

Abstract
Background
Lactobacillus casei is widely used in the dairy and pharmaceutical industries and a promising candidate for use as cell factories. Recently, genome sequencing and functional genomics provide the possibility for reducing L. casei genome. However, it was still limited by the inefficient and laborious genome deletion methods.
Results
Here, we proposed a genome minimization strategy based on LCABL_13040-50-60 recombineering and Cre-lox site-specific recombination system in L. casei. The LCABL_13040-50-60 recombineering system was used to introduce two lox sites (lox66 and lox71) into 5′ and 3′ ends of the targeted region. Subsequently, the targeted region was excised by Cre recombinase. The robustness of the strategy was demonstrated by single-deletion of a nonessential ~ 39.3 kb or an important ~ 12.8 kb region and simultaneous deletion of two non-continuous genome regions (5.2 and 6.6 kb) with 100% efficiency. Furthermore, a cyclical application of this strategy generated a double-deletion mutant of which 1.68% of the chromosome was sequentially excised. Moreover, biological features (including growth rate, electroporation efficiency, cell morphology or heterologous protein productivity) of these mutants were characterized.
Conclusions
To our knowledge, this strategy is the first instance of sequential deletion of large-scale genome regions in L. casei. We expected this efficient and inexpensive tool can help for rapid genome streamlining and generation restructured L. casei strains used as cell factories.
Electronic supplementary material
The online version of this article (10.1186/s12934-018-0872-4) contains supplementary material, which is available to authorized users.
Keywords: Lactobacillus casei; Large-scale genome deletion, genome engineering; Recombineering; Cre-lox system
Background
Lactic acid bacteria (LAB) are native to food-related habitats, including animal and vegetables niches and critical for the production of fermented beverages and foods [1]. In recent years, fast growing numbers of the whole genome sequences and functional genomics of lactic acid bacterial strains have provided an abundant of information for further understanding LAB, including their gene organization, their biological properties, and their ecological roles in animal or human health as well as their environmental interactions [2–5]. The whole genome sequences also offered the possibility for us to engineer LAB strains with custom-design and reduced genomes to serve as cell factories [6]. Therefore, the next widely significant breakthrough in genetic engineering is the ability to simply and efficiently reduce the genomes of LAB.
Of all the LAB stains, Lactobacillus casei plays key roles in dairy fermentations and pharmaceutical industries and is also the ideal cell factories for production of high-value metabolites [7–11]. However, the number of highly and satisfactorily engineered L. casei strains for biotechnological production purposes is still relatively low. Though the whole genome of L. casei BL23 has been sequenced and annotated in the GenBank database, it is still far from being well understood. By searching the genome of L. casei BL23 (GenBank: FM177140.1), almost 40% of its genes encoded for proteins with no assigned function [12]. Moreover, annotation of the genome relying on comparative genomics may result in inaccurate metabolic pathway or essential gene predictions [13, 14], limiting the processes on constructing novel strains with a markedly reduced genome that produce desired products efficiently. One of the best strategies for the veracity of L. casei BL23 functional genomics which is especially important for the whole genomics field is to simultaneously carry out one or a small number of genes deletion.
A high-efficiency genome editing tool in L. casei based on prophage-derived recombinases and Cre-lox system was established for construction of deletion mutants in our previous study [15]. This recombineering system consisted of a presumptive 5′–3′ exonuclease LCABL_13060, a ssDNA annealing protein LCABL_13050 and a predicted host nuclease inhibitor LCABL_13040 analogous to Exo, Beta and Gam, respectively. However, the hurdle for large-scale chromosomal deletion (> 6 kb) was the low efficiency of homologous recombination mediated by prophage-derived recombinases LCABL_13040-50-60. Therefore, construction of large-scale chromosomal deletion mutations was still based on a variety of conditional replication plasmids mediated by RecA dependent homologous recombination [16, 17]. These methods are useful but inefficient and laborious for large-scale region deletions, especially for the regions containing gene(s) impairing cell growth which are named important gene(s) in this study. Therefore, it is very instant to develop of a powerful and high-efficiency large-scale genome deletion method for identifying essential genetic information and engineering novel L. casei strains used for cell factories through experimental reduction of the genome to its minimal gene set in L. casei.
Large-scale genome engineering systems using for genome reduction have been described in bacteria [18–22]. Several of them are mainly based on the Cre-loxP site-specific recombination system of the bacteriophage P1 [18–20]. The Cre-loxP system was composed of two components: the 343 amino acid Cre recombinase facilitates the site-specific recombination and a 34 bp loxP site containing an asymmetric 8 bp core sequence and two 13 bp inverted repeats at which recombination takes place [23]. The Cre recombinase could excise any chromosomal region flanked by two loxP sites in which the 8 bp core sequence in same orientation, leaving one loxP site which limits the repeated use of the strategy [24, 25]. In order to efficiently and cyclically engineer large-scale genome, a pair of mutant loxP sites (lox66 and lox71) was used for excision of the targeted region in the genome of L. casei BL23 strain.
Mobile genetic elements (MGEs) containing bacteriophages, transposable elements and genomic islands present in bacteria with continuous challenges to genomic stability, promoting evolution through horizontal gene transfer [26]. Comparative genome analysis showed that an insertion island (LCABL_12890 to LCABL_13480) was exclusive for L. casei BL23, and the majority of genes were predicted to encode prophage-related proteins, suggesting the region is a prophage remnant [27]. In this study, the insertion island (LCABL_12890 to LCABL_13480) was deleted coupling the LCABL_13040-50-60 recombineering to Cre-lox system, and the properties of the resulting mutant strain (including growth rate and electroporation efficiency) were characterized. Moreover, the applicability or reproducibility of the method was demonstrated by the construction of a single deletion containing an important gene mutant or a double deletion mutant. This markerless large DNA deletion strategy represents a significant improvement to existing methods, and will accelerate the development of L. casei in genome streamlining and synthetic biology.
Results
Scheme for deletion of a large-scale genome region in L. casei
To develop a large genome region deletion strategy in L. casei, the Cre-lox site-specific recombination system was used in this study (Fig. 1). However, the targeted large chromosomal DNA region needs to be introduced two parallel mutant loxP (lox66 and lox71) sites (8 bp core sequence in same orientation) at 5′ and 3′ ends. Therefore, LCABL_13040-50-60 expression plasmid pMSP456 (Additional file 1: Figure S2A) and Cre recombinase expression plasmid pMSPcre (Additional file 1: Figure S2B) were employed, which the LCABL_13040-50-60 and cre were both driven by an inducible PnisA promoter, respectively.
Fig. 1.
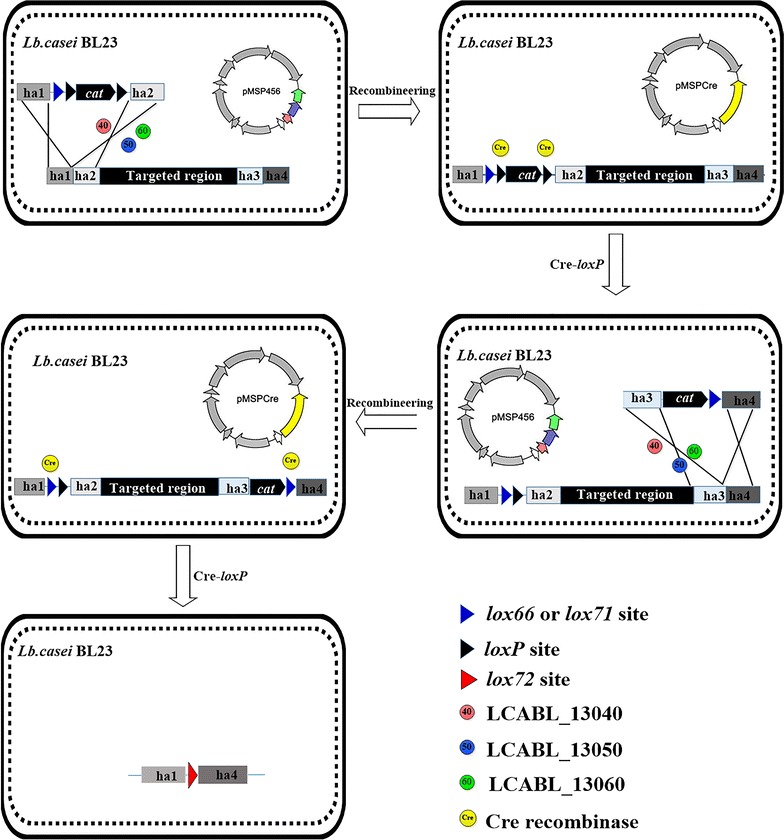
Scheme for markerless deletion of a large-scale genome region in L. casei BL23. Ha indicated four ~ 1 kb homology arms
Because the LCABL_13040-50-60 recombineering system was not used for editing multiplex sites simultaneously, a two-step procedure was employed to sequentially introduce two parallel mutant loxP sites at 5′ and 3′ ends of the targeted region. Unfortunately, by two rounds of LCABL_13040-50-60 recombineering tests, it was difficult to insert second mutant loxP site (lox72) into the 3′ end using the linear donor disruption cassette H-cat-lox71-H as the substrates with 2 × 5 or 4 × 5 mg/ml chloramphenicol selection. While the first mutant loxP site (lox66) was easy to insert into the 5′ end using h-lox66-cat-h as the substrates with 5 mg/ml chloramphenicol selection.
Therefore, an enhanced three-step strategy was explored to supply the inadequacy of the above two-step procedure. Initially, the first linear donor disruption cassette h-lox66-loxP-cat-loxP-h was used to insert the lox66-loxP-cat-loxP into the 5′ end of the targeted region by LCABL_13040-50-60 recombineering. Secondly, the plasmid-free recombinant was transformed with pMSPcre, then induced to express Cre recombinase to excise the chloramphenicol resistance gene cat from the chromosome and leave lox66-loxP at the 5′ end of the targeted region. Finally, the lox71 site resident in the second cassette h-cat-lox71-h was inserted into the 3′ end of the targeted region by another round of LCABL_13040-50-60 recombineering. A large-scale genome region flanked with lox66 and lox71 sites (8 bp core sequence in same orientation) will be excised in the presence of Cre recombinase.
High-efficiency markerless deletion of a large-scale genome region in L. casei
To test the scheme for markerless deletion of a large genome region, we initially searched the prophage-like genes in L. casei BL23 genome because prophages or prophage remnants may not always be necessary for the strain growth in the laboratory or industrial condition. Previously, a 38.9 kb genome region [27] which composed of main prophage remnants was identified and the genetic organization of this region was shown in Fig. 2a. This region contained 59 open reading frames (ORFs) and comprised approximately 1.27% of its genome. Among them, 19 genes have been annotated and ∼ 67.8% of the corresponding ORFs encoding proteins of unknown function. To assess the functions of this 38.9 kb DNA region, the mutants with the 38.9 kb genome region deletion were achieved by inducing with nisin to express Cre and subjecting to chloramphenicol resistance test. The sizes of homology regions ha1, ha2, ha3 and ha4 were 1187, 1114, 962 and 1121 bp in size, respectively. Colony PCR was subsequently used to further confirm the excision event with primer pair H1F and H2R. The expected size of PCR products amplified from the mutants should be ~ 2 kb (Fig. 2a). After induced with nisin for 12 h, all colonies tested were sensitivity to chloramphenicol (Fig. 2b). Results of colony PCR were coincident with the expected size using primers H1F and H4R (Fig. 2c). DNA sequencing further confirmed that gene cat has been taken out and a single lox72 site has been successfully replaced by the 38.9 kb DNA fragment. Thus, these results clearly demonstrated that this strategy developed in this study could be used for construction of a 38.9 kb deletion mutant BLD1 with 100% efficiency.
Fig. 2.
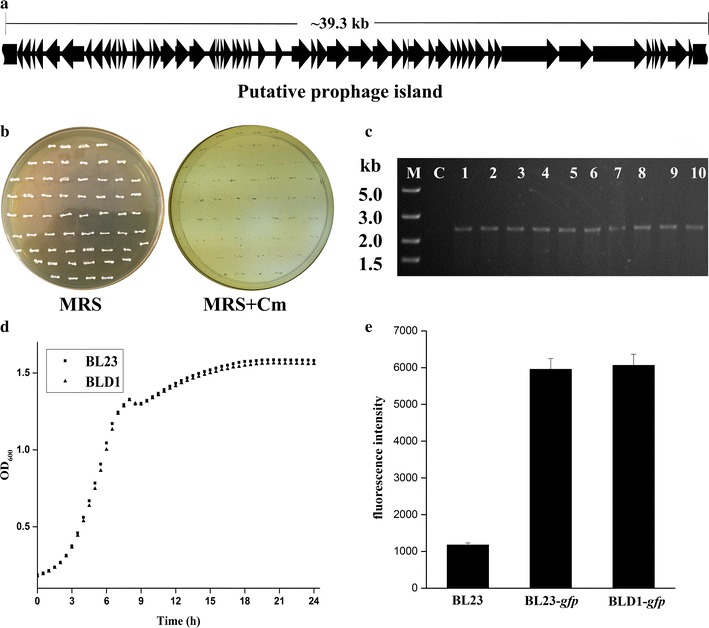
Deletion of large-scale genome region in L. casei BL23. a Layout and genetic context of the ~ 39.3 kb prophage island located on the chromosome. b Chloramphenicol resistance detection of BLD1 mutants. c Colony PCR results. M: DNA marker; C: wild-type strain; 1–10: chloramphenicol resistance colonies. d Growth of L. casei BL23 and the deletion strain L. casei BLD1 on MRS. e Comparison of fluorescence intensity of GFP in BL23, BL23-gfp and BLD1-gfp. Data represent the mean of three independent experiments
Moreover, biological features of the mutant strain BLD1 (including growth rate, electroporation efficiency and heterologous protein productivity) were characterized. The growth profiles of strains were monitored by the optical density at 600 nm (OD600) of cells in MRS media. As shown in Fig. 2d, the deletion mutant BLD1 showed a slightly increased in both of growth rate and final OD600 compared to the wild-type strain BL23. Electroporation efficiencies of the mutant BLD1 and wild type strain BL23 were compared for two plasmids with different replication modes (pSec:Leiss:Nuc [28] and pTRKH2 [29]) and a temperature sensitive plasmid pG+Host9 [17] (Table 1). The electroporation efficiency of all the three tested plasmids for mutant BLD1 was approximately threefold higher than that for strain BL23. Moreover, the mutant BLD1 was evaluated as cell factories for heterologous protein production with the green fluorescent protein (GFP) as a reporter. Unexpectedly, the fluorescence intensity of the mutant BLD1-gfp was very similar to that of the strain BL23-gfp (Fig. 2e).
Table 1.
The electroporation efficiency in BL23 and BLD1
| Plasmids | Average number of transformants (CFU/µg plasmid DNA) | |
|---|---|---|
| BL23 | BLD1 | |
| pTRKH2 | 4.91 × 103 | 1.64 × 104 |
| pG+host9 | 4.62 × 103 | 1.39 × 104 |
| pSec:Leiss:Nuc | 7.21 × 104 | 1.77 × 105 |
Deletion of a large-scale genome region containing an important gene in L. casei
The high excision efficiency of Cre in L. casei BL23 encouraged us to test its activity in removal a large-scale genome region containing an important gene in L. casei BL23. Therefore, the 12.8 kb genome region which equipped with the putative important gene galE was selected as a target to be excised (Additional file 1: Figure S3A) by the above methods. The size of DNA fragment between two mutant loxP sites was around 12.8 kb. Twenty of the chloramphenicol sensitive colonies were picked and the excision of 12.8 kb DNA fragment was confirmed by PCR using primers H5F and H8R (Additional file 1: Figure S3B), the experimental data were coincident with the expected results. DNA sequencing of PCR products further proved the accurate excision (Biosune Company, Shanghai, China). Quantitative growth curves analysis indicated that the 12.8 kb deletion mutant BLD2 showed a decreased growth rate in MRS medium (Additional file 1: Figure S3C), suggesting that the deletion altered the strain’s phenotype. These results clearly demonstrated that this method could be used to delete a large-scale genome region containing an important gene in L. casei BL23.
Simultaneous deletion of two non-contiguous regions in L. casei
To investigate whether this procedure could be used for simultaneous deletion of two non-contiguous regions, the above 12.8 kb genome region was distributed into two DNA fragments by gene galE and the sizes of the two non-contiguous regions were 5.2 and 6.6 kb, respectively. For the simultaneous deletion of these two non-contiguous regions, we prepared the second disruption cassette h-cat-lox71-galE-h for replacing h-cat-lox71-h in the second round of LCABL_13040-50-60 recombineering test. Subsequently, expression of the Cre recombinase would replace the 12.8 kb genome region with lox72-galE, resulting in the simultaneous deletion of these two non-contiguous regions, generating strain BLD3. Two randomly picked mutants were confirmed by PCR using primers H5F and H8R (Fig. 3a) and DNA sequencing. As shown in Fig. 3b, a mutant strain BLD3 with the targeted two non-contiguous region deletions was successfully constructed. Expectedly, only slight differences in growth rate was observed between the deletion strain BLD3 and the wild type strain BL23 (Fig. 3c).
Fig. 3.

Simultaneously deletion of two non-continuous large-scale genome regions in L. casei BL23. a Layout and genetic context of the ~ 12.8 kb genome region and the sizes of PCR products amplified with primers H1F and H2R from BLD3 strains. b Colony PCR results. M: DNA marker; 1–2: random picked chloramphenicol resistance colonies. c Growth of L. casei BL23 and the deletion strain L. casei BLD3 on MRS
Second and successive deletion of large-scale genome regions in L. casei
As a result of the first 39.3 kb genomic region deletion, the mutant strain BLD1 retains a single lox72 site. To test whether this strategy has the capability of generating multiple deletions in L. casei, two mutant loxP sites aiming at excising the 12.8 kb region (Additional file 1: Figure S3A) was introduced into the mutant strain BLD1 by the same procedure (Fig. 1). After the recombination and excision step, markerless double-deletion of the two regions (1.68% of the genome size) was confirmed by PCR using pairs of primers (H1F/H4R and H5F/H8R) specific to the endpoints of the targeted region and sequencing (Fig. 4a, b). Therefore, this strategy described in this study was capable of creating successive chromosomal deletions in L. casei strain.
Fig. 4.
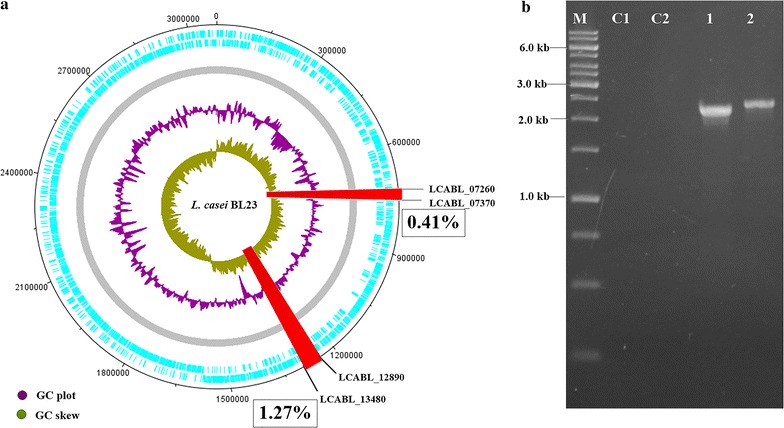
Sequential deletion of double large-scale genome regions in L. casei BL23. a Genomic deletions in the double-deletion mutants of L. casei BL23. The red boxes represent the 1.68% deleted regions. b Colony PCR results. M: DNA marker; C1: L. casei BL23 (12.8 kb); C2: L. casei BL23 (39.3 kb); 1: random picked chloramphenicol resistance colonies (12.8 kb); 2: random picked chloramphenicol resistance colonies (39.3 kb)
Discussion
In recent years, large-scale genome engineering has been beneficial to the therapeutic and industrial applications, as well as a role in basic research into the origin and evolution of life and mechanisms of cellular metabolism [30–34]. However, it is still time-consuming and laborious to engineering a reduced L. casei genome. In this study, we proposed a novel genome engineering method for sequentially generating multiple markerless large-scale chromosomal deletions in L. casei BL23. To our knowledge, this is the first time to couple the recombineering to Cre-loxP system for simplified and programmable construction large-scale chromosomal deletions in L. casei BL23. First step in the strategy is to introduce two mutant loxP sites into both ends of the targeted region by LCABL_13040-50-60 recombineering system and the Cre-loxP recombination system. Second step is to excise the large DNA by Cre recombinase.
The ~ 39.3 kb region (LCABL_12890 to LCABL_13480) is a prophage remnant island for L. casei BL23 [27]. Prophage remnant islands, member of mobile genetic elements (MGEs), present in bacteria with continuous challenges to genomic stability [35]. However, our results strongly indicated that the deletion of this island increased the growth rate in MRS, suggesting that the reduced genome decrease the redundancy among genes and regulatory circuits [36]. This finding is in contrast to experiments targeting integrated mobile genetic elements, which resulted in a significantly longer generation time during log phase [35], possibly because this targeted region did not contain any core genes. Moreover, the length of the deleted region was larger than that in Lc. lactis [6, 37]. Considering the high-efficiency of deletion ~ 39.3 kb, larger-scale genomic regions could be excised for genome reduction using this strategy. However, our results showed that the heterologous GFP protein productivity of the engineered strain BLD1 with ~ 39.3 kb deletion was only slightly higher than that of the parental strain BL23, suggesting further studies should be done to identify and delete more nonessential DNA regions to reduce the genome and use the genome reduction strains as cell factories for enhanced heterologous protein expression.
Surprisingly, the efficiency of the ~ 12.8 kb genome region deletion which showed a strongly reduced growth rate when it was grown in a liquid medium containing glucose was nearly 100%. In Lc. lactis MG1363, deletion of galE gene would result in the reduced growth rate and long-chain phenotype [38]. Similarly, morphology analysis showed that the growth defect of the mutant cells was also be linked to formation of long chains of cells (Additional file 1: Figure S4). Our results also showed that mutant with the 12.8 kb deletion except for galE showed a very similar growth rates to the wild-type strain, suggesting that GalE catalyzing UDP-glucose to UDP-galactose plays a key role in the regulation of cell autolysis in L. casei BL23.
Previously, two regions separated by an essential gene should be excised independently in L. casei. Nevertheless, with this deletion strategy using a modified disruption cassette, simultaneous deletion of two noncontiguous genomic regions and excision of a genomic region that contains an essential gene could be performed rapidly. Additionally, this simultaneous noncontiguous deletion strategy also opens a new way for replacement deletion regions with available information and re-engineering large sections of the genome [20]. However, due to the low efficiency of DNA replacement mediated by prophage-derived recombinases LCABL_13040-50-60, deletion of targeted regions that contained an essential gene larger than 5.0 kb was not achieved in this study. Our results also showed that the galKTRM deletion strain L. casei BLD3 holds a very similar growth rate to L. casei BL23 in CDM medium supplemented with galactose (Additional file 1: Figure S5), suggesting that the major contributor to the catabolism of galactose was the tagatose-6-phosphate (T6P) pathway rather than the Leloir pathway in L. casei BL23 [39].
Currently, there is an increasing interest of using the CRISPR/Cas9 system for large-scale genome deletions in bacteria, including lactic acid bacteria [35]. However, the targeted regions were limited to be flanked by the homologous IS elements [35]. The application of the CRISPR/Cas9 or CRISPR-Cas9D10A in L. casei for large-scale genome deletion (> 5 kb) has not been reported [40]. Instead, chromosomal deletions in L. casei have often been generated by using conditional replication plasmids [17, 41] or counterselection marker [16]. The new genome engineering strategy developed in this study has several practical advantages over those methods. First of all, both the efficiency of insertion a ~ 1 kb DNA fragment into the chromosomal locus mediated by LCABL_13040-50-60 recombineering and excision the large-scale genome region between lox66 and lox71 sites were close to 100%. Therefore, the efficiency of this strategy was higher than those above methods in which only 50% of the double crossover colonies carrying the expected deletion in theory [16, 17, 41]. Secondly, this strategy does not require chemically defined or semi-defined media for counterselection [16], but only simply commercial MRS broth.
With this method developed here, the sequential introduced into the Δ39.3 and Δ12.8 kb double-deletion was performed, and the loss of the vector could be selected without erythromycin. Previously, it was successfully achieved the deletion of large regions of Escherichia coli K-12 chromosome using a combination of Cre-loxP and λ Red has been reported [42]. However, it was not used for successive deletion of large DNA segments due to the high-efficiency of site-specific recombination between two loxP sites (one loxP site from the first genomic excision and the new inserted loxP site) [42]. To address this problem, two mutant loxP sites were used in this study. Because the low-efficiency site-specific recombination between lox72 and loxP, the cyclical genomic deletions could be achieved [43]. The repetition of the method on the double-deletion mutant as well as the establishment of further multiple-deletion mutants could allow further increases in the genome reduction beyond the 1.68% already obtained, leading towards determining a functional essential core of the genome under the specified conditions.
Conclusions
In conclusion, we firstly proposed the targeted large-scale deletion method for genome streamline based on LCABL_13040-50-60 recombineering and Cre-loxP site-specific recombination system in L. casei. With the modification of the linear donor disruption cassette, our strategy can be used for a variety of genome modifications. We also expected that this possible multiplexing method could provide a new way for rapid genome streamlining and generation restructured lactic acid bacteria strains served as cell factories with the development of the novel recombineering system in other lactic acid bacteria.
Methods
Bacterial strains, plasmids and growth conditions
Bacterial strains and plasmids used in this study are listed in Table 2. As a cloning host in this study, E. coli DH5α was grown in Luria–Bertani (LB) medium at 37 °C under agitation. Unless otherwise specified, lactobacilli and their derivatives used in this study were cultured in deMan Rogosa Sharpe (MRS) broth (Oxoid) at 37 °C under static conditions. When needed, the antibiotics were supplemented as followed: 5 μg/ml erythromycin or chloramphenicol for lactobacilli, 100 μg/ml ampicillin, 30 μg/ml kanamycin and 10 μg/ml chloramphenicol for E. coli DH5α, respectively.
Table 2.
Plasmids and bacterial strains used in this study
| Strain or plasmid | Characteristic(s) | Source |
|---|---|---|
| Strains | ||
| Escherichia coli DH5α | F− supE44 ∆lacU169 Ф80lacZ ∆M15 hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Novagen |
| Lactobacillus casei BL23 | Derivative of L. casei ATCC 393 (pLZ15−) | [44] |
| Plasmids | ||
| pG+host9 | Ermr; temperature-sensitive vector | [17] |
| pUC19 | Ampr; cloning vector | This study |
| pSec:Leiss:Nuc | pWV01 replicon; expresses Nuc under PnisA control; Cmr | [28] |
| pET-28a | Kanr; cloning vector | This study |
| pTRKH2 | Ermr; theta-replicating vector | [29] |
| pMSP456 | Expression LCABL_13040-50-60 under PnisA control | This study |
| pMSPcre | Expression Cre under PnisA control | This study |
| pUCgalK | Source of fragment loxP-cat-loxP | [15] |
| pCD4033-gfp | Gene gfp as a reporter in the downstream of Pldh | [45] |
DNA manipulation
Plasmid Mini Kits (Omega) was used for E. coli plasmid DNA isolation and Gel Extraction Kits (Omega) or Cycle-Pure Kits (Omega) was used for the linear DNA purification. All the restriction enzymes, T4 DNA ligase and DNA polymerases used in this study were purchased from TaKaRa and strictly in accordance with the manufactural instructions for processing. PCR reactions for expressed purpose were generated with 2 × Primestar Max while PCR amplifications for screened purpose were performed by rTaq DNA polymerase.
Construction of the linear donor disruption cassettes
All primers used for PCR amplification are listed in Table 3. To prepare of linear donor cassette (h1-lox66-loxP-cat-loxP-h2) for disruption the upstream of the ~ 39.3 kb region (Additional file 1: Figure S1), the chloramphenicol-resistance gene cat with its promoter region flanked by two loxP sites (loxP-cat-loxP) was obtained by general PCR from the vector pUCgalK [15] using primers catF1 and catR1. The upstream and downstream homology arms (h1 and h2) were PCR amplified from the chromosomal DNA of Lb. casei BL23 using primer pairs H1F/H1R and H2F/H2R. The restriction site SalI responsible for ligating to loxP-cat-loxP was introduced by the primers H2F. The two resultant fragments were spliced by an overlap extension PCR using primer pair H1F and H2R. The fusing product h1-lox66-h2 was digested with BglII and XhoI and ligated into the BglII and XhoI sites of pET-28a. The yielding vector pUD1 or loxP-cat-loxP were respectively digested with SalI or XhoI and ligated to create pHA1. Finally, the linear donor disruption cassette h1-lox66-loxP-cat-loxP-h2 was generated by PCR from vector pHA1 using primers H1F and H2R.
Table 3.
Oligonucleotide primers used in this study
| Primer | Sequence (5′–3′)a | Restriction site |
|---|---|---|
| H1F | GAAGAAGATCATTTACTCGAG | XhoI |
| H1R | TACCGTTCGTATAGCATACATTATACGAAGTTATCAGAACGCAAATCTCTTCTA | |
| H2F | TGTATGCTATACGAACGGTAGTCGACTTACTTAATGCTATTCATTA | SalI |
| H2R | TTAAGATCTACATCGAGTTCAGCAAGCTAA | BglII |
| H3F | ATATGTCGACATTACAGTTACAAGCCATATAC | SalI |
| H3R | TCAATATTCTTCTCCGTCCC | |
| H4F | GGGACGGAGAAGAATATTGAAGATCTCACTGCAGTATATCAGGTAACAAAAAGT | BglII, SalI |
| H4R | ACATGCATGCTTCCCTCTCCAAATATGCAC | SphI |
| H5F | CGGGATCCAATAGCTCAGATTTTTAACAACA | BamHI |
| H5R | TACCGTTCGTATAGCATACATTATACGAAGTTATTAATTTATCAAAAACCTTATTC | |
| H6R | CCCAAGCTTGTACATCATACTGTTCATGCC | HindIII |
| H6F | TGTATGCTATACGAACGGTACTCGAGAAGCCATCGCTGCTAACAAA | XhoI |
| H7F | TTTTGGCCAAGCTGGTTTT | |
| H7R | TTGTTTTTTCGCGGTACTGA | |
| H8F | TCAGTACCGCGAAAAAACAAGATCTCACACTCGAGTGTAGTCGATAAAAATT | BglII, XhoI |
| H8R | GCA ACATGCATGCAGACCATGAGACTATTTCAG | SphI |
| PrF | TATAGCATACATTATACGAAGTTATCCAGCTAGGCCTAGTGTCCGT | |
| PrR | CGTCAATACCTCCTAATTGA | |
| galEF | TCAATTAGGAGGTATTGACGATGACAATTGCAGTTTTAGG | |
| galER | TTACTCGAGTCAATTCCGGTCACCAAATC | XhoI |
| catF1 | CCGCTCGAGATAACTTCGTATAATGTATGCTATA | XhoI |
| catR1 | CCGCTCGAGATAACTTCGTATAGCATACATTATA | XhoI |
| catF2 | CCGCTCGAGATAACTTCGTATAGCATACATTATACGAACGGTACGAAAGTCGACGGCAATAG | XhoI |
| catR2 | GGAAGATCTCTGTAATATAAAAACCTTCTTC | BglII |
aThe restriction sites in the primer sequences are underlined
To prepare of linear donor cassette (h3-cat-lox71-h4) for disruption the downstream of the ~ 39.3 kb region, the chloramphenicol-resistance gene cat with its promoter region flanked by a lox71 sites (cat-lox71) was obtained by general PCR from the vector pUCgalK [15] using primers catF2 and catR2. The upstream and downstream homology arms (h3 and h4) were PCR amplified from the chromosomal DNA of Lb. casei BL23 using primer pairs H3F/H3R and H4F/H4R. The restriction sites BglII and SalI responsible for ligating to cat-lox71 was introduced by the primers H4F. The two resultant fragments were spliced by an overlap extension PCR using primer pair H3F and H4R. The fusing product h3–h4 was digested with SalI and SphI and ligated into the SphI and XhoI sites of pET-28a. The yielding vector pUD2 or cat-lox71 were respectively digested with SalI or XhoI and BglII and ligated to create pHA2. Finally, the linear donor disruption cassette h3-cat-lox71-h4 was generated by PCR from vector pHA2 using primers H3F and H4R. The upstream or downstream linear donor disruption cassettes of the ~ 12.8 kb region was prepared by the similar method except for the skeleton plasmid pET-28a or pUC19.
Integration of lox sites in L. casei BL23
Integration of lox sites in desired locus was achieved by the LCABL_13040-50-60 recombineering. The LCABL_13040-50-60 recombineering steps were carried out according to our previously study [15]. Briefly, L. casei strains harboring plasmid pMSP456 were grown in 5 ml MRS medium supplemented with 1% glycine and 0.75 M sorbital and cultured at 37 °C statically. Electrocompetent cells was prepared after induction LCABL_13040-50-60 recombinases expression by 5 ng/ml nisin at initial OD600 of 0.25–0.30 until OD600 of 0.60–0.65. The electroporation was performed utilizing a BioRad Genepulser and 2 mm electroporation cuvette after the mixture of linear donor dsDNA disruption cassette and electrocompetent cells was kept on ice for 10 min. After electroporation, 1 ml of SMRS was added to the cuvette and recovered at 37 °C for 1 h. Subsequently, the recover was plated on MRS plates containing chloramphenicol.
Markerless deletion of a large-scale genomic region in L. casei BL23
The Cre-lox site-specific recombination system was used for markerless deletion of a selected regions. The steps were also carried out according to our previously study [15]. Briefly, plasmid-free mutants with two mutant loxP sites were grown and electrocompetent cells were prepared as described above. pMSPcre was introduced to the cells by electroporation and erythromycin resistant colonies were selected at 37 °C. The Cre recombinase expression was induced at OD600 of 0.4–0.8 with 10 ng/ml nisin for 24 h. After plated onto MRS plates, marker-free mutants were tested by PCR. Subsequently, the plasmid pMSPcre was cured at 37 °C without erythromycin for 24 h and streaked on MRS plates to obtain plasmid-free large DNA deletion mutants.
Quantitative growth curves
Lactobacillus casei BL23 and its derivates were first grown at 37 °C in MRS for 24 h. Samples were harvested by centrifugation at 10,000g for 3 min. After being resuspended twice with MRS medium. 200 μl of bacterial suspension was subsequently loaded into a 96-well plate (Costar) in six wells per strain. The 96-well plate was incubated in a plate reader (Epoch2, BioTek) statically at 37 °C. Cell growth was detected at an absorbance of 600 nm, with readings obtained at 30 min intervals for 24. All liquid cultivations were conducted with six biological replicates and the growth curves were obtained for each well.
Fluorescence assay
Fluorescence intensity of recombinant strains harboring the pCD4033-gfp [45] was determined according to our previous work [46]. Briefly, samples for measurement were taken out after 24 h and harvested by centrifugation at 10,000g for 3 min. After being resuspended twice with PBS buffer (pH7.4), 200 μl of bacterial suspension was transferred into a 96-well plate in which OD600 and fluorescence were read with excitation at 485 nm and emission at 528 nm using a Multi-Detection Microplate Reader, Synergy HT (BioTek). For each sample, three repetitions were performed with PBS as a blank.
Additional file
Additional file 1: Figure S1. Strategies for obtaining linear donor disruption cassette H1-lox66-loxP-cat-loxP-H2. Figure S2. Structure of plasmids used in this study. Arrow heads show genes and their direction. (A) LCABL_13040-50-60-expression plasmid pMSP456. (B) Cre-expression plasmid pMSPCre. LCABL_13040-50-60 and Cre were transcribed from PnisA that is controlled by the product of nisin. Figure S3. Deletion of large-scale genome region containing important gene(s) in L. casei BL23. (A) Layout and genetic context of the ~ 12.8 kb genome region and the size of PCR products amplified with primers H1F and H2R from BLD2 strains. (B) Colony PCR results. M: DNA marker; C: wild-type strain; 1–20: chloramphenicol resistance colonies. (C) Growth of L. casei BL23 and the deletion strain L. casei BLD2 on MRS. Figure S4. Cellular morphologies of L. casei strains BL23 and BLD2. Figure S5. Growth of L. casei BL23 and the deletion strain L. casei BLD3 on CDM medium containing galactose.
Authors’ contributions
YPX and JK conceived and designed the experiments. YPX carried out the experimental work. YPX, TTG, YLM and JK wrote and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank S. Hazebrouck for his generous gift of Lactobacillus casei BL23.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grants 31400077 and 31471715) and the Public Service Sectors (Agriculture) Special and Scientific Research Projects Hi-Tech Research and Development Program of China (Grant 201503134).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- LAB
lactic acid bacteria
- MGEs
mobile genetic elements
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12934-018-0872-4) contains supplementary material, which is available to authorized users.
Contributor Information
Yongping Xin, Email: 1355596467@qq.com.
Tingting Guo, Email: guotingting@sdu.edu.cn.
Yingli Mu, Email: 1343554013@qq.com.
Jian Kong, Phone: +86 531 88362318, Email: kongjian@sdu.edu.cn.
References
- 1.Leroy F, De Vuyst L. Lactic acid bacteria as functional starter cultures for the food fermentation industry. Trends Food Sci Technol. 2004;15:67–78. doi: 10.1016/j.tifs.2003.09.004. [DOI] [Google Scholar]
- 2.Johnson BR, Klaenhammer TR. Impact of genomics on the field of probiotic research: historical perspectives to modern paradigms. Antonie Van Leeuwenhoek. 2014;106:141–156. doi: 10.1007/s10482-014-0171-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Douillard FP, de Vos WM. Functional genomics of lactic acid bacteria: from food to health. Microb Cell Fact. 2014;13(Suppl 1):S8. doi: 10.1186/1475-2859-13-S1-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makarova K, Slesarev A, Wolf Y, Sorokin A, Mirkin B, Koonin E, Pavlov A, Pavlova N, Karamychev V, Polouchine N, et al. Comparative genomics of the lactic acid bacteria. Proc Natl Acad Sci USA. 2006;103:15611–15616. doi: 10.1073/pnas.0607117103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfeiler EA, Klaenhammer TR. The genomics of lactic acid bacteria. Trends Microbiol. 2007;15:546–553. doi: 10.1016/j.tim.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Zhu D, Fu Y, Liu F, Xu H, Saris PE, Qiao M. Enhanced heterologous protein productivity by genome reduction in Lactococcus lactis NZ9000. Microb Cell Fact. 2017;16:1. doi: 10.1186/s12934-016-0616-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vinogradov E, Sadovskaya I, Grard T, Chapot-Chartier MP. Structural studies of the rhamnose-rich cell wall polysaccharide of Lactobacillus casei BL23. Carbohydr Res. 2016;435:156–161. doi: 10.1016/j.carres.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Nadal I, Rico J, Perez-Martinez G, Yebra MJ, Monedero V. Diacetyl and acetoin production from whey permeate using engineered Lactobacillus casei. J Ind Microbiol Biotechnol. 2009;36:1233–1237. doi: 10.1007/s10295-009-0617-9. [DOI] [PubMed] [Google Scholar]
- 9.Bosma EF, Forster J, Nielsen AT. Lactobacilli and pediococci as versatile cell factories—evaluation of strain properties and genetic tools. Biotechnol Adv. 2017;35:419–442. doi: 10.1016/j.biotechadv.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 10.De Boeck R, Sarmiento-Rubiano LA, Nadal I, Monedero V, Perez-Martinez G, Yebra MJ. Sorbitol production from lactose by engineered Lactobacillus casei deficient in sorbitol transport system and mannitol-1-phosphate dehydrogenase. Appl Microbiol Biotechnol. 2010;85:1915–1922. doi: 10.1007/s00253-009-2260-9. [DOI] [PubMed] [Google Scholar]
- 11.Overbeck T, Steele JL, Broadbent JR. Fermentation of de-oiled algal biomass by Lactobacillus casei for production of lactic acid. Bioprocess Biosyst Eng. 2016;39:1817–1823. doi: 10.1007/s00449-016-1656-z. [DOI] [PubMed] [Google Scholar]
- 12.Maze A, Boel G, Zuniga M, Bourand A, Loux V, Yebra MJ, Monedero V, Correia K, Jacques N, Beaufils S, et al. Complete genome sequence of the probiotic Lactobacillus casei strain BL23. J Bacteriol. 2010;192:2647–2648. doi: 10.1128/JB.00076-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu M, Nauta A, Francke C, Siezen RJ. Comparative genomics of enzymes in flavor-forming pathways from amino acids in lactic acid bacteria. Appl Environ Microbiol. 2008;74:4590–4600. doi: 10.1128/AEM.00150-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siezen RJ, van Enckevort FH, Kleerebezem M, Teusink B. Genome data mining of lactic acid bacteria: the impact of bioinformatics. Curr Opin Biotechnol. 2004;15:105–115. doi: 10.1016/j.copbio.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 15.Xin Y, Guo T, Mu Y, Kong J. Identification and functional analysis of potential prophage-derived recombinases for genome editing in Lactobacillus casei. FEMS Microbiol Lett. 2017;364:fnx243. doi: 10.1093/femsle/fnx243. [DOI] [PubMed] [Google Scholar]
- 16.Song L, Cui H, Tang L, Qiao X, Liu M, Jiang Y, Cui W, Li Y. Construction of upp deletion mutant strains of Lactobacillus casei and Lactococcus lactis based on counterselective system using temperature-sensitive plasmid. J Microbiol Methods. 2014;102:37–44. doi: 10.1016/j.mimet.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 17.Maguin E, Duwat P, Hege T, Ehrlich D, Gruss A. New thermosensitive plasmid for gram-positive bacteria. J Bacteriol. 1992;174:5633–5638. doi: 10.1128/jb.174.17.5633-5638.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ullrich S, Schuler D. Cre-lox-based method for generation of large deletions within the genomic magnetosome island of Magnetospirillum gryphiswaldense. Appl Environ Microbiol. 2010;76:2439–2444. doi: 10.1128/AEM.02805-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki N, Okayama S, Nonaka H, Tsuge Y, Inui M, Yukawa H. Large-scale engineering of the Corynebacterium glutamicum genome. Appl Environ Microbiol. 2005;71:3369–3372. doi: 10.1128/AEM.71.6.3369-3372.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishnakumar R, Grose C, Haft DH, Zaveri J, Alperovich N, Gibson DG, Merryman C, Glass JI. Simultaneous non-contiguous deletions using large synthetic DNA and site-specific recombinases. Nucleic Acids Res. 2014;42:e111. doi: 10.1093/nar/gku509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leprince A, de Lorenzo V, Voller P, van Passel MW, Martins dos Santos VA. Random and cyclical deletion of large DNA segments in the genome of Pseudomonas putida. Environ Microbiol. 2012;14:1444–1453. doi: 10.1111/j.1462-2920.2012.02730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu BJ, Kang KH, Lee JH, Sung BH, Kim MS, Kim SC. Rapid and efficient construction of markerless deletions in the Escherichia coli genome. Nucleic Acids Res. 2008;36:e84. doi: 10.1093/nar/gkn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sternberg N, Hamilton D. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J Mol Biol. 1981;150:467–486. doi: 10.1016/0022-2836(81)90375-2. [DOI] [PubMed] [Google Scholar]
- 24.Araki K, Araki M, Yamamura K. Targeted integration of DNA using mutant lox sites in embryonic stem cells. Nucleic Acids Res. 1997;25:868–872. doi: 10.1093/nar/25.4.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoess RH, Wierzbicki A, Abremski K. The role of the loxP spacer region in P1 site-specific recombination. Nucleic Acids Res. 1986;14:2287–2300. doi: 10.1093/nar/14.5.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darmon E, Leach DR. Bacterial genome instability. Microbiol Mol Biol Rev. 2014;78:1–39. doi: 10.1128/MMBR.00035-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen C, Ai L, Zhou F, Wang L, Zhang H, Chen W, Guo B. Complete genome sequence of the probiotic bacterium Lactobacillus casei LC2W. J Bacteriol. 2011;193:3419–3420. doi: 10.1128/JB.05017-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Loir Y, Gruss A, Ehrlich SD, Langella P. A nine-residue synthetic propeptide enhances secretion efficiency of heterologous proteins in Lactococcus lactis. J Bacteriol. 1998;180:1895–1903. doi: 10.1128/jb.180.7.1895-1903.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Sullivan DJ, Klaenhammer TR. High- and low-copy-number Lactococcus shuttle cloning vectors with features for clone screening. Gene. 1993;137:227–231. doi: 10.1016/0378-1119(93)90011-Q. [DOI] [PubMed] [Google Scholar]
- 30.Yu BJ, Sung BH, Koob MD, Lee CH, Lee JH, Lee WS, Kim MS, Kim SC. Minimization of the Escherichia coli genome using a Tn5-targeted Cre/loxP excision system. Nat Biotechnol. 2002;20:1018–1023. doi: 10.1038/nbt740. [DOI] [PubMed] [Google Scholar]
- 31.Kolisnychenko V, Plunkett G, 3rd, Herring CD, Feher T, Posfai J, Blattner FR, Posfai G. Engineering a reduced Escherichia coli genome. Genome Res. 2002;12:640–647. doi: 10.1101/gr.217202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goryshin IY, Naumann TA, Apodaca J, Reznikoff WS. Chromosomal deletion formation system based on Tn5 double transposition: use for making minimal genomes and essential gene analysis. Genome Res. 2003;13:644–653. doi: 10.1101/gr.611403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hashimoto M, Ichimura T, Mizoguchi H, Tanaka K, Fujimitsu K, Keyamura K, Ote T, Yamakawa T, Yamazaki Y, Mori H, et al. Cell size and nucleoid organization of engineered Escherichia coli cells with a reduced genome. Mol Microbiol. 2005;55:137–149. doi: 10.1111/j.1365-2958.2004.04386.x. [DOI] [PubMed] [Google Scholar]
- 34.Anderson JC, Clarke EJ, Arkin AP, Voigt CA. Environmentally controlled invasion of cancer cells by engineered bacteria. J Mol Biol. 2006;355:619–627. doi: 10.1016/j.jmb.2005.10.076. [DOI] [PubMed] [Google Scholar]
- 35.Selle K, Klaenhammer TR, Barrangou R. CRISPR-based screening of genomic island excision events in bacteria. Proc Natl Acad Sci USA. 2015;112:8076–8081. doi: 10.1073/pnas.1508525112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Posfai G, Plunkett G, 3rd, Feher T, Frisch D, Keil GM, Umenhoffer K, Kolisnychenko V, Stahl B, Sharma SS, de Arruda M, et al. Emergent properties of reduced-genome Escherichia coli. Science. 2006;312:1044–1046. doi: 10.1126/science.1126439. [DOI] [PubMed] [Google Scholar]
- 37.Wan X, Usvalampi AM, Saris PE, Takala TM. A counterselection method for Lactococcus lactis genome editing based on class IIa bacteriocin sensitivity. Appl Microbiol Biotechnol. 2016;100:9661–9669. doi: 10.1007/s00253-016-7828-6. [DOI] [PubMed] [Google Scholar]
- 38.Grossiord BP, Luesink EJ, Vaughan EE, Arnaud A, de Vos WM. Characterization, expression, and mutation of the Lactococcus lactis galPMKTE genes, involved in galactose utilization via the Leloir pathway. J Bacteriol. 2003;185:870–878. doi: 10.1128/JB.185.3.870-878.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Q, Shah NP. The potential of species-specific tagatose-6-phosphate (T6P) pathway in Lactobacillus casei group for galactose reduction in fermented dairy foods. Food Microbiol. 2017;62:178–187. doi: 10.1016/j.fm.2016.10.027. [DOI] [PubMed] [Google Scholar]
- 40.Song X, Huang H, Xiong Z, Ai L, Yang S. CRISPR-Cas9D10A nickase-assisted genome editing in Lactobacillus casei. Appl Environ Microbiol. 2017;83:e01259-17. doi: 10.1128/AEM.01259-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu D, Zhao K, Xu H, Zhang X, Bai Y, Saris PEJ, Qiao M. Construction of thyA deficient Lactococcus lactis using the Cre-loxP recombination system. Ann Microbiol. 2014;65:1659–1665. doi: 10.1007/s13213-014-1005-x. [DOI] [Google Scholar]
- 42.Fukiya S, Mizoguchi H, Mori H. An improved method for deleting large regions of Escherichia coli K-12 chromosome using a combination of Cre/loxP and λ Red. FEMS Microbiol Lett. 2004;234:325–331. doi: 10.1016/j.femsle.2004.03.042. [DOI] [PubMed] [Google Scholar]
- 43.Albert H, Dale EC, Lee E, Ow DW. Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J. 1995;7:649–659. doi: 10.1046/j.1365-313X.1995.7040649.x. [DOI] [PubMed] [Google Scholar]
- 44.Hazebrouck S, Pothelune L, Azevedo V, Corthier G, Wal JM, Langella P. Efficient production and secretion of bovine beta-lactoglobulin by Lactobacillus casei. Microb Cell Fact. 2007;6:12. doi: 10.1186/1475-2859-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun Z, Kong J, Kong W. Characterization of a cryptic plasmid pD403 from Lactobacillus plantarum and construction of shuttle vectors based on its replicon. Mol Biotechnol. 2010;45:24–33. doi: 10.1007/s12033-010-9242-0. [DOI] [PubMed] [Google Scholar]
- 46.Xin Y, Guo T, Mu Y, Kong J. Development of a counterselectable seamless mutagenesis system in lactic acid bacteria. Microb Cell Fact. 2017;16:116. doi: 10.1186/s12934-017-0731-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Strategies for obtaining linear donor disruption cassette H1-lox66-loxP-cat-loxP-H2. Figure S2. Structure of plasmids used in this study. Arrow heads show genes and their direction. (A) LCABL_13040-50-60-expression plasmid pMSP456. (B) Cre-expression plasmid pMSPCre. LCABL_13040-50-60 and Cre were transcribed from PnisA that is controlled by the product of nisin. Figure S3. Deletion of large-scale genome region containing important gene(s) in L. casei BL23. (A) Layout and genetic context of the ~ 12.8 kb genome region and the size of PCR products amplified with primers H1F and H2R from BLD2 strains. (B) Colony PCR results. M: DNA marker; C: wild-type strain; 1–20: chloramphenicol resistance colonies. (C) Growth of L. casei BL23 and the deletion strain L. casei BLD2 on MRS. Figure S4. Cellular morphologies of L. casei strains BL23 and BLD2. Figure S5. Growth of L. casei BL23 and the deletion strain L. casei BLD3 on CDM medium containing galactose.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article.