

Abstract

Acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) plays an important role in triglyceride synthesis and is a target of interest for the treatment of metabolic disorders. Herein we describe the structure–activity relationship of a novel tetralone series of DGAT1 inhibitors and our strategies for overcoming genotoxic liability of the anilines embedded in the chemical structures, leading to the discovery of a candidate compound, (S)-2-(6-(5-(3-(3,4-difluorophenyl)ureido)pyrazin-2-yl)-1-oxo-2-(2,2,2-trifluoroethyl)-1,2,3,4-tetrahydronaphthalen-2-yl)acetic acid (GSK2973980A, 26d). Compound 26d is a potent and selective DGAT1 inhibitor with excellent DMPK profiles and in vivo efficacy in a postprandial lipid excursion model in mice. Based on the overall biological and developability profiles and acceptable safety profiles in the 7-day toxicity studies in rats and dogs, compound 26d was selected as a candidate compound for further development in the treatment of metabolic disorders.
Keywords: DGAT1 inhibitors, tetralones, GSK2973980A, acyltransferase, metabolic disorders, aniline, genotoxicity, mutagenicity, Ames
Synthesis of triglycerides (TG) is a fundamental biochemical pathway important for nutrient utilization and energy storage. Excess TG has been linked to human diseases such as obesity, insulin resistance, dyslipidemia, and nonalcoholic steatohepatitis (NASH). Acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) catalyzes the final step in the TG biosynthetic pathway. Mice lacking DGAT1 are viable and show reduced plasma and tissue TG levels associated with increased sensitivity to insulin and leptin, and increased expression of leptin-regulated genes compared to wild type mice.1 DGAT1 deficiency protected against hepatic steatosis by reducing TG synthesis and increasing fatty acid oxidation in adipose and skeletal muscle.2,3 Furthermore, DGAT1 deficient mice are resistant to diet-induced obesity.4 Since DGAT1 is an attractive therapeutic target for the treatment of metabolic disorders, there has been intense interest over recent years in identifying DGAT1 inhibitors.5−7
Representative DGAT1 inhibitors (LCQ908,8,9 PF-04620110,10 and AZD768711,12), which have been evaluated in clinical trials, are shown in Figure 1. All three compounds possessed a phenyl cyclohexyl acetic acid functionality that is a privileged scaffold for DGAT1 inhibitors. Pradigastat (LCQ908) is the most advanced DGAT1 inhibitor and is currently in Phase III evaluation for the treatment of familial chylomicronemia syndrome.8,9 Herein, we report our efforts that led to the discovery of a candidate compound, GSK2973980A (26d), a novel, potent, and highly selective DGAT1 inhibitor, as a potential therapeutic agent for the treatment of metabolic disorders.
Figure 1.

Structures of representative DGAT1 inhibitors.
Our initial discovery effort was focused on tetrahydronaphthalene and a tetralone series of DGAT1 inhibitors (Figure 2).13 Tetrahydronapthalene (compound A) and tetralone (compound B) emerged as interesting initial hits, which showed IC50 values of 5.1 and 2.8 μM, respectively, in the human DGAT1 (hDGAT1) fluorescence-based biochemical assay (CPM assay).
Figure 2.

Tetrahydronaphthalene (compound A) and tetralone (compound B) as DGAT1 hits.
Limited structure–activity relationship (SAR) explorations suggested that meta-substitution on ring D improved potency in both compounds A and B and that tetralones tend to have better potency than tetrahydronaphthalenes. Therefore, tetralone (compound B) was selected for lead optimization. There were two perceived challenges associated with the monosubstituted tetralone AB ring system: (1) the presence of an enolizable ketone could result in racemization, and (2) the ketone functionality could be labile and subjected to nucleophilic attack. We envisioned resolving these issues by introducing a quaternary center at the α-position of ketone in the AB ring with the goal of improving stability of the compound. In addition, we explored the SAR of the ring D substitutions with the goal of improving potency and pharmacokinetic (PK) profiles of DGAT1 inhibitors.
Tetralone derivatives were synthesized using standard conditions as described in Scheme 1. Treatment of 6-methoxytetralone 1 with glyoxalic acid in diglyme followed by sulfuric acid under heating conditions provided olefin 2. Reduction of olefin 2 under Zn/AcOH conditions followed by esterification resulted in ester 4. Alkylation of compound 4 followed by demethylation and subsequent ester hydrolysis afforded acid 6. Re-esterification of acid 6 followed by conversion of the phenol to its corresponding triflate led to the triflate 8. Suzuki coupling of triflate 8 with p-nitrophenyl boronic acid gave nitro compound 9, which was reduced to its corresponding aniline 10. Aniline 10 was then converted to urea 11 upon treatment with an appropriately substituted phenyl isocyanate. Finally, hydrolysis of esters of urea 11 with LiOH provided the corresponding tetralone derivatives 12.
Scheme 1. General Synthesis of Tetralone Derivatives.

Reagents and conditions: (a) glyoxalic acid, H2SO4, diglyme; (b) Zn, AcOH; (c) MeSO3H, EtOH; (d) alkyl halide, NaH, DMF; (e) aq. HBr; (f) CH3SO3H, EtOH; (g) Tf2O, CH2Cl2, Et3N; (h) 4-nitrophenyl boronic acid, Pd(PPh3)4, Cs2CO3, dioxane-H2O; (i) Fe-NH4Cl, EtOH-H2O; (j) R1PhNCO, Et3N, THF; (k) LiOH, THF-H2O.
As shown in Table 1, we evaluated both the steric and electronic effects at the α-position of the tetralones as racemic mixtures. Interestingly, electron donating groups such as methyl (12o), ethyl (12p), and n-propyl (12q) substitutions showed significant improvement in enzyme potency, while electron withdrawing fluoro group (12r) was less active. Interestingly trifluoroethyl substitution (12s) resulted in a very potent compound. We selected methyl and ethyl substitutions at the α-position as preferred substituents for further optimization because of lower lipophilicity (clogP) compared to n-propyl substitution and lower molecular weight (MW) compared to trifluoroethyl substitution. In addition, compounds 12o and 12p have reasonable solubility and acceptable potency. Further exploration of the D ring with these α-substituents is shown in Table 2.
Table 1. SAR of the α-Substituent of Tetralones 12o–s.
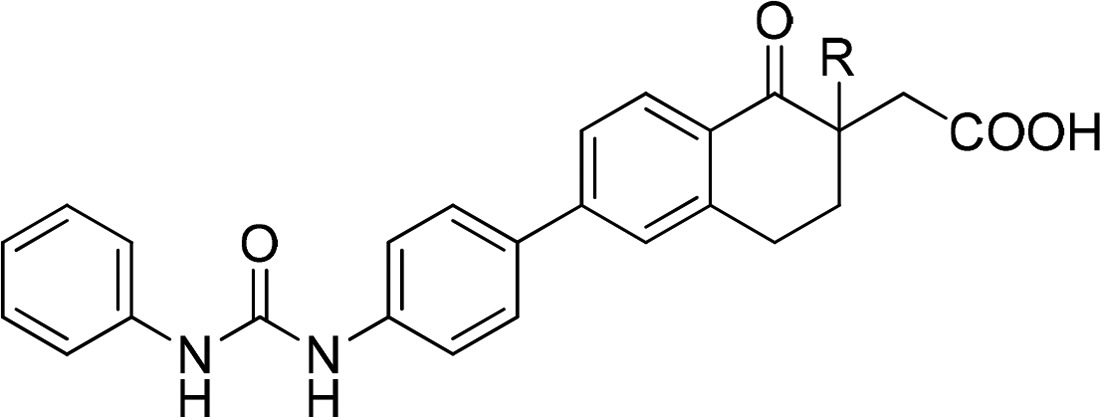
| compd # | R | MW | clogPa | hDGAT1 CPM IC50 (nM)b | solubility (μg/mL)b |
|---|---|---|---|---|---|
| B | H | 414.5 | 4.6 | 2820 | 58 |
| 12o | Me | 428.5 | 5.1 | 126 | 54 |
| 12p | Et | 442.5 | 5.6 | 63 | 34 |
| 12q | n-Pr | 456.5 | 6.2 | 16 | 74 |
| 12r | F | 432.5 | 4.8 | 84% @ 10 μM | 31 |
| 12s | CF3CH2 | 496.5 | 5.5 | 3 | 30 |
ClogP is a calculated LogP value using Daylight model.
Value is an average of at least n = 2.
Table 2. SAR of the D Ring Substituent of the Tetralones 12a–n.
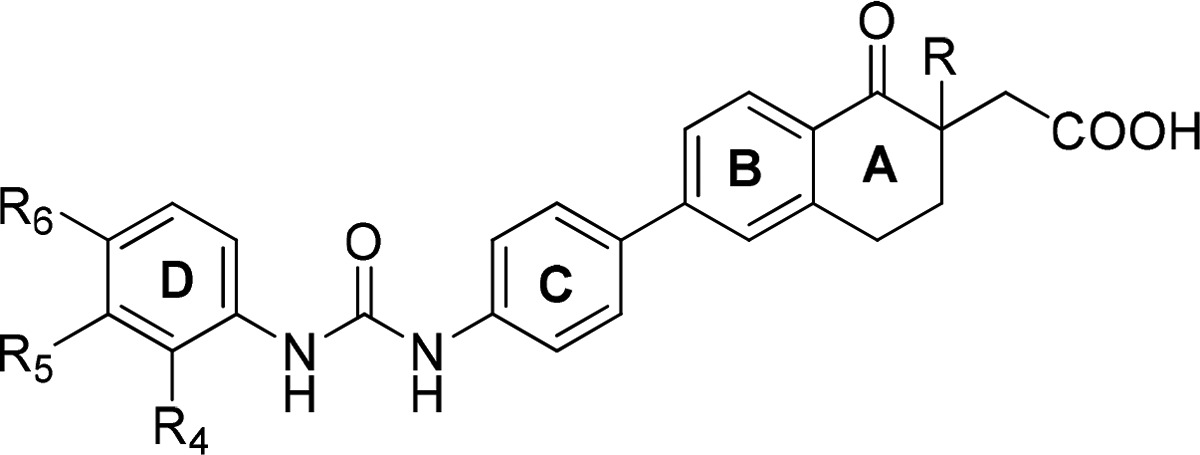
| compd # | R | R4 | R5 | R6 | hDGAT1 CPM IC50 (nM)a | solubility (μg/mL)a |
|---|---|---|---|---|---|---|
| 12a | Et | H | CF3 | H | 3 | 39 |
| 12b | Et | H | Me | H | 16 | 6 |
| 12c | Et | H | Cl | H | 6 | 101 |
| 12d | Et | H | OMe | H | 50 | 6 |
| 12e | Et | H | F | H | 32 | 38 |
| 12f | Et | H | H | CF3 | 20 | 46 |
| 12g | Et | H | H | Cl | 2 | 100 |
| 12h | Et | H | H | Me | 10 | 36 |
| 12i | Et | CF3 | H | H | 126 | 26 |
| 12j | Me | H | CF3 | H | 32 | 72 |
| 12k | Me | H | Me | H | 63 | 103 |
| 12l | Me | H | Cl | H | 16 | 75 |
| 12m | Me | H | OMe | H | 200 | 40 |
| 12n | Me | H | F | H | 63 | 13 |
Value is an average of at least n = 2.
We first explored the electronic effect of small substituents at the R5meta-position of the D ring, based on our prior knowledge, for both α-methyl and α-ethyl tetralones. α-Ethyl analogues (12a–e) were consistently more potent against hDGAT1 than the corresponding α-methyl analogues (12j–n) regardless of the electronic effect of the substituents. In addition, analogues with electron-withdrawing substituents (CF3 and Cl) showed significant improvement in potency over the electron-donating substituents (Me and −OMe) for the corresponding α-methyl analogues (12j and 12l vs 12k and 12m) and α-ethyl analogues (12a and 12c vs 12b and 12d). We also noted that para-substitution (12f–g) maintained similar hDGAT1 potency to the corresponding meta-substitution (12a–c); however, the ortho-substituted CF3 analogue (12i) showed significant reduction in hDGAT1 potency when compared to the corresponding para- (12f) and meta-substituted analogue (12a).
All the tetralone derivatives tested thus far in the biological assays were racemates. Therefore, in order to determine if there was any stereopreference, the most potent racemic analogues (12a and 12g) were chosen for separation. The R and S enantiomers of 12a and 12g were obtained by separation of the corresponding ethyl ester precursors 11a and 11g by chiral semiprep-HPLC followed by ester hydrolysis of esters (R-11a, S-11a, and R-11g, S-11g) to acids (R-12a, S-12a, and R-12g, S-12g). The absolute stereochemistry was determined and assigned by the ab initio vibrational circular dichroism (VCD) analysis of the corresponding ethyl ester precursors (R-11a, S-11a, and R-11g, S-11g).14 Biological activities of the racemates (12a and 12g) and their corresponding enantiomers in S and R configurations are shown in Table 3.
Table 3. DGAT1 Activities and Solubility of Racemates 12a and 12g and Their Corresponding Enantiomers (S-12a, R-12a, and S-12g, R-12g).
| compd # | hDGAT1 CPM IC50 (nM)a | hDGAT1 LE IC50 (nM)a | rDGAT1 LE IC50 (nM)a | C2C12 Inh. at 300 nM (%)a | solubility (μg/mL)a |
|---|---|---|---|---|---|
| 12a (Rac) | 3 | 8 | 158 | 62 | 39 |
| S-12a | 2.5 | 1 | 126 | 75 | 78 |
| R-12a | 126 | 251 | 631 | 27 | 88 |
| 12g (Rac) | 2 | NDb | NDb | 72 | 100 |
| S-12g | 2 | 8 | 158 | 83 | 5 |
| R-12g | 158 | NDb | 1260 | NDb | 0.4 |
Value is an average of at least n = 2.
ND = not determined.
Interestingly, the S enantiomers were approximately 50- to 80-fold more potent than their corresponding R enantiomers (Table 3). Since hDGAT1 potency measured by fluorescence-based CPM assay was similar for analogues S-12a and S-12g, we evaluated these compounds in a more sensitive radiometric LE assay in order to further discriminate between these two compounds. In hDGAT1 LE assay, S-12a was ∼8-fold more potent than S-12g. In addition, compounds were more potent against the human DGAT1 compared with the rat DGAT1 enzyme in the LE assays. Both S enantiomers showed similar inhibition in a cell-based assay using C2C12 mouse myoblast cells, and the data was consistent with the potency observed in the rat DGAT1 LE assay. Compound S-12a has better solubility compared to S-12g; therefore, S-12a was selected for further profiling. Compound S-12a showed >1000-fold selectivity over human DGAT2, ACAT1 (SOAT1), and ACAT2 (SOAT2) in the LE assay. Based on the overall potency, selectivity, and developability profiles, compound S-12a was selected for further profiling for DMPK properties and in vivo pharmacodynamic models. Table 4 summarized the PK properties of S-12a in mouse, rat, and dog. Compound S-12a was a low to moderate clearance compound with excellent oral bioavailability across species.
Table 4. Pharmacokinetics Properties of Compound S-12a in Male Mouse, Rat, and Dog (n = 3).
| species
(strain) |
|||
|---|---|---|---|
| parameter | mouse (C57BL/6J) | rat (Sprague–Dawley) | dog (Beagle) |
| dose IV/PO (mg/kg) | 2.2/2.7 | 1/2 | 1.8/5.3 |
| CLp (mL/min/kg) | 42.3 | 6.3 | 4.6 |
| VSS (L/kg) | 2.4 | 1.0 | 0.6 |
| IV T1/2 (h) | 2.6 | 3.9 | 4.6 |
| oral bioavailability (%F) | 54 | 87 | 149 |
To investigate the in vivo effects of DGAT1 inhibition, compound S-12a was evaluated in an acute postprandial lipid excursion model, which measured plasma triglycerides (TG) following a corn oil bolus challenge in male Swiss Albino mice (Figure 3).15 Overnight fasted animals were orally administered with various doses (0.03, 0.1, 0.3, 1, 3, and 10 mg/kg) of compound S-12a. Thirty minutes later, mice were challenged with oral bolus of corn oil (5 mL/kg), and the plasma TG was measured after 2.5 h following the corn oil challenge. As shown in Figure 3, compound S-12a was highly efficacious, resulting in statistically significant reduction in plasma TG levels in a dose-dependent manner with an estimated ED50 of 0.23 mg/kg. This inhibition occurred at low plasma concentration of compound S-12a (7.89 ng/mL at 3 h post-dose with 0.3 mg/kg, a dose closest to the estimated ED50 dose). This is likely due to relatively higher local unbound concentration in the gut compared to the plasma concentration, resulting in greater DGAT1 inhibition in enterocytes than what would be anticipated from the plasma concentration. PF-04620110 was used as a reference and showed 94% inhibition of TG levels at 0.3 mg/kg.
Figure 3.
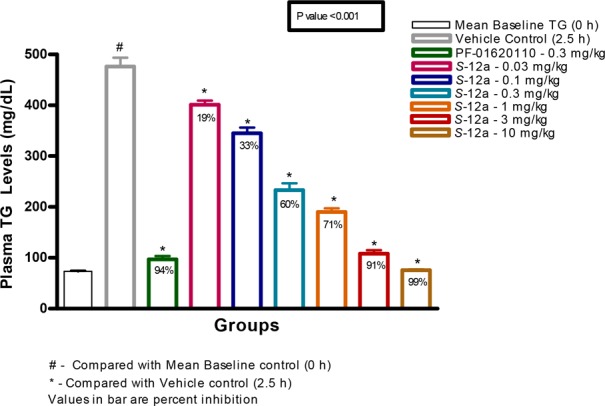
Dose response of compound S-12a and PF-01620110 in reducing TG following lipid challenge in mice.15
Since compound S-12a showed excellent selectivity, stability, and in vivo efficacy, additional selectivity profiling of compound S-12a was initiated. These included cardiac ion channels (hERG, Nav1.5, and Cav1.2) and a panel of 43 targeted pharmacologic receptors, ion channels, and transporter assays. Compound S-12a showed overall good selectivity profile (>500-fold over hDGAT1 CPM IC50) across panels of assays evaluated.
One concern about the scaffold is the potential hydrolysis or degradation of the urea functional group to the corresponding anilines, which could be mutagenic (Figure 4).16
Figure 4.
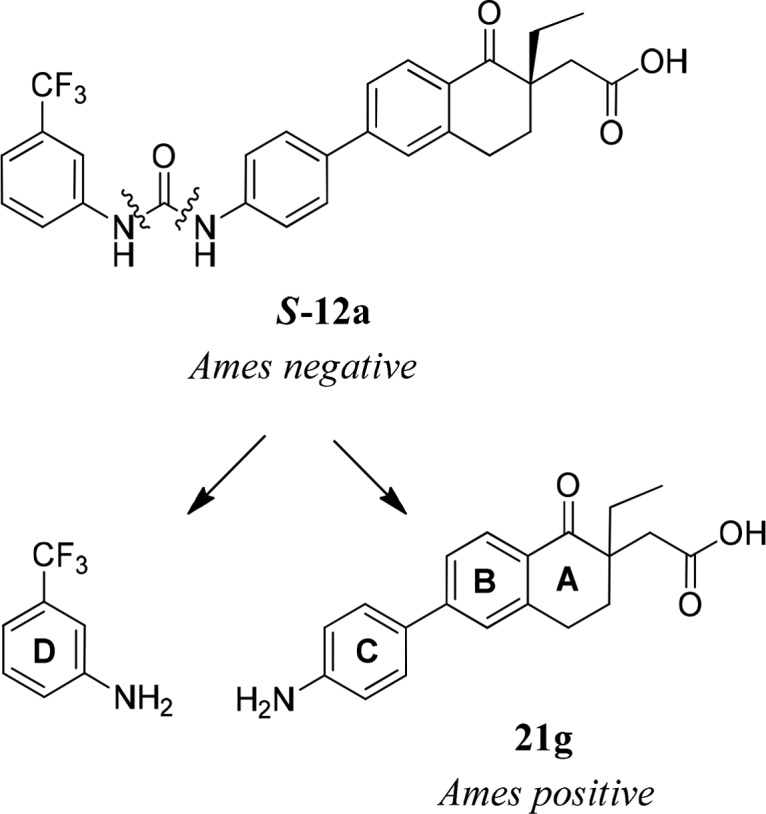
Hydrolysis of urea S-12a and the corresponding anilines.
Compound S-12a and the left-hand side (LHS) of m-trifluoromethyl aniline were not mutagenic when tested in the bacterial mutation screening assay (Ames test) with Salmonella typhimurium TA1535, TA1537, TA98, TA100, and Escherichia coli WP2uvrA (pKM101) in the presence and absence of the metabolic activating S9-mix.17 However, the right-hand side (RHS) of aniline 21g (racemic, potential degradant of racemic 12a) was mutagenic with and without S9 in the Ames test. S-Enantiomer of α-ethyl aniline 21g was not soluble enough to be tested in the Ames test. However, 21g was predicted to be positive in the in silico DEREK screen. In addition, S-enantiomer of similar aniline (the α-methyl analogue) was positive in both Ames test and DEREK screen. While we did not observe aniline 21gin vivo, we could not rule out with certainty that metabolism-driven hydrolysis would not occur in human. Therefore, we opted to address a potential genotoxicity issue of the anilines by identifying Ames negative aniline fragments.
It is known that electron deficient anilines reduce the risk of compounds being mutagenic.16 We decided to explore substitution of the C-ring with electron-withdrawing groups and replacement of C-ring with heteroaryls. In addition, we decided to re-evaluate α-trifluoroethyl substitution for mutagenicity potential as compound 12s was a potent DGAT1 inhibitor (Table 1). In order to expedite discovery of nonmutagenic anilines, we utilized in silico mutagenicity prediction models (DEREK and eHOMO) to prioritize synthesis and evaluation of RHS anilines in Ames assay.18 Since screening anilines in all five bacterial strains in Ames assay are time and resource demanding, we initiated screening of anilines in a single bacterial strain (TA98), which is known to be sensitive to aromatic amines, to get a quick go/no go decision. This approach allowed us to increase throughput of the assay, to reduce compound amount needed for testing, and to speed up turnaround time for generating the data. If aniline was tested negative in TA98, it would then be evaluated in the full Ames panel to confirm its nonmutagenic profile. Since the RHS aniline (21g) was positive in Ames with and without S9, it suggested that the mutagenicity of the aniline may not be related to eHOMO measurement as eHOMO required oxidation.
As shown in Table 5, there was poor correlation between in silico prediction and the TA98 Ames result. In general, α-trifluoroethyl analogues (21b, 21e, 21f, 24b, and 24d) were predicted to be negative in DEREK. Chloro-containing anilines (21c–e), pyridinyl anilines (24a–b), and pyridazinyl anilines (24c–d) were predicted to be negative based on eHOMO measurement. Pyridazinyl anilines (24c–d) were consistent in both DEREK and eHOMO prediction and were predicted to be nonmutagenic. Unfortunately, most analogues tested were positive in the TA98 Ames test regardless of the results from the in silico prediction with the exception of two chloro-containing anilines (21c and 21d) and one pyridazinyl aniline (24d). TA98 negative anilines (21c, 21d, and 24d) were further evaluated in the full panel of Ames assay and were confirmed to be nonmutagenic. Although in silico mutagenicity predictions were not correlative to the Ames result, our approaches including using TA98 as a triage for Ames testing did result in the discovery of nonmutagenic anilines in a timely manner.
Table 5. In Silico DEREK and eHOMO Prediction and TA98 Ames Results of RHS Anilines (21a–g, 24a–d).
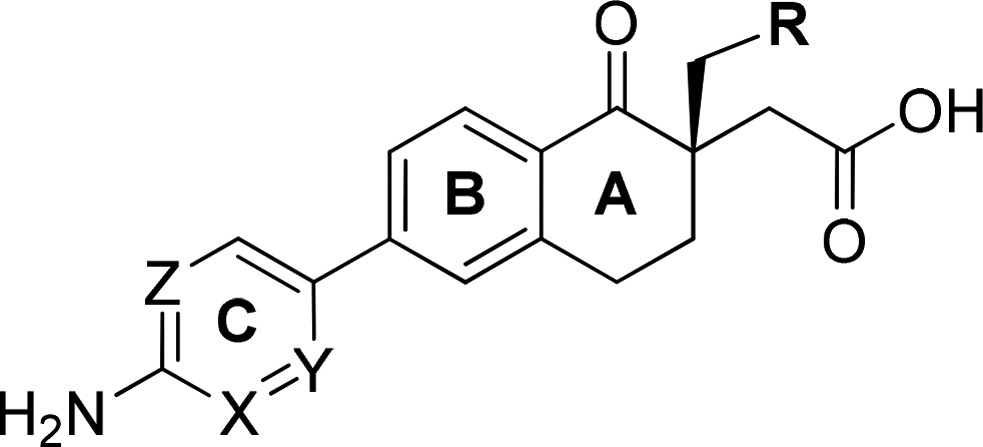
| compd # | R | X | Y | Z | DEREK/eHOMO/TA98 |
|---|---|---|---|---|---|
| 21g (Rac) | CH3 | CH | CH | CH | +/+/+ |
| 21f | CF3 | CH | CH | CH | −/+/+ |
| 21a | CH3 | C–F | CH | CH | +/+/+ |
| 21b | CF3 | C–F | CH | CH | −/+/+ |
| 21c | H | CH | C–Cl | CH | +/−/− |
| 21d | CH3 | CH | C–Cl | CH | +/−/− |
| 21e | CF3 | CH | C–Cl | CH | –/–/+ |
| 24a | CH3 | CH | N | CH | +/−/+ |
| 24b | CF3 | CH | N | CH | –/–/+ |
| 24c | CH3 | CH | N | N | –/–/+ |
| 24d | CF3 | CH | N | N | –/–/– |
Armed with the nonmutagenic RHS anilines in hand, we then performed a mix-and-match effort with the Ames negative LHS anilines to identify potential precandidates without genotoxic liabilities. α-Ethyl (21d) and α-trifluoroethyl RHS anilines (24d) were selected for mix-and-match because of the potency improvement over the α-methyl RHS aniline (21c). The most potent compounds with nonmutagenic anilines are shown in Table 6.
Table 6. DGAT1 Activities of Ames Negative Aniline Containing Tetralones 26a–d.
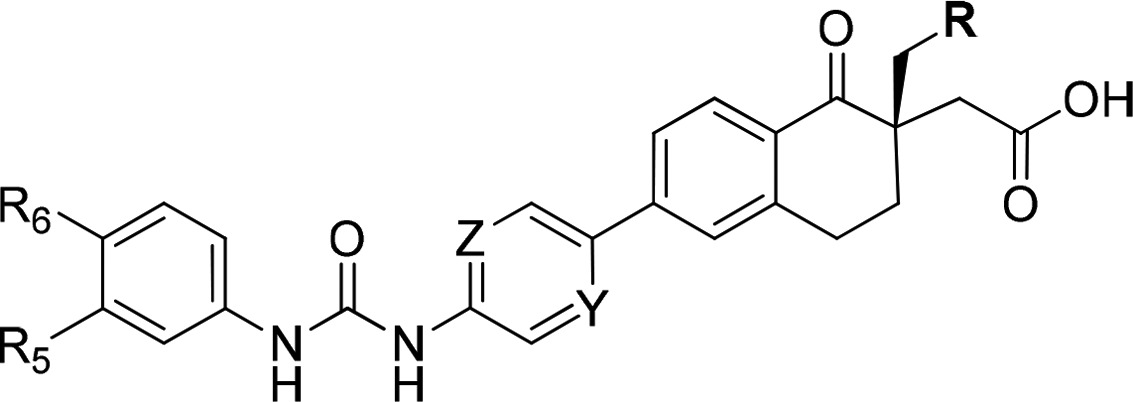
| compd # | R | Y | Z | R5 | R6 | hDGAT1 LE IC50 (nM)a | solubility (μg/mL)a |
|---|---|---|---|---|---|---|---|
| 26a | CH3 | C–Cl | CH | CF3 | H | 8 | 151 |
| 26b | CH3 | C–Cl | CH | F | F | 20 | 125 |
| 26c | CF3 | N | N | CF3 | H | 3 | 56 |
| 26d | CF3 | N | N | F | F | 3 | 134 |
Value is an average of at least n = 2.
All the compounds showed good potency against hDGAT1 enzyme. Compounds 26a and 26c exhibited poor oral bioavailability (23 and 25 %F, respectively), while compounds 26b and 26d demonstrated modest to good oral bioavailability (51 and 48 %F, respectively) in Sprague–Dawley male rats. Compounds 26b and 26d were evaluated for inhibition of TG in a postprandial lipid excursion model in Swiss Albino mice.15 Compound 26d showed excellent inhibition of TG in the model with near complete inhibition at 1 mg/kg when dosed orally.19 Specifically, as shown in Table 1S in the Supporting Information, 26d showed 76% TG inhibition at 0.3 mg/kg with terminal plasma drug level of 36 ng/mL, and 97.6% TG inhibition at 1 mg/kg with terminal plasma drug level of 106 ng/mL.
Compound 26d was confirmed to be nonmutagenic in the Ames test. Since 26d was a potent human DGAT1 inhibitor with good solubility, pharmacokinetic properties, and in vivo efficacy, additional selectivity and developability data were generated to evaluate its suitability as a precandidate compound. Compound 26d exhibited >2900-fold selectivity over human DGAT2, ACAT1 (SOAT1), and ACAT2 (SOAT2) enzymes (IC50 values of >10 μM) in radiometric assays, and an IC50 value of 77 nM in the cell-based C2C12 assay. In addition, 26d had an excellent cardiac ion channel profile with IC50 values of >10 μM in the hERG, Nav1.5, and Cav1.2 QPatch assays and exhibited good selectivity profile (>700-fold over hDGAT1 LE IC50) across a panel of selectivity assays. Compound 26d showed minimal CYP inhibition with IC50 values of >10 μM across CYP1A2 (phenacetin), CYP2C9 (diclofenac), CYP2C19 (S-mephenytoin), CYP2D6 (DEX), and CYP3A4 (midazolam), and demonstrated no time-dependent CYP inhibition at up to 50 μM of 26d and no PXR activation with EC50 > 50 μM. Compound 26d did not form glutathione conjugates in the presence of 5 mM GSH containing NADPH in human liver microsomes at up to 10 μM 26d. Moreover, 26d had high protein binding across species with plasma protein binding of 99.98%, 99.9%, and 99.95% in human, mouse, and rat, respectively. Compound 26d exhibited good PK profiles in rat (CL = 3.3 mL/min/kg, Vdss = 0.42 L/kg, iv T1/2 = 2.6 h, %F = 48). In addition, it was a low clearance compound with plasma clearances of 13.3 and 5.1 mL/min/kg in mouse and dog, respectively, and had good oral bioavailability of 41 and 68 %F in mouse and dog, respectively. Based on the overall developability profiles, compound 26d (GSK2973980A) was nominated as a development candidate for the treatment of obesity. Full in vitro and in vivo biological profiles of compound 26d have been reported recently.19
Conclusion
Starting with tetralone compound B with low micromolar human DGAT1 potency, we successfully identified a promising lead S-12a with potency improvement over 1000-fold, along with excellent selectivity, pharmacokinetic profiles, and in vivo efficacy. Unfortunately, compound S-12a contained RHS mutagenic Ames-positive aniline, which was a synthetic intermediate and a potential degradant. To expedite discovery of nonmutagenic anilines, we utilized in silico DEREK and eHOMO prediction to help prioritize anilines for synthesis and testing in Ames. We then employed a mini-screen of a single bacterial strain (TA98), which is known to be sensitive to aromatic amines, as a first pass to improve turnaround time for mutagenicity evaluation. TA98-negative anilines were then assessed in full Ames panel for nonmutagenic potential. Although in silico mutagenicity predictions were not predictive of the Ames outcome, we were able to quickly identify nonmutagenic anilines using this approach. This led us to the identification of (S)-2-(6-(5-(3-(3,4-difluorophenyl)ureido)-pyrazin-2-yl)-1-oxo-2-(2,2,2-trifluoroethyl)-1,2,3,4-tetrahydronaphthalen-2-yl)acetic acid (GSK2973980A, 26d) as a development candidate compound with suitable biological and developability profile for further progression. Full biological profiles of 26d have been reported recently.19
Acknowledgments
We thank Collaborative Research at the GVK Biosciences for the syntheses and profiling of compounds in biological and DMPK assays. We thank Amanda Giddings, Angela White, Jordi Munoz-Muriedas, Bibi Satter, Paul Hastwell, and Sharon Robinson for supporting in silico and in vitro genotoxicity evaluation of compounds. We thank Brian Donovan for providing the cardiac ion channel selectivity data and Dan Hassler for providing DGAT2, ACAT1, and ACAT2 selectivity data. We thank Douglas Minick for the VCD stereochemistry determination of the compounds.
Glossary
ABBREVIATIONS
- ACAT
acyl-coenzyme A cholesterol acyltransferase
- CL
clearance
- CPM
7-diethylamino-3-(4-maleimidophenyl)-4-methylcoumarin
- CYP
cytochrome P450
- DAG
diacylglycerol
- DGAT
acyl-CoA:diacylglycerol acyltransferase
- DEREK
deductive estimation of risk from existing knowledge
- DMPK
distribution, metabolism, and pharmacokinetics
- eHOMO
energy of the highest occupied molecular orbital
- GSH
glutathione
- hERG
human ether-à-go-go-related gene
- iv
intravenous
- LE
lipid extraction
- MC
methylcellulose
- NADPH
nicotinamide adenine dinucleotide phosphate
- PK
pharmacokinetics
- SOAT
sterol O-acyltransferase
- T1/2
half life
- TG
triglyceride or triglycerol or triacylglycerol
- VCD
vibrational circular dichroism
- Vdss
volume of distribution at steady state
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00450.
Experimental section on assays and methods, and procedures for the synthesis of all compounds described herein. PK (%F in rats) and in vivo efficacy (% inhibition of TG and terminal plasma level in mice) of tetralones 26a–d (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was supported by GlaxoSmithKline Pharmaceutical Company.
The authors declare the following competing financial interest(s): M.C., J.L.A., and S.K. are currently employed by and stockholders of GlaxoSmithKline.
Supplementary Material
References
- Chen H. C.; Farese R. V. Jr. Fatty acids, Triglycerides, and Glucose Metabolism: Recent Insights from Knockout Mice. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 359–363. 10.1097/00075197-200207000-00002. [DOI] [PubMed] [Google Scholar]
- Chen H. C.; Farese R. V. Jr. Inhibition of Triglyceride Synthesis as a Treatment Strategy for Obesity: Lessons from DGAT1-deficient Mice. Arterioscler., Thromb., Vasc. Biol. 2005, 25, 482–486. 10.1161/01.ATV.0000151874.81059.ad. [DOI] [PubMed] [Google Scholar]
- Villanueva C. J.; Monetti M.; Shih M.; Zhou P.; Watkins S. M.; Bhanot S.; Farese R. V. Jr. Specific Role for Acyl CoA:Diacylglycerol Acyltransferase 1 (Dgat1) in Hepatic Steatosis due to Exogenous Fatty Acids. Hepatology 2009, 50, 434–442. 10.1002/hep.22980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. J.; Cases S.; Jensen D. R.; Chen H. C.; Sande E.; Tow B.; Sanan D. A.; Raber J.; Eckel R. H.; Farese R. V. Jr. Obesity Resistance and Multiple Mechanisms of Triglyceride Synthesis in Mice Lacking Dgat. Nat. Genet. 2000, 25, 87–90. 10.1038/75651. [DOI] [PubMed] [Google Scholar]
- DeVita R. J.; Pinto S. Current Status of the Research and Development of Diacylglycerol O-Acyltransferase 1 (DGAT1) Inhibitors. J. Med. Chem. 2013, 56, 9820–9825. 10.1021/jm4007033. [DOI] [PubMed] [Google Scholar]
- Naik R.; Obiang-Obounou B. W.; Kim M.; Choi Y.; Lee H. S.; Lee K. Therapeutic Strategies for Metabolic Diseases: Small-Molecule Diacylglycerol Acyltransferase (DGAT) Inhibitors. ChemMedChem 2014, 9, 2410–2424. 10.1002/cmdc.201402069. [DOI] [PubMed] [Google Scholar]
- Ohshiro T.; Tomoda H. Acyltransferase Inhibitors: A Patent Review (2010-Present). Expert Opin. Ther. Pat. 2015, 25, 145–158. 10.1517/13543776.2014.989833. [DOI] [PubMed] [Google Scholar]
- Meyers C.; Gaudet D.; Tremblay K.; Amer A.; Chen J.; Aimin F. The DGAT1 Inhibitor LCQ908 Decreases Triglyceride Levels in Patients with the Familial Chylomicronemia Syndrome. J. Clin. Lipidol. 2012, 6, 266–267. 10.1016/j.jacl.2012.04.034. [DOI] [Google Scholar]
- Meyers C. D.; Serrano-Wu M.; Amer A.; Chen J.; Erik R.; Commerford R.; Hubbard B.; Brousseau M.; Li L.; Meihui P.; Chatelain R.; Dardik B. The DGAT1 Inhibitor Pradigastat Decreases Chylomicron Secretion and Prevents Postprandial Triglyceride Elevation in Humans. J. Clin. Lipidol. 2013, 7, 285. 10.1016/j.jacl.2013.03.093. [DOI] [Google Scholar]
- Dow R. L.; Li J.-C.; Pence M. P.; Gibbs E. M.; LaPerle J. L.; Litchfield J.; Piotrowski D. W.; Munchhof M. J.; Manion T. B.; Zavadoski W. J.; Walker G. S.; McPherson R. K.; Tapley S.; Sugarman E.; Guzman-Perez A.; DaSilva-Jardine P. Discovery of PF-04620110, a Potent, Selective and Orally Bioavailable Inhibitor of DGAT-1. ACS Med. Chem. Lett. 2011, 2, 407–412. 10.1021/ml200051p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denison H.; Nilsson C.; Kujacic M.; Lofgren L.; Karlsson C.; Knutsson M.; Eriksson J. W. Proof of Mechanism for the DGAT1 Inhibitor AZD7687: Results from a First-time-in-human Single-dose Study. Diabetes, Obes. Metab. 2013, 15, 136–143. 10.1111/dom.12002. [DOI] [PubMed] [Google Scholar]
- Denison H.; Nilsson C.; Lofgren L.; Himmelmann A.; Martensson G.; Knutsson M.; Al-Shurbaji A.; Tornqvist H.; Eriksson J. W. Diacylglycerol Acyltransferase 1 Inhibition with AZD7687 Alters Lipid Handling and Hormone Secretion in the Gut with Intolerable Side Effects: A Randomized Clinical Trial. Diabetes, Obes. Metab. 2014, 16, 334–343. 10.1111/dom.12221. [DOI] [PubMed] [Google Scholar]
- Christensen S. B. IV; Qin D.; Joshi H.; Tangirala R.. Novel Compounds as Diacylglycerol Acyltransferase Inhibitors. PCT. Int. Appl. WO2012162127, 2012.
- Vogt F. G.; Spoors G. P.; Su Q.; Andemichael Y. W.; Wang H.; Potter T. C.; Minick D. J. A Spectroscopic and Computational Study of Stereochemistry in 2-Hydroxymutilin. J. Mol. Struct. 2006, 797, 5–24. 10.1016/j.molstruc.2006.03.015. [DOI] [Google Scholar]
- Zhang X. D.; Yan J. W.; Yan G. R.; Sun X. Y.; Ji J.; Li Y. M.; Hu Y. H.; Wang H. Y. Pharmacological inhibition of diacylglycerol acyltransferase 1 reduces body weight gain, hyperlipidemia, and hepatic steatosis in db/db mice. Acta Pharmacol. Sin. 2010, 31, 1470–1477. 10.1038/aps.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch A. M.; Groombridge S.; Law R.; Leach A. G.; Mee C. D.; Schramm C. Rationally Designing Safer Anilines: The Challenging Case of 4-Aminobiphenyls. J. Med. Chem. 2012, 55, 3923–3933. 10.1021/jm3001295. [DOI] [PubMed] [Google Scholar]
- Maron D. M.; Ames B. N. Revised Methods for the Salmonella Mutagenicity Test. Mutat. Res. 1983, 113, 173–215. 10.1016/0165-1161(83)90010-9. [DOI] [PubMed] [Google Scholar]
- Fioravanzo E.; Bassan A.; Pavan M.; Mostrag-Szlichtyng A.; Worth A. P. Role of In Silico Genotoxicity Tools in the Regulatory Assessment of Pharmaceutical Impurities. SAR QSAR Environ. Res. 2011, 23, 257–277. 10.1080/1062936X.2012.657236. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Tirunagaru V. G.; Ariazi J.; Awasthi A.; Jayaraman V. B.; Arumugam P.; Yanamandra M.; Mitra S.; Tiwari S.; Tangirala R. S.; Werner T.; Thomson D.; Bergamini G.; Cheung M. A Novel Acyl-CoA: Diacylglycerol Acyltransferase 1 (DGAT1) inhibitor, GSK2973980A, Inhibits Postprandial Triglycerides and Reduces Body Weight in a Rodent Diet-Induced Obesity Model. J. Pharm. Res. Int. 2017, 18, 1. 10.9734/JPRI/2017/36835. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.