

Abstract
Although dengue virus (DENV) antibodies can neutralize or enhance Zika virus (ZIKV) infection in vitro, their contribution to ZIKV infection in vivo remains unclear. In a recent issue of Science, Bardina et al. (2017) explore the protective versus pathogenic roles of DENV-immune antibodies in ZIKV infection using a mouse model.
The genus Flavivirus comprises more than 70 members and includes several medically relevant viruses, such as dengue (DENV), yellow fever (YFV), West Nile (WNV), and Zika (ZIKV). DENV infects 390 million people annually and causes disease ranging from mild dengue fever to severe dengue hemorrhagic fever/dengue shock syndrome (Bhatt et al., 2013). DENV exists as four similar, but distinct, serotypes (DENV1–DENV4) that share 65%–70% amino acid homology. The preexisting antibody response induced by one DENV serotype can enhance infection and disease severity upon secondary infection with heterologous DENV serotype via the phenomenon known as antibody-dependent enhancement (ADE) (Halstead, 2007). The envelope (E) protein of flaviviruses is the major target of the antibody response. The E proteins of DENV1–DENV4 and ZIKV are 54%–59% identical at the amino acid level, and these flaviviruses share high structural similarity (Dejnirattisai et al., 2016). As the 2015–2016 ZIKV outbreak in South America occurred mainly in DENV-endemic countries and territories, the frequency of DENV-exposed individuals was high in most areas affected by ZIKV. Considering this co-circulation of these two viruses and their close antigenic relationship at the antibody level, the severe consequences of ZIKV infection—congenital Zika syndrome and Guillain-Barre syndrome—may in part be due to the influence of prior DENV antibodies during subsequent ZIKV infection. As ZIKV spreads further into DENV-endemic regions and the geographic ranges of these viruses continue to merge, studies evaluating the impact of prior DENV infection on the outcome of ZIKV infection are urgently required.
However, epidemiologic studies (both retrospective and prospective) will take several years to perform. Therefore, multiple laboratories have used cell culture models of ZIKV infection and observed that, in vitro, DENV and ZIKV cross-reactive antibodies can reciprocally neutralize or promote ADE of ZIKV and DENV (Harrison, 2016), suggesting that pre-existing antibodies in DENV-immune individuals might contribute to ZIKV pathogenesis under certain conditions. Using a mouse model of experimental ZIKV infection, Bardina et al. (2017) provide experimental evidence for ADE of ZIKV infection and pathogenesis mediated by DENV-immune antibodies.
Specifically, Bardina et al. (2017) harvested convalescent plasma from large numbers of DENV- or WNV-infected people (141 DENV and 146 WNV cases) and evaluated neutralization and enhancement capacities of these plasma samples against ZIKV in vitro and in vivo. In vitro studies using the human erythroleukemia cell line K562 showed that only the highly cross-reactive DENV-immune plasma samples were capable of neutralizing ZIKV in vitro, whereas both DENV- and WNV-immune plasma enhanced ZIKV infection, with higher ADE effects observed with DENV-immune plasma relative to WNV-immune plasma. Inhibitor- or antibody-mediated FcγR blockade experiments using purified IgG from plasma and IgG-depleted plasma confirmed that the in vitro ADE induced by the DENV- and WNV-immune human plasma required the IgG-FcγR interaction. To evaluate whether ADE could occur during ZIKV infection in vivo, Bardina et al. (2017) passively transferred Stat2−/− C57BL/6 mice—which, unlike wild-type mice, can support significant levels of ZIKV replication and manifest lethal disease due to the absence of a functional interferon response—with pooled immune plasma from control or DENV-exposed donors and then challenged them with ZIKV. Relative to mice that received control plasma and survived the ZIKV challenge after exhibiting weight loss and clinical disease, mice administered a low amount of DENV-immune plasma (20 or 2 μL/mouse) manifested greater weight loss and clinical disease and succumbed to ZIKV challenge, while those dosed with a higher amount of DENV-immune plasma (200 μL/mouse) showed no disease or weight loss. These results demonstrate that the same antibodies can mediate both protection and enhancement depending on the concentration (Figure 1), in agreement with studies that demonstrated ADE of DENV in mouse models (Balsitis et al., 2010; Zellweger et al., 2010). This finding has important implications for the DENV/ZIKV pathogenesis and vaccine development fields, as it suggests that, under certain conditions, antibodies induced by prior DENV infection or vaccination may precipitate severe ZIKV disease manifestations and vice versa (Richner et al., 2017). The precise features of the flaviviral antibody response (in terms of antibody concentration, iso-type, affinity, specificity, in vitro binding and neutralization titer, and effector functions, such as complement fixation and antibody-dependent cellular cytotoxicity) that contribute to protection versus pathogenesis remain to be fully understood.
Figure 1.
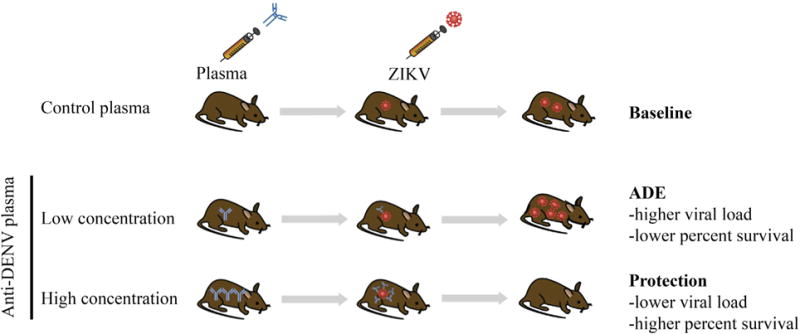
Relationship between DENV-Immune Human Plasma Level and ZIKV Disease Outcome in Mice
By revealing the impact of DENV-immune antibodies on mediating both protection against and pathogenesis of ZIKV infection in vivo, Bardina et al. (2017) have addressed a critical question about the potential role of flavivirus cross-reactive antibodies in modulating the outcome of ZIKV infection. In doing so, they have set the stage to answer the next central question related to naturally occurring flavivirus infections: what is the role of prior DENV humoral versus cell-mediated immunity during subsequent ZIKV infection in the same host? As the precise contributions of cellular versus humoral immunity cannot be fully dissected in humans and epidemiologic studies evaluating complex interplays between antibody and T cell responses to flaviviruses in people will take years, animal models are again poised to provide key insights about flaviviral immunity. Although studies to date have focused on the antibody response to ZIKV, experimental evidence revealing a protective role for CD8 T cells against ZIKV (Elong Ngono et al., 2017) and cross-reactivity between DENV- and ZIKV-immune CD8 T cells (Wen et al., 2017) have recently become available, implicating roles for both antibodies and T cells in protection versus pathogenesis in ZIKV infection.
By providing direct experimental evidence for ADE of ZIKV infection and disease, similar to that for DENV (Balsitis et al., 2010; Zellweger et al., 2010), Bardina et al. (2017) raise another critical question in the flavivirus field: what are the molecular and cellular mechanisms of ADE? As we wait for epidemiologic study results, animal models, in combination with primary human cell culture models, will again play an important role in investigating many ADE-related questions that need to be addressed urgently. The net impact of the global research effort through clinical cases, epidemiologic studies, and vaccine and antiviral trials, together with experimental studies using animal and primary human cell culture models, should decipher mechanisms of DENV/ZIKV cross-immunity and pathogenesis.
References
- Balsitis SJ, Williams KL, Lachica R, Flores D, Kyle JL, Mehlhop E, Johnson S, Diamond MS, Beatty PR, Harris E. PLoS Pathog. 2010;6:e1000790. doi: 10.1371/journal.ppat.1000790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardina SV, Bunduc P, Tripathi S, Duehr J, Frere JJ, Brown JA, Nachbagauer R, Foster GA, Krysztof D, Tortorella D, et al. Science. 2017;356:175–180. doi: 10.1126/science.aal4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, et al. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejnirattisai W, Supasa P, Wongwiwat W, Rouvinski A, Barba-Spaeth G, Duangchinda T, Sakuntabhai A, Cao-Lormeau VM, Malasit P, Rey FA, et al. Nat Immunol. 2016;17:1102–1108. doi: 10.1038/ni.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elong Ngono A, Vizcarra EA, Tang WW, Sheets N, Joo Y, Kim K, Gorman MJ, Diamond MS, Shresta S. Cell Host Microbe. 2017;21:35–46. doi: 10.1016/j.chom.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead SB. Lancet. 2007;370:1644–1652. doi: 10.1016/S0140-6736(07)61687-0. [DOI] [PubMed] [Google Scholar]
- Harrison SC. Nat Immunol. 2016;17:1010–1012. doi: 10.1038/ni.3539. [DOI] [PubMed] [Google Scholar]
- Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, et al. Cell. 2017;169:176. doi: 10.1016/j.cell.2017.03.016. [DOI] [PubMed] [Google Scholar]
- Wen J, Tang WW, Sheets N, Ellison J, Sette A, Kim K, Shresta S. Nat Microbiol. 2017;2:17036. doi: 10.1038/nmicrobiol.2017.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger RM, Prestwood TR, Shresta S. Cell Host Microbe. 2010;7:128–139. doi: 10.1016/j.chom.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]