. It was suggested that the hydroxamate group of HDACi may have been hydrolyzed to the corresponding carboxylic acid or subjected to glucuronidation and sulfation to generate the inactive metabolites before they could reach solid tumor sites.10−14 Developing acyl derivatives of 1 and other hydroxamate-based HDACi enhances cell permeability and hydrolytic stability, which suggests that the alkylation of hydroxamate of an HDACi is an effective strategy in improving their pharmacokinetic (PK) properties.15 In another strategy, a similar carbamate prodrug (4, Figure 1) of the hydroxamates improves druglike properties, especially cellular permeability.16 However, both of these strategies rely on hydrolysis of prodrugs in vivo to release the active drug and do not improve drug–target specificity for selected disease states or sites of disease. Recently, a new prodrug of 1 (SAHA-TAP, 5, Figure 1) was developed by appending a promoiety, sensitive to thiol, to the hydroxamic acid.17 The authors envision that SAHA-TAP can become activated in the presence of thiols such as glutathione in its reduced form (GSH), which is frequently more abundant at tumor sites. Furthermore, a biorthogonal precursor (6, Figure 1) of vorinostat (1) was reported to be triggered to release 1 by palladium-functionalized resins which could be designed as nanodevices with cell-targeting capabilities.18
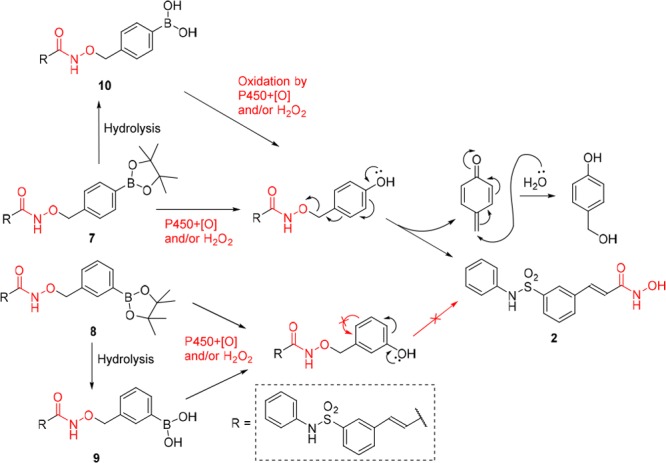
It has long been observed that cancer cells often have elevated levels of hydrogen peroxide compared to noncancerous cells,19−22 and the boronate and boronic acid molecules can be efficiently oxidized into the phenols by hydrogen peroxide.23−25 Thus, a strategy was employed to design drug conjugates with a p-boronate benzyl moiety which can undergo a selective H2O2-triggered self-immolation release and the formation of byproduct quinone methide,26−30 which was shown to be a cytotoxic agent in its own right.31−33 Our previous studies also found that the introduction of boron significantly enhanced the bioavailability of drugs.34−38 More importantly, we found that oxidative cleavage of the boron–carbon bond occurred metabolically without requiring an elevated level of H2O2.34−38 The unique biocompatibility of boronic acid was further illustrated in a report by Chang et al. where a small–molecule imaging agent attached to an aryl boronic acid moiety was able to travel throughout the body of living mice to reach deep tumor tissues.39 Therefore, to address the challenge that HDACi drugs have not proven to be efficacious against solid tumors in clinical trials even with high dosages reaching 1000 mg/m2/d as monotherapy40,41 or combination agent,42 we introduced boronic acid or their ester moiety to the scaffold of hydroxamate-based HDACi. For this prodrug strategy to work, the benzyl boronate moiety needs to be sufficiently labile under physiological conditions to allow conversion to belinostat once in vivo. We envision that this can be achieved by either metabolic oxidation or exposure to potentially higher level of H2O2 in tumor tissues, or both. Thus, as illustrated in Figure 2, new conjugates 7 and 8 were developed by connecting a benzyl group with boronate to the hydroxyl of 2 to be potent HDAC inhibitors with better pharmacokinetic properties.
Figure 2.
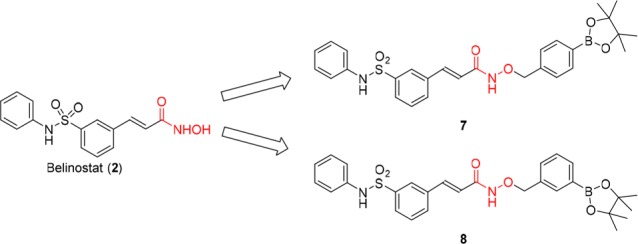
Design of boron-containing prodrugs (7 and 8) of belinostat.
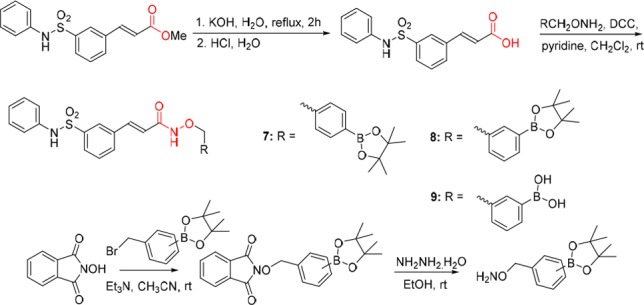
As shown in Scheme 1, following the hydrolysis of methyl (E)-3-(3-(N-phenylsulfamoyl)phenyl)acrylate, the designed boron-containing prodrugs 7 and 8 were prepared by N,N′-dicyclohexylcarbodiimide (DCC)-induced condensation of (E)-3-(3-(N-phenylsulfamoyl)phenyl)acrylic acid with O-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl)hydroxyl amine or O-(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl)hydroxylamine which was obtained from N-hydroxyphthalimide in two steps following the procedure of reference (43). Prodrug 9 was obtained as a minor product during the purification of 8 by flash chromatography. It was noted that 8 tends to be more easily converted to its free boronic acid form 9 than 7 to 10 (Scheme 2).
Scheme 1. Synthesis of Boron-Containing Prodrugs 7–9.
Scheme 2. Proposed Oxidative Release of Active Belinostat from Boron-Containing Prodrug 7.
We first evaluated the antiproliferative activity of prodrugs 7–9 against human breast adenocarcinoma MDA-MB-231, human lung carcinoma A549, and human cervical cancer HeLa cell lines. The results in Table 1 show that prodrug 7 with IC50 values of 0.303, 0.453, and 0.273 μM is 3–6 times less potent than 2 against these three cell lines, respectively. Interestingly, prodrug 8 appears to be significantly weaker than 7 (7–30 fold), with IC50 values ranging from 2.03 to 8.32 μM in inhibiting the same cancer cell lines. The boronic ester prodrug 8 and its free boronic acid 9 have comparable antiproliferation activities in the same cytotoxicity assays. Further in vitro assays reveal that 7 and vorinostat (1) also have similar activities against human melanoma SK-MEL-28, human lung carcinoma NCI-H460, and MDA-MB-231 cells. The activity (IC50 of 1.46 μM) of 7 against human breast adenocarcinoma MCF-7 is 15 times less potent than that of 2 with an IC50 of 0.096 μM. Both prodrug 7 and 2 have low toxicity toward normal mammary epithelial cells (MCF-10A) at 10 μM (Figure S1).
Table 1. Antiproliferation Activities of Boron-Containing Prodrugs 7–9 against Various Human Cancer Cell Linesa.
| IC50 (μM) |
||||||
|---|---|---|---|---|---|---|
| compd | MDA-MB-231 | A549 | HeLa | MCF-7 | SK-MEL-28 | NCI-H460 |
| 1 | 0.201 ± 0.116 | nd | nd | nd | 0.482 ± 0.064 | 0.425 ± 0.033 |
| 2 | 0.062 ± 0.006 | 0.077 ± 0.002 | 0.087 ± 0.009 | 0.096 ± 0.016 | nd | nd |
| 7 | 0.303 ± 0.024 | 0.453 ± 0.020 | 0.273 ± 0.043 | 1.46 ± 0.68 | 0.678 ± 0.039 | 0.430 ± 0.088 |
| 8 | 2.03 ± 0.54 | 3.60 ± 0.84 | 8.32 ± 2.27 | nd | nd | nd |
| 9 | 2.20 ± 1.39 | 6.58 ± 1.44 | 12.90 ± 2.20 | nd | nd | nd |
nd: not determined. ±: standard deviation (SD) of triplicate experiments.
The marked differences in cytotoxicity between prodrug 7 and prodrugs 8 or 9 could be understood in light of the transformations depicted in Scheme 2. Prodrug 7 which possesses a para-substituted aryl boronic ester connected to the hydroxyl position of 2 releases active 2 upon oxidation either by P450 enzymes or by hydrogen peroxide. However, the release of 2 from prodrugs 8 and 9 was blocked in step 2.
To confirm if the prodrugs undergo release of belinostat 2, we analyzed the cell culture media for concentrations of the prodrugs and the active form 2 in MDA-MB-231 and HeLa cells. The results show that pinacol ester prodrugs 7 and 8 rapidly hydrolyzed to their boronic acid forms 10 and 9 (Scheme 2). Using HPLC coupled to an Orbitrap mass spectrometer, we were able to separate, identify, and quantify 10, 9, and the active form 2. As shown in Table 2, the concentrations of 2 and 10 were measured at 30.7 and 331.8 ng/mL, respectively, in the culture media after 1 day incubation of prodrug 7 with MDA-MB-231 cells, indicating that 7 has been completely converted to 10 and partially to 2. From day 1 to day 6, the concentration of 10 decreased gradually from 331.8 to 130.9 ng/mL, while the concentration of 2 remained nearly constant. Similar results were observed in HeLa cells where the concentrations of 2 and 10 were measured at 8.4 and 282.7 ng/mL in the media after 1 day incubation, 3.8 and 49.6 ng/mL on day 6, respectively. Incubation of prodrug 8 with MDA-MB-231 cells did not yield detectable 2 while the concentrations of 9 were measured at 359.1 and 155.8 ng/mL after 1 day and 6 days of incubation with MDA-MB-231 cells, respectively. Similar results were observed when HeLa cells were treated with prodrug 8, where 303.2 and 66.6 ng/mL of 9 were recovered after 1 day and 6 days separately without the peak of 2. These results confirmed that prodrug 7 could be partially transformed to active 2, but prodrug 8 could not release 2 (Scheme 2), which may be the reason why prodrugs 7–9 displayed significant differences in their cytotoxicity against cancer cell lines when compared with 2 (Table 1). Thus, the relative in vitro cytotoxic potency is observed to follow the order belinostat 2 > prodrug 7 ≫ prodrug 8 ∼ prodrug 9.
Table 2. Concentration (ng/mL) of 2, 9, and 10 in the Culture Media after Incubation of 7 and 8 with Cancer Cell Linesa.
| prodrug 7 |
prodrug 8 |
|||||||
|---|---|---|---|---|---|---|---|---|
| MDA-MB-231 |
HeLa |
MDA-MB-231 |
HeLa |
|||||
| day | 2 | 10 | 2 | 10 | 2 | 9 | 2 | 9 |
| 1 | 30.7 ± 0.4 | 331.8 ± 4.5 | 8.4 ± 0.1 | 282.7 ± 14.2 | NF | 359.1 ± 13.7 | NF | 303.2 ± 24.1 |
| 2 | 27.7 ± 1.3 | 268.5 ± 2.2 | 6.6 ± 0.1 | 147.4 ± 3.3 | NF | 262.9 ± 10.9 | NF | 188.6 ± 3.9 |
| 3 | 20.3 ± 0.5 | 246.9 ± 11.2 | 13.4 ± 0.5 | 144.9 ± 0.8 | NF | 199.0 ± 16.1 | NF | 147.9 ± 8.3 |
| 4 | 19.8 ± 0.9 | 171.8 ± 3.8 | 7.8 ± 0.1 | 107.0 ± 0.7 | NF | 198.8 ± 13.5 | NF | 134.7 ± 3.1 |
| 5 | 28.5 ± 1.0 | 145.8 ± 3.2 | 3.7 ± 0.2 | 82.7 ± 3.6 | NF | 168.5 ± 3.8 | NF | 97.2 ± 3.8 |
| 6 | 27.8 ± 0.5 | 130.9 ± 5.4 | 3.8 ± 0.2 | 49.6 ± 0.9 | NF | 155.8 ± 6.5 | NF | 66.6 ± 1.3 |
NF: no belinostat found. ±: standard error (SEM) of triplicate experiments.
To elucidate the anticancer mechanism of prodrug 7, we investigated the histone deacetylase (HDAC) inhibitory activity of 7 with the histone deacetylase activity assay kit (Fluorometric) ab156064 (Abcam, Cambridge, UK). As shown in Table 3, prodrug 7 displays an EC50 value of 0.35 μM after 20 min incubation following the protocol for the assay kit, which is higher than the EC50 values of vorinostat 1 (0.18 μM) and belinostat 2 (0.031 μM). The difference in EC50 values among prodrug 7, 1, and 2 is consistent with their difference in IC50 values against cancer cell lines (Table 1), which suggests that the cytotoxicity of prodrug 7 may also be related to its HDAC inhibitory activity. These results are expected by design in that release of the active drug 2 from the prodrug 7 is only partial in in vitro systems, hence the reduced potency of 7. The inhibitory activities of prodrug 7 against HDAC isoforms 1–11 were also determined, where 7 inhibited HDAC6 and HDAC8 with EC50 of 20.2 and 341 μM, respectively (Table S1).
Table 3. HDAC Inhibitory Activity of Prodrug 7.
| compd | EC50 (μM) |
|---|---|
| 1 (vorinostat) | 0.18 |
| 2 (belinostat) | 0.03 |
| 7 | 0.35 |
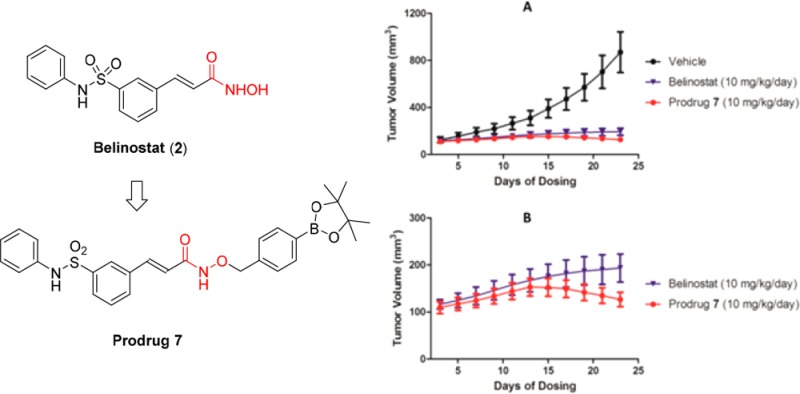
We next investigated the in vivo efficacy of prodrug 7 in mice. A head-to-head comparative study with a dosage of 10 mg/kg/day of 7 and belinostat 2 by subcutaneous injection in an MCF-7 xenograft model in mice was designed to test the tumor inhibitory efficacy of the two compounds. Figure 3A indicates that both 2 and 7 have potent inhibition activity against the growth of tumor compared with rapid increase in tumor volume in the vehicle group. In the enlarged view of tumor growth curves displayed in Figure 3B, the difference in tumor growth between the treatment groups of 7 and 2 becomes clear after 2 weeks’ dosing at 10 mg/kg. Prodrug 7 treatment not only inhibited the growth of tumor but also resulted in tumor remission in this MCF-7 xenograft model. In the belinostat treatment group, average tumor volume continued its slow increase from 167 mm3 on day 12 to 194 mm3 on day 22, whereas tumor volume in mice treated with 7 decreased from 153 mm3 to 127 mm3 in the same period of time. Moreover, due to its heavier molecular weight, the molar concentration of 7 is lower than 2 given at the same dosage in mg/kg, which adds to the observed efficacy of 7. Taken together, prodrug 7 afforded a significantly greater efficacy than 2 in the in vivo assay, with 85.4% and 77.7% inhibition of tumor growth (TGI) and 14.6% and 22.3% tumor volume ratio (T/C), respectively. Importantly, in all of the in vitro assays, prodrug 7 demonstrated a consistently lower potency than 2 against a panel of cancer cell lines. This is dramatically reversed once the prodrug enters an in vivo system where it showed a greater efficacy than 2. Statistical analysis by t test shows that the tumor weights of the 7-treated group are significantly different from those of the 2-treated group with p-value of 0.0045 (<0.01) (Table S2). However, compared with the vehicle group, the body weights of both 7-treated and 2-treated mice were unaffected.
Figure 3.
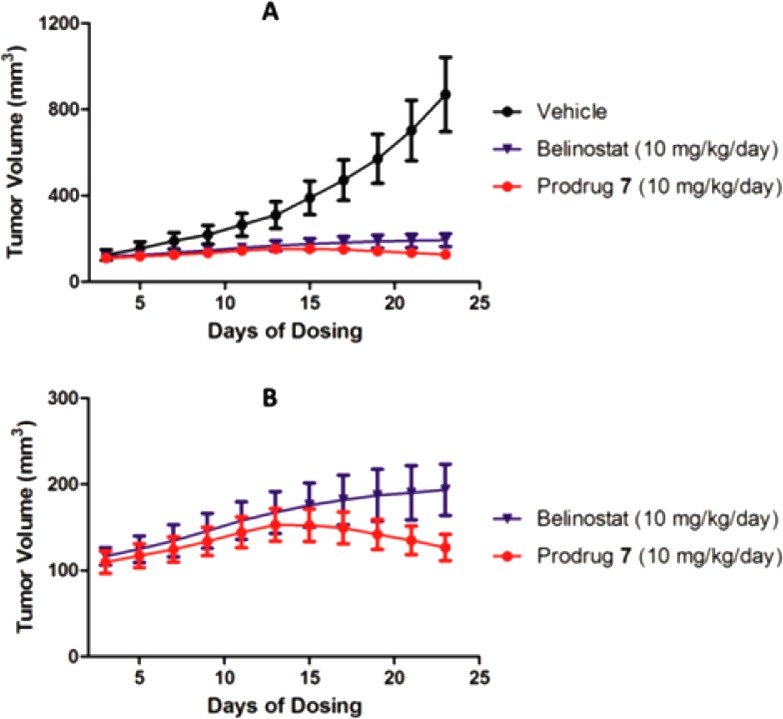
Inhibition of tumor growth in an MCF-7 tumor xenograft model in mice. (A) Tumor volumes of the vehicle, belinostat, or prodrug 7-treated groups by subcutaneous injection. (B) The enlarged section of tumor volumes of belinostat and prodrug 7-treated groups.
To better understand the in vivo efficacy demonstrated by prodrug 7, we sought to measure the drug concentrations in tumor tissues after 22 days of treatment with 2 or 7. Mice were sacrificed and tumors were surgically removed, homogenized, and extracted for HPLC–MS/MS analysis for 2 and 10 (the corresponding free boronic acid of 7). As shown in Table 4, in the 2-treated group, the mean concentration of 2 in tumor tissues was 23.4 ng/g, while in the 7-treated group, 2 was present at the concentration of 57.0 ng/g, a level over 2-fold higher than that in belinostat treated tumors. Moreover, the metabolic product of 7, the free boronic acid form (10) was found at the concentration of 51.0 ng/g in tumor tissue. As a precursor of 2, the boronic acid 10 may act as a reservoir for release of additional active ingredient (2) in a prolonged time-course, thereby further increase the drug exposure to the target tumor tissues. In addition, it should be noted that, at equal weight dosage (10 mg/kg), prodrug 7 was administered at about half of the molar dosage compared to 2, yet the 7-treated group has a higher concentration of 2 in tumor tissues than the belinostat treated group. These results confirmed that prodrug 7 has better bioavailability than belinostat 2 in an MCF-7 tumor xenograft mode, as demonstrated by its higher exposure to the target tumor tissues than achieved by belinostat. Therefore, the dramatic reversal in the in vivo efficacy of 7 could be attributed to its superior biocompatibility with the active target site.
Table 4. Average Terminal Tumor Volume, Weight, And Tissue Concentration of Drugs after 22 Days of Treatmenta.
| conc
in tumor tissue (ng/g) |
||||||
|---|---|---|---|---|---|---|
| groups | avg tumor volume (mm3) | avg tumor weight (mg) | T/C (%) | TGI (%) | 2 | 10 |
| vehicle | 869 ± 173 | 743 ± 142 | NA | NA | NA | NA |
| 2 | 194 ± 30 | 167 ± 24 | 22.3 | 77.7 | 23.4 ± 1.6 | NA |
| 7 | 127 ± 15 | 109 ± 12 | 14.6 | 85.4 | 57.0 ± 15.2 | 51.0 ± 42.0 |
| tumor
size (mm3) of each mouse on last day of measurement | ||||
|---|---|---|---|---|
| mice | 1 | 2 | 3 | 4 |
| 2 | 222.2 | 216.3 | 171.9 | 164.3 |
| 7 | 138.6 | 110.6 | 140.8 | 116.6 |
NA: not available. ±: standard deviation (SD) of four mice.
In summary, two boron-containing prodrugs 7 and 8 of belinostat were designed and synthesized for the purpose of enhancing the therapeutic efficacy of the HDAC inhibitor against solid tumors such as metastatic breast cancer. The large difference in the antiproliferative activities of 7 and 8 may be attributed to the blockage of the transformation of 8 to the active form, belinostat (2). Prodrug 7 demonstrates a slightly lower potency than 2 against a panel of cancer cell lines. However, when administered to tumor-bearing mice, prodrug 7 showed significantly greater efficacy than 2 in the MCF-7 xenograft model where 7 not only inhibited tumor growth but also reduced tumor volumes after 3 weeks of treatment. This marked difference between in vitro and in vivo activities of the prodrug 7 is consistent with the observation that the conversion of 7 to its active form of belinostat is incomplete in cell models. However, once in vivo, the superior bioavailability of prodrug 7 enabled the release of a significantly higher systemic level of belinostat than could be achieved by the direct administration of belinostat, thereby exerting a greater in vivo efficacy. Data of drug distribution in tumor tissues support this rationale. Taken together, our study suggests that prodrug 7 of belinostat may have promising clinical potential in the treatment of solid tumors.
Acknowledgments
This work was supported by NIH-NIMHD through Grant No. 2G12MD007595 and in part by Louisiana Cancer Research Consortium.
Glossary
Abbreviations
- HDACi
histone deacetylase inhibitor
- SAHA
vorinostat
- CTCL
cutaneous T-cell lymphoma
- MTD
maximum tolerated dose
- DCC
N,N′-dicyclohexylcarbodiimide
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00504.
Experimental protocols and characterization data (PDF)
Author Contributions
S.Z., J.L., and G.W. conceived the project and designed the experiments. S.Z. synthesized the compounds. Q.Z. (Zhong) performed in vitro cell assay. S.G. and L.Y. performed HDAC activity assay and in vivo efficacy assay and PK study. C.Z. analyzed the samples of in vitro and in vivo assays. Q.Z. (Zhang) performed HRMS analysis. S.Z., J.L., and G.W. analyzed the data and wrote the manuscript.
The authors declare the following competing financial interest(s): The authors declare that a patent application was filed on compound 7.
Supplementary Material
References
- Glozak M. A.; Seto E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- Ropero S.; Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. 10.1016/j.molonc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckschlager T.; Plch J.; Stiborova M.; Hrabeta J. histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maolanon A. R.; Madsen A. S.; Olsen C. A. Innovative strategies for selective inhibition of histone deacetylases. Cell Chem. Bio. 2016, 23, 759–768. 10.1016/j.chembiol.2016.06.011. [DOI] [PubMed] [Google Scholar]
- Bolden J. E.; Peart M. J.; Johnstone R. W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discovery 2006, 5, 769–784. 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Mottamal M.; Zheng S.; Huang T. L.; Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P.; Xu W. S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T. A.; Witter D. J.; Belvedere S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5097–5116. 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M.; Clarke C.; Marks P. A. Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- Rubin E. H.; Agrawal N. G.; Friedman E. J.; Scott P.; Mazina K. E.; Sun L.; Du L.; Ricker J. L.; Frankel S. R.; Gottesdiener K. M. A study to determine the effects of food and multiple dosing on the pharmacokinetics of vorinostat given orally to patients with advanced cancer. Clin. Cancer Res. 2006, 12, 7039–7045. 10.1158/1078-0432.CCR-06-1802. [DOI] [PubMed] [Google Scholar]
- Du L.; Musson D. G.; Wang A. Q. Stability studies of vorinostat and its two metabolites in human plasma, serum and urine. J. Pharm. Biomed. Anal. 2006, 42, 556–564. 10.1016/j.jpba.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Mulder G. J.; Meerman J. Sulfation and glucuronidation as competing pathways in the metabolism of hydroxamic acids: the role of N, O-sulfonation in chemical carcinogenesis of aromatic amines. Environ. Health Persp. 1983, 49, 27. 10.1289/ehp.834927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flipo M.; Charton J.; Hocine A.; Dassonneville S.; Deprez B.; Deprez-Poulain R. Hydroxamates: relationships between structure and plasma stability. J. Med. Chem. 2009, 52, 6790–6802. 10.1021/jm900648x. [DOI] [PubMed] [Google Scholar]
- Sanderson L.; Taylor G. W.; Aboagye E. O.; Alao J. P.; Latigo J. R.; Coombes R. C.; Vigushin D. M. Plasma pharmacokinetics and metabolism of the histone deacetylase inhibitor trichostatin a after intraperitoneal administration to mice. Drug Metab. Dispos. 2004, 32, 1132–1138. 10.1124/dmd.104.000638. [DOI] [PubMed] [Google Scholar]
- Miller T. A.; Witter D. J.; Belvedere S.. Histone deacetylase inhibitor prodrugs. US8227636B2, 2012.
- Schlimme S.; Hauser A. T.; Carafa V.; Heinke R.; Kannan S.; Stolfa D. A.; Cellamare S.; Carotti A.; Altucci L.; Jung M. Carbamate prodrug concept for hydroxamate HDAC inhibitors. ChemMedChem 2011, 6, 1193–1198. 10.1002/cmdc.201100007. [DOI] [PubMed] [Google Scholar]
- Daniel K. B.; Sullivan E. D.; Chen Y.; Chan J. C.; Jennings P. A.; Fierke C. A.; Cohen S. M. Dual-Mode HDAC Prodrug for Covalent Modification and Subsequent Inhibitor Release. J. Med. Chem. 2015, 58, 4812–4821. 10.1021/acs.jmedchem.5b00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Ruiz B.; Weiss J. T.; Unciti-Broceta A. Efficient palladium-triggered release of vorinostat from a bioorthogonal precursor. J. Med. Chem. 2016, 59, 9974–9980. 10.1021/acs.jmedchem.6b01426. [DOI] [PubMed] [Google Scholar]
- Szatrowski T. P.; Nathan C. F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [PubMed] [Google Scholar]
- Trachootham D.; Alexandre J.; Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach?. Nat. Rev. Drug Discovery 2009, 8, 579–591. 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- Zieba M.; Suwalski M.; Kwiatkowska S.; Piasecka G.; Grzelewska-Rzymowska I.; Stolarek R.; Nowak D. Comparison of hydrogen peroxide generation and the content of lipid peroxidation products in lung cancer tissue and pulmonary parenchyma. Resp. Med. 2000, 94, 800–805. 10.1053/rmed.2000.0825. [DOI] [PubMed] [Google Scholar]
- Lim S. D.; Sun C.; Lambeth J. D.; Marshall F.; Amin M.; Chung L.; Petros J. A.; Arnold R. S. Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 2005, 62, 200–207. 10.1002/pros.20137. [DOI] [PubMed] [Google Scholar]
- Kuivila H. G.; Armour A. G. Electrophilic displacement reactions. IX. Effects of substituents on rates of reactions between hydrogen peroxide and benzeneboronic Acid1–3. J. Am. Chem. Soc. 1957, 79, 5659–5662. 10.1021/ja01578a020. [DOI] [Google Scholar]
- Lippert A. R.; Van de Bittner G. C.; Chang C. J. Boronate oxidation as a bioorthogonal reaction approach for studying the chemistry of hydrogen peroxide in living systems. Acc. Chem. Res. 2011, 44, 793–804. 10.1021/ar200126t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q.; Zhong Q.; Zhang Q.; Zheng S.; Wang G. Boron-based 4-Hydroxytamoxifen bioisosteres for treatment of de Novo Tamoxifen resistant breast cancer. ACS Med. Chem. Lett. 2012, 3, 392–396. 10.1021/ml3000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major Jourden J. L.; Cohen S. M. Hydrogen peroxide activated matrix metalloproteinase inhibitors: a prodrug approach. Angew. Chem., Int. Ed. 2010, 49, 6795–6797. 10.1002/anie.201003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broaders K. E.; Grandhe S.; Fréchet J. M. A biocompatible oxidation-triggered carrier polymer with potential in therapeutics. J. Am. Chem. Soc. 2011, 133, 756–758. 10.1021/ja110468v. [DOI] [PubMed] [Google Scholar]
- Kuang Y.; Balakrishnan K.; Gandhi V.; Peng X. Hydrogen peroxide inducible DNA cross-linking agents: targeted anticancer prodrugs. J. Am. Chem. Soc. 2011, 133, 19278–19281. 10.1021/ja2073824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen H.; Marzenell P.; Jentzsch E.; Wenz F.; Veldwijk M. R.; Mokhir A. Aminoferrocene-based prodrugs activated by reactive oxygen species. J. Med. Chem. 2012, 55, 924–934. 10.1021/jm2014937. [DOI] [PubMed] [Google Scholar]
- Wang M.; Sun S.; Neufeld C. I.; Perez-Ramirez B.; Xu Q. Reactive Oxygen Species-Responsive Protein Modification and Its Intracellular Delivery for Targeted Cancer Therapy. Angew. Chem., Int. Ed. 2014, 53, 13444–13448. 10.1002/anie.201407234. [DOI] [PubMed] [Google Scholar]
- Hulsman N.; Medema J. P.; Bos C.; Jongejan A.; Leurs R.; Smit M. J.; de Esch I. J.; Richel D.; Wijtmans M. Chemical insights in the concept of hybrid drugs: the antitumor effect of nitric oxide-donating aspirin involves a quinone methide but not nitric oxide nor aspirin. J. Med. Chem. 2007, 50, 2424–2431. 10.1021/jm061371e. [DOI] [PubMed] [Google Scholar]
- Dufrasne F.; Gelbcke M.; Neve J.; Kiss R.; Kraus J.-L. Quinone methides and their prodrugs: A subtle equilibrium between cancer promotion, prevention, and cure. Curr. Med. Chem. 2011, 18, 3995–4011. 10.2174/092986711796957301. [DOI] [PubMed] [Google Scholar]
- Noh J.; Kwon B.; Han E.; Park M.; Yang W.; Cho W.; Yoo W.; Khang G.; Lee D. Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nat. Commun. 2015, 6, 6907. 10.1038/ncomms7907. [DOI] [PubMed] [Google Scholar]
- Zhong Q.; Zhang C.; Zhang Q.; Miele L.; Zheng S.; Wang G. Boronic prodrug of 4-hydroxytamoxifen is more efficacious than tamoxifen with enhanced bioavailability independent of CYP2D6 status. BMC Cancer 2015, 15, 625. 10.1186/s12885-015-1621-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Zhong Q.; Zhang Q.; Zheng S.; Miele L.; Wang G. Boronic prodrug of endoxifen as an effective hormone therapy for breast cancer. Breast Cancer Res. Treat. 2015, 152, 283–291. 10.1007/s10549-015-3461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Zheng S.; Akerstrom V. L.; Yuan C.; Ma Y.; Zhong Q.; Zhang C.; Zhang Q.; Guo S.; Ma P.; Skripnikova E. V.; Bratton M. R.; Pannuti A.; Miele L.; Wiese T. E.; Wang G. Fulvestrant-3 boronic acid (ZB716): an orally bioavailable selective estrogen receptor downregulator (SERD). J. Med. Chem. 2016, 59, 8134–8140. 10.1021/acs.jmedchem.6b00753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Zheng S.; Guo S.; Zhang C.; Zhong Q.; Zhang Q.; Ma P.; Skripnikova E. V.; Bratton M. R.; Wiese T. E.; Wang G. Rational Design of a Boron-Modified Triphenylethylene (GLL398) as an Oral Selective Estrogen Receptor Downregulator. ACS Med. Chem. Lett. 2017, 8, 102–106. 10.1021/acsmedchemlett.6b00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Guo S.; Yang L.; Liu J.; Zheng S.; Zhong Q.; Zhang Q.; Wang G. metabolism, pharmacokinetics, and bioavailability of zb716, a steroidal selective estrogen receptor downregulator (SERD). Oncotarget 2017, 8, 103874–103889. 10.18632/oncotarget.21808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Bittner G. C.; Dubikovskaya E. A.; Bertozzi C. R.; Chang C. J. In vivo imaging of hydrogen peroxide production in a murine tumor model with a chemoselective bioluminescent reporter. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 21316–21321. 10.1073/pnas.1012864107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele N. L.; Plumb J. A.; Vidal L.; Tjørnelund J.; Knoblauch P.; Rasmussen A.; Ooi C. E.; Buhl-Jensen P.; Brown R.; Evans T. R. J.; DeBono J. S. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 804–810. 10.1158/1078-0432.CCR-07-1786. [DOI] [PubMed] [Google Scholar]
- Gimsing P.; Hansen M.; Knudsen L. M.; Knoblauch P.; Christensen I. J.; Ooi C. E.; Buhl-Jensen P. A phase I clinical trial of the histone deacetylase inhibitor belinostat in patients with advanced hematological neoplasia. Eur. J. Haematol. 2008, 81, 170–176. 10.1111/j.1600-0609.2008.01102.x. [DOI] [PubMed] [Google Scholar]
- Vitfell-Rasmussen J.; Judson I.; Safwat A.; Jones R. L.; Rossen P. B.; Lind-Hansen M.; Knoblauch P.; Krarup-Hansen A. A Phase I/II Clinical Trial of Belinostat (PXD101) in combination with doxorubicin in patients with soft tissue sarcomas. Sarcoma 2016, 2016, 1–9. 10.1155/2016/2090271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.-Z.; Xu H.; Liu T.-W.; Feng Q.; Yu S.-J.; Wang S.-H.; Li Z.-M. Design, synthesis and antifungal activities of novel pyrrole alkaloid analogs. Eur. J. Med. Chem. 2011, 46, 1463–1472. 10.1016/j.ejmech.2011.01.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.