

Abstract
Aim
The aim of this study was to study potential cytochrome P450 (CYP) induction by dicloxacillin.
Methods
We performed an open‐label, randomized, two‐phase, five‐drug clinical pharmacokinetic cocktail crossover study in 12 healthy men with and without pretreatment with 1 g dicloxacillin three times daily for 10 days. Plasma and urine were collected over 24 h and the concentration of all five drugs and their primary metabolites was determined using a liquid chromatography coupled to triple quadrupole mass spectrometry method. Cryopreserved primary human hepatocytes were exposed to dicloxacillin for 48 h and changes in gene expression and the activity of CYP3A4, CYP2C9, CYP2B6 and CYP1A2 were investigated. The activation of nuclear receptors by dicloxacillin was assessed using luciferase assays.
Results
A total of 10 days of treatment with dicloxacillin resulted in a clinically and statistically significant reduction in the area under the plasma concentration–time curve from 0 to 24 h for omeprazole (CYP2C19) {geometric mean ratio [GMR] [95% confidence interval (CI)]: 0.33 [0.24, 0.45]}, tolbutamide (CYP2C9) [GMR (95% CI): 0.73 (0.65, 0.81)] and midazolam (CYP3A4) [GMR (95% CI): 0.54 (0.41, 0.72)]. Additionally, other relevant pharmacokinetic parameters were affected, indicating the induction of CYP2C‐ and CYP3A4‐mediated metabolism by dicloxacillin. Investigations in primary hepatocytes showed a statistically significant dose‐dependent increase in CYP expression and activity by dicloxacillin, caused by activation of the pregnane X receptor.
Conclusions
Dicloxacillin is an inducer of CYP2C‐ and CYP3A‐mediated drug metabolism, and we recommend caution when prescribing dicloxacillin to users of drugs with a narrow therapeutic window.
What is Already Known about this Subject
Dicloxacillin is known to reduce the therapeutic efficacy of warfarin treatment but the mechanism is unknown.
What this Study Adds
This study shows that dicloxacillin is a clinically relevant inducer of cytochrome P450 (CYP2C9)‐, CYP2C19‐ and CYP3A4‐mediated expression and activity. In vitro results show that this is caused by pregnane X receptor activation, leading to increased expression and activity of these enzymes. Clinicians prescribing dicloxacillin should be aware that dicloxacillin induces CYP2C9‐, CYP2C19‐ and CYP3A4‐mediated metabolism.
Introduction
Dicloxacillin belongs to the group of narrow‐spectrum isoxazolyl beta‐lactam penicillins (Figure 1) and is primarily used for skin‐, soft tissue‐, or bone infections caused by Staphylococcus aureus 1. As dicloxacillin exhibits strong time‐dependent antibacterial activity, the time above the minimal inhibitory concentration (0.125 μg ml–1) is crucial for its clinical effect 2. Due to the short elimination half‐life of dicloxacillin (60–90 min after oral ingestion 3, 4), administration every 6–8 h is necessary to sustain sufficient bactericidal concentrations. Following an oral dose of 500 mg dicloxacillin administered four times daily, plasma concentrations fluctuate from 2 μM, the minimum plasma concentration (Cmin), to 57 μM, the maximum plasma concentration (Cmax) 4. Data on the utilization patterns of beta‐lactamase‐resistant penicillins in general, and dicloxacillin specifically, are scarce 5. Dicloxacillin is the dominant choice of isoxazolyl penicillin in both Denmark and Norway, with annual prevalences of 25 6 and 20 (www.reseptregisteret.no) per 1000 inhabitants, respectively.
Figure 1.
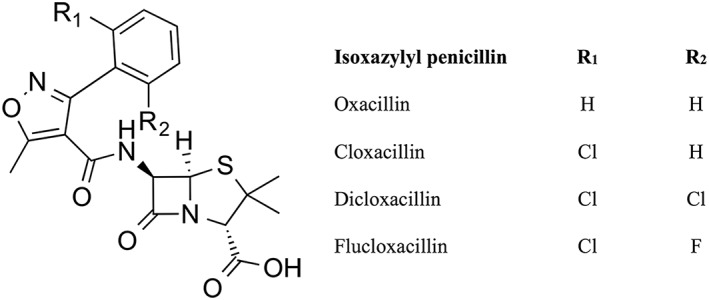
The chemical structure of isoxazolyl penicillins
In humans, dicloxacillin is primarily excreted unchanged renally, but also metabolized to some extent to the 5‐hydroxy metabolite 7, although it is unclear which cytochrome P450 (CYP) enzymes are involved in the metabolism. Dicloxacillin is a substrate of P‐glycoprotein (P‐gp, ABCB1) 8, and concomitant intake of rifampicin leads to lower plasma concentrations of dicloxacillin, accompanied by increased plasma concentrations of the 5‐hydroxy metabolite. This is likely to be caused by a combination of induction of CYP enzymes relevant for dicloxacillin metabolism and induction of intestinal P‐gp 9.
Several case reports have described that initiation of either dicloxacillin or cloxacillin treatment leads to a decreased international normalized ratio (INR), a proxy biomarker of clinical efficacy, during treatment with the vitamin K antagonist, warfarin 10, 11, 12, 13. In a recent observational study of 236 patients, we confirmed that initiation of dicloxacillin treatment resulted in markedly decreased INR values during warfarin therapy 14, leading to subtherapeutic INR levels in 60% of the treated patients. Additional case reports have described that initiation of treatment with another isoxazolyl penicillin, flucloxacillin, resulted in decreased plasma levels of voriconazole 15 and quinidine 16. The mechanistic basis for drug–drug interactions with isoxazolyl penicillins is unknown. Sparse in vitro data have indicated that dicloxacillin activates pregnane X receptor (PXR), causing increased CYP3A4‐mediated testosterone metabolism in human hepatocytes 17 and induction of CYP2C9 18. PXR activation leading to upregulation of CYP enzymes and subsequent increased metabolism may provide an explanation for the above‐mentioned drug–drug interactions, but this has not been confirmed in clinical pharmacokinetic studies.
The objective of the present study was to determine the effect of treatment with a clinically relevant treatment course of dicloxacillin on the metabolic capacity of CYP3A4, CYP2C9, CYP2C19, CYP1A2 and CYP2D6 in healthy volunteers. We investigated the underlying mechanism in vitro using cryopreserved human hepatocytes to assess changes in the expression and activity of CYP enzymes and nuclear receptor activation by dicloxacillin.
Methods
Clinical study
The study was designed as an open‐label, randomized, two‐phase, five‐drug clinical pharmacokinetic cocktail study to assess the potential of dicloxacillin to induce CYP enzymes. We used a validated and previously used five‐drug cocktail 19, 20, 21 to assess metabolic capacity of major drug‐metabolizing enzymes (CYP2C9: tolbutamide; CYP3A4: midazolam; CYP2C19: omeprazole; CYP1A2: caffeine; and CYP2D6: dextromethorphan).
Pharmacokinetic cocktail studies are used to assess the metabolic capacity of CYP3A4, CYP2C19, CYP2D6 and CYP1A2 simultaneously by pharmacokinetic assessment of CYP‐specific probes. We used a modified Cooperstown cocktail (midazolam, omeprazole, dextromethorphan and caffeine) 22. As (S)‐warfarin (the most pharmacologically active isomer of racemic warfarin) is metabolized by CYP2C9, we added tolbutamide, a well‐validated substrate and biomarker for CYP2C9 metabolism 23, to the cocktail. This combination of biomarker drugs has previously been validated 19 and used to evaluate the changes in drug metabolism caused by drug–drug interactions 20, 21.
Study medication and dose
Tolbutamide (Arcosal® one 500 mg tablet, Meda AS, Allerød, Denmark), dextromethorphan (Dexofan® one 30 mg tablet, Takeda Pharma, Taastrup, Denmark), omeprazole (Omestad® one 20 mg enteric capsule, Stada Nordic ApS, Herlev, Denmark) and caffeine (two 100 mg caffeine tablets produced at Glostrup Hospital Pharmacy, Copenhagen, Denmark) were all administered orally. Midazolam was administered as a buccal solution of 2.5 mg (Buccolam®, Shire Sweden AB, Stockholm, Sweden) between the cheek and the gums slowly, at least 3 min after the oral drugs were ingested. Dicloxacillin was administered as two 500 mg capsules (Dicloxacillin ‘Alternova’®, Alternova A/S, Odense, Denmark) three times daily for 10 days; at least 1 h before or at least 2 h after a meal. This is the dicloxacillin dosage regimen for the treatment of Staphylococcus aureus in Denmark, as recommended by the Danish Physicians' Desk Reference (www.pro.medicin.dk).
Design and study criteria
Twelve healthy men were recruited for this two‐phase study and ingested the five‐drug cocktail in both phases after 12 h of fasting. Women were excluded owing to a putative clinically relevant drug–drug interaction between oral contraceptives and dicloxacillin. In phase A, no concomitant drugs were used. In phase B, dicloxacillin was administered as 1 g three times daily for 10 days prior to the study day. On the morning of day 11, the five‐drug cocktail was ingested as in phase A. There was a washout period of at least 6 weeks between the two phases (Figure 2). Plasma samples were drawn at 0, 0.5, 1, 2, 3, 4, 6, 8, 10, 12 and 24 h. Urine was collected at 0–12 h and 12–24 h for determination of metabolites. The participants were given a meal 3 h after ingesting the drugs. The inclusion criteria for the study were: nonsmoking healthy men aged 18–55 years, body mass index (BMI) 18.5–29.9 kg m–2 and estimated glomerular filtration rate (eGFR), alanine transaminase (ALAT), bilirubin, haemoglobin and glycosylated haemoglobin A1c (HbA1c) within the respective reference ranges. Exclusion criteria for the study were: known allergies to any of the used drugs; known penicillin allergy or type 1 reaction to cephalosporines; known allergy to sulphonylureas; intake of prescription drugs, over‐the‐counter drugs, herbal medicines or supplements known to affect drug pharmacokinetics; chronic or daily alcohol abuse; and participation in other intervention trials. Health status and use of drugs were assessed by answers to the following five questions: Are you healthy? Do you suffer from any chronic diseases? Do you ingest drugs on a daily basis? Do you occasionally use prescription drugs? Do you use any over‐the‐counter or herbal medicines or supplements? A data manager performed block randomization using Sealed Envelope, which is freely available at www.sealedenvelope.com. The list was then made accessible to trial investigators using REDCap 24.
Figure 2.
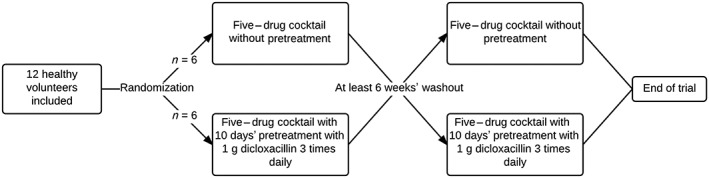
Flowchart of clinical study with 12 healthy men. All individuals completed the study
Study approval
The clinical study was conducted in accordance with the Helsinki Declaration and Good Clinical Practice (GCP) and monitored by the GCP Unit, Odense University Hospital, Odense, Denmark. The study protocol was approved by the Danish Medicines Agency (identifier 2 016 043 478), registered in the EudraCT database (identifier 2016–001334‐10) and the Regional Scientific Ethical Committee of Southern Denmark (identifier S‐20160073), and all subjects consented to participate in the study. The trial was registered at http://www.clinicaltrials.gov (identifier NCT02983890).
Determination of drugs and their metabolites in plasma and urine
The analysis for the five probe drugs for major CYP isoenzymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4) was performed in plasma and urine samples. The samples were analysed for caffeine, paraxanthine, omeprazole, 5‐hydroxyomeprazole, tolbutamide, 4‐hydroxytolbutamide, dextromethorphan, dextrorphan, midazolam and α‐hydroxymidazolam. The analysis of plasma was performed according to the method described by Wohlfarth et al. 19 with minor modification, using isotope dilution, solid‐phase extraction (SPE) and liquid chromatography coupled to triple quadrupole mass spectrometry (LC–MS/MS). Urine samples were deglucuronidated prior to the addition of the isotope‐labelled internal standard and dilution with the mobile phase before injection into the LC–MS/MS system. Analysis of omeprazole and its metabolite in urine was performed prior to the deglucuronidation procedure as these compounds are not stable during this acidic process. The LC–MS/MS system consisted of a Dionex Ultimate 3000 UHPLC system equipped with an EQuan MAX autosampler unit; a Dionex Ultimate 3000 RS column compartment; and an Ultimate 3000 RS Pump, connected to a TSQ Quantiva Triple Quadrupole Mass Spectrometer, with heated‐electrospray ionization operated in positive mode (Thermo Scientific, San José, CA, USA). Data acquisition was performed in single reaction monitoring (SRM) mode, with transitions optimized for all the compounds. Calibration curves, blanks and quality control samples were included in each batch of samples analysed. The method was linear, with r > 0.9992. The between‐day reproducibility and intra‐day repeatability was <5.2% and <5.5%, respectively, for all compounds. The accuracy reported as bias ranged from −2.6% to 1.8%. The limit of detection ranged from 0.05 ng ml–1 to 0.75 ng ml–1, and the limit of quantification ranged from 0.1 ng ml–1 to 1.5 ng ml–1 19.
Genotyping
Briefly, a Maxwell™ 16 Blood DNA kit (Promega, Madison, WI, USA) was used to isolate DNA from whole‐blood samples according to the manufacturer's protocol. Single nucleotide polymorphisms [CYP2C9*2/*3, CYP2C19*2/*3/*17 and CYP2D6*3/*4/*6/copy number variation (*N), (Table S1)] were genotyped using predesigned TaqMan SNP genotyping assays on a StepOne Plus real‐time instrument (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's protocol.
Statistics and pharmacokinetic analysis
The primary endpoint of the study was the difference in the area under the plasma concentration–time curve from 0 to 24 h (AUC0–24 h) for tolbutamide with and without dicloxacillin. To detect a difference of ≥25% in tolbutamide AUC0–24 h with 80% power, a two‐sided significance level of 5% and, allowing a dropout rate of 20%, a total of 12 healthy individuals were required. Demographic data are shown as medians with interquartile ranges (IQR; 25th–75th percentiles). Pharmacokinetic endpoints are presented as medians with IQR and geometric mean ratios, with 95% confidence intervals. Statistical significance was determined using paired t‐tests and accepted at P < 0.05. Pharmacokinetic data were analysed by noncompartmental analysis using the R package NCAPPC 25. The AUC0‐t was estimated using the linear‐up/logarithmic‐down method. The formation clearance (CLf) and renal clearance (CLR) were estimated using the following equations:
Due to the short elimination half‐lives of omeprazole and midazolam, 0–12 h CLf and CLR were calculated, while 0–24 h values were calculated for caffeine, dextromethorphan and tolbutamide.
In vitro
Cell culture
The methods for cell culture, gene expression and CYP activities in human hepatocytes have been described in detail previously 26. Briefly, cryopreserved human hepatocytes [male lots: HU1765 (ThermoFisher catalogue #HMCPIS (ThermoFisher Scientific, Waltham, MA, USA)), HUM4034 (Triangle Research Laboratories catalogue #HUCPI (Morrisville, NC, USA)); female lots: HH1057 (In Vitro ADMET Laboratories (Columbia, MD, USA) catalogue #82006), OII (Bioreclamation IVT (Westbury, NY, USA) catalogue #F00995‐P)] were plated at 70 000 cells/well in collagen‐coated 96‐well plates in CHRM™ medium (ThermoFisher Scientific, Waltham, MA, USA) to obtain a monolayer of cells. After 4 h of incubation, the cells were overlaid with cold incubation media containing 0.35 mg ml–1 GelTrex® (ThermoFisher Scientific, Waltham, MA, USA). The following day, the medium was replaced with fresh medium and incubated for another 24 h. On day 3, the cells were incubated for 48 h with dicloxacillin, flucloxacillin, rifampicin (PXR agonist, CYP3A and CYPC regulation), phenobarbital [constitutive androstane receptor (CAR) agonist, CYP2B6 regulation] or 3‐methylcholanthrene [‐aryl hydrocarbon receptor (AhR) agonist, CYP1A regulation] in increasing concentrations or with vehicle [0.1% dimethyl sulfoxide (DMSO)].
Gene expression and activity of CYP enzymes
Changes in gene expression for a panel of drug‐metabolizing enzymes and several transporters were determined using a QuantiGene Plex 2.0 assay kit (ThermoFisher Scientific, Waltham, MA, USA). The activity of relevant CYP enzymes was assessed using well‐validated probe drugs (CYP3A4: 100 μM testosterone; CYP2C9: 100 μM diclofenac; CYP2B6: 500 μM bupropion; and CYP1A2: 100 μM phenacetin). After incubation with dicloxacillin, flucloxacillin and positive controls, hepatocytes were incubated with probe drugs for 90 min and metabolite levels were measured using an established LC–MS/MS method 27. Activity and gene expression values are presented as activity/expression relative to vehicle (0.1% DMSO).
Nuclear receptor activity
Activation of PXR, CAR and AhR was assessed by Puracyp (Carlsbad, CA, USA), as previously described 28, 29. Briefly, a HepG2‐derived cell line stably transfected with nuclear receptors and corresponding response elements were seeded in 96‐well plates. After 24 h, the cells were exposed to six different concentrations of dicloxacillin and flucloxacillin, and then incubated for another 24 h. Receptor activity was assessed using ONE‐Glo® luciferase assays (Promega Corporation, Madison, WI, USA), and cytotoxicity was assessed using the CellTiter‐Fluor® assay (Promega Corporation). Rifampicin, 6‐(4‐Chlorophenyl)imidazo[2,1‐b][1,3]thiazole‐5‐carbaldehyde O‐(3,4‐dichlorobenzyl)oxime (CITCO) and 3‐methylcholanthrene were used as positive controls for PXR, CAR and AhR activation, respectively. Data for nuclear receptor activity were adjusted for viability and expressed relative to vehicle control.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 30, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 31, 32, 33.
Results
Clinical study
The study was an open, two‐phase, randomized, clinical pharmacokinetic crossover study in 12 healthy men (Figure 2). The median age was 22 years (IQR 21–24; range 21–29 years) and the median BMI was 24.3 kg m–2 (IQR 23.3–25.6; range 20.4–28.7). A five‐drug cocktail of midazolam (CYP3A4) (2.5 mg buccal), tolbutamide (CYP2C9) (500 mg tablet), omeprazole (CYP2C19) (20 mg enteric capsule), caffeine (CYP1A2) (two 100 mg tablets) and dextromethorphan (CYP2D6) (30 mg tablet) was administered before and after 10 days of treatment with 1 g dicloxacillin three times daily. The sequence of study phases was randomized to avoid period effects, and a washout period of at least 6 weeks was required between the two phases, to avoid carryover effects. Two subjects suffered nausea and dizziness following multiple attempts at insertion of the cubital vein cannula. Following treatment with dicloxacillin, one subject described dyspepsia and stomach pain, and one subject had an erythematous rash on the extremities, all of which are well‐described adverse reactions 34. All adverse reactions were transient and not deemed serious.
Pharmacogenetics
Subjects were genotyped for the most common functional genetic variants in CYP2D6 (*3; *4; *6 and copy number variation *N), CYP2C9 (*2; *3) and CYP2C19 (*2; *3; *17) after conclusion of the study. Genotypes for the 12 subjects are shown in Table S1. One CYP2D6 poor metabolizer, two CYP2C19 ultrarapid metabolizers and two CYP2C9 poor metabolizers were included in the study. The main analysis presented here is intent‐to‐treat, including all subjects, regardless of genotype. A sensitivity analysis which excluded individuals carrying altered function variants did not affect the conclusions of the study (Table S2).
Pharmacokinetics
AUC0–24 h, Cmax, elimination half‐life (T½) and CLf for the main metabolite of all five probe drugs with and without dicloxacillin exposure are shown in Table 1 and concentration–time curves for the five probes with and without dicloxacillin are depicted in Figure 3. Individual values of CLf with and without dicloxacillin exposure are shown in Figure 4. Detailed pharmacokinetic results are shown in Table S3, which contains pharmacokinetic data for all probe drugs and their metabolites. Full pharmacokinetic profiles were available for all 12 individuals, for all drugs except caffeine; two subjects were excluded from the caffeine analysis as they violated the protocol by ingesting caffeine during the trial.
Table 1.
Noncompartmental pharmacokinetic analysis of probes
| Drug | Parameter | Without dicloxacillin | With dicloxacillin | GMR (95% CI) |
|---|---|---|---|---|
|
Tolbutamide
(CYP2C9) |
AUC0–24 h (ng*h ml–1) | 621 507 (505775–744 162) | 404 391 (355015–582 480) | 0.73 (0.65, 0.81) |
| Cmax (ng ml–1) | 51 706 (48907–54 467) | 49 130 (47739–52 615) | 0.93 (0.87, 1.00) | |
| T½ (h) | 9.0 (6.8–13.5) | 6.1 (4.8–9.2) | 0.72 (0.65, 0.79) | |
| CLf (l h–1) | 0.06 (0.05–0.12) | 0.12 (0.09–0.16) | 1.64 (1.44, 1.87) | |
|
Omeprazole
(CYP2C19) |
AUC0–24 h (ng*h ml–1) | 228.4 (188.8–343.7) | 80.2 (63.7–101.1) | 0.33 (0.24, 0.45) |
| Cmax (ng ml–1) | 149.5 (105.5–242.2) | 58.0 (42.8–77.3) | 0.40 (0.25, 0.66) | |
| T½ (h) | 1.5 (1.2–3.5) | 3.5 (2.2–5.6) | 1.62 (0.71, 3.71) | |
| CLf (l h–1) | 1.9 (0.5–4.3) | 3.7 (1.7–7.5) | 2.01 (0.69, 5.89) | |
|
Midazolam
(CYP3A4) |
AUC0–24 h (ng*h ml–1) | 37.4 (31.9–44.0) | 23.5 (14.6–29.1) | 0.54 (0.41, 0.72) |
| Cmax (ng ml–1) | 12.0 (9.6–13.7) | 8.9 (6.2–12.5) | 0.77 (0.59, 1.01) | |
| T½ (h) | 7.6 (5.5–9.9) | 7.7 (6.3–8.5) | 1.08 (0.87, 1.33) | |
| CLf (l h–1) | 38 (31–43) | 67 (42–80) | 1.59 (1.23, 2.06) | |
|
Dextromethorphan
(CYP2D6) |
AUC0–24 h (ng*h ml–1) | 12.0 (5.4–24.5) | 4.5 (2.7–11.2) | 0.52 (0.32, 0.83) |
| Cmax (ng ml–1) | 1.15 (0.74–2.5) | 0.76 (0.65–0.95) | 0.69 (0.54, 0.89) | |
| T½ (h) (n = 6) | 13.9 (12.4–18.2) | 21.4 (11.8–25.1) | 1.25 (0.53, 2.93) | |
| CLf (l h–1) | 771 (275–1927) | 1561 (483–2800) | 1.57 (0.97, 2.55) | |
|
Caffeine
a
(CYP1A2) |
AUC0–24 h (ng*h ml–1) | 22 447 (20671–27 837) | 19 358 (14563–23 851) | 0.81 (0.71, 0.92) |
| Cmax (ng m–1l) | 3185 (2936–3890) | 3213 (2838–3422) | 0.92 (0.81, 1.04) | |
| T½ (h) | 4.5 (3.9–4.6) | 4.0 (3.0–4.8) | 0.88 (0.79, 0.99) | |
| CLf (1 h–1) | 0.45 (0.33–0.50) | 0.47 (0.38–0.62) | 1.17 (0.98, 1.39) |
Data are shown as medians with interquartile ranges. AUC0–24 h, area under the plasma concentration–time curve from 0 to 24 h; CI, confidence interval; Cmax, maximum plasma concentration; CLf: formation clearance of the main metabolite; CYP, cytochrome P450; GMR, geometric mean ratio; T½, half‐life
Pharmacokinetic data for only 10 individuals for caffeine are shown
Figure 3.
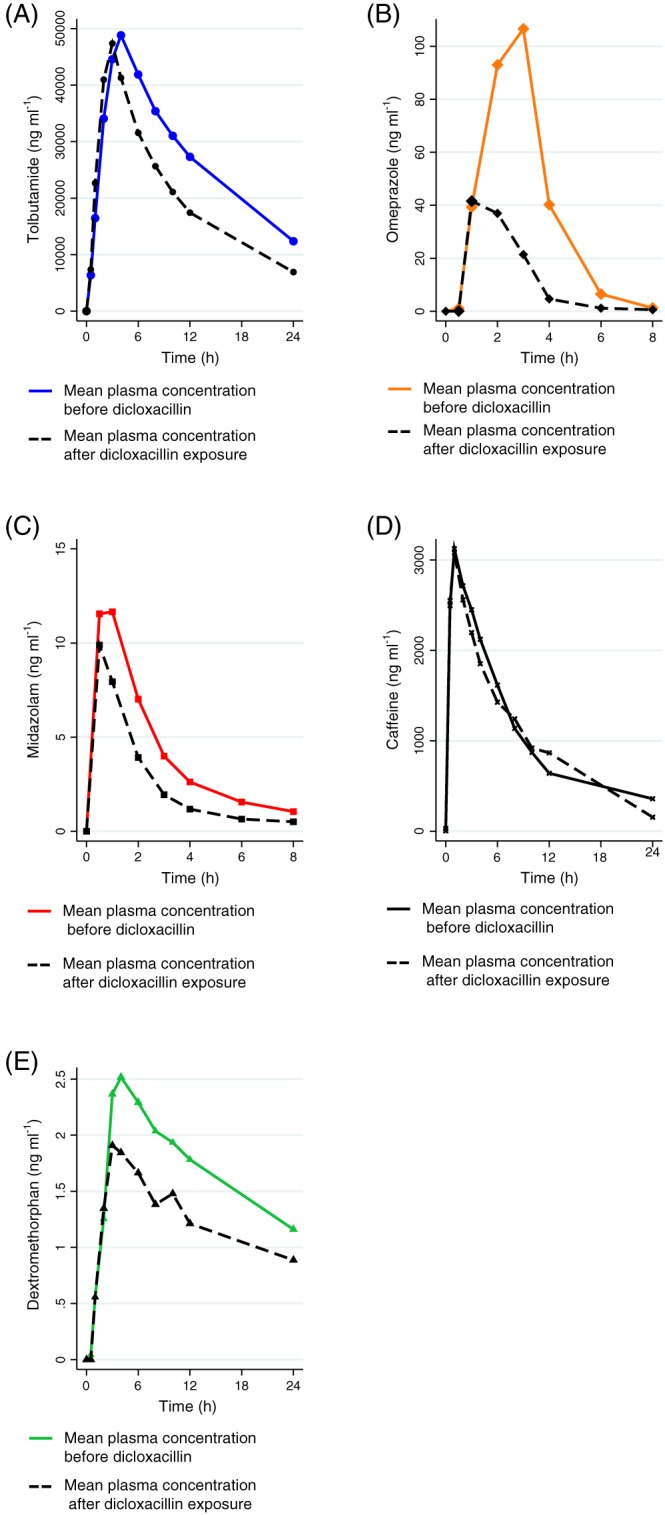
Plasma concentration–time curves for tolbutamide (A), omeprazole (B), midazolam (C) caffeine (D) and dextromethorphan (E) with and without dicloxacillin. Only 10 individuals were included in the caffeine group owing to a violation of the protocol
Figure 4.
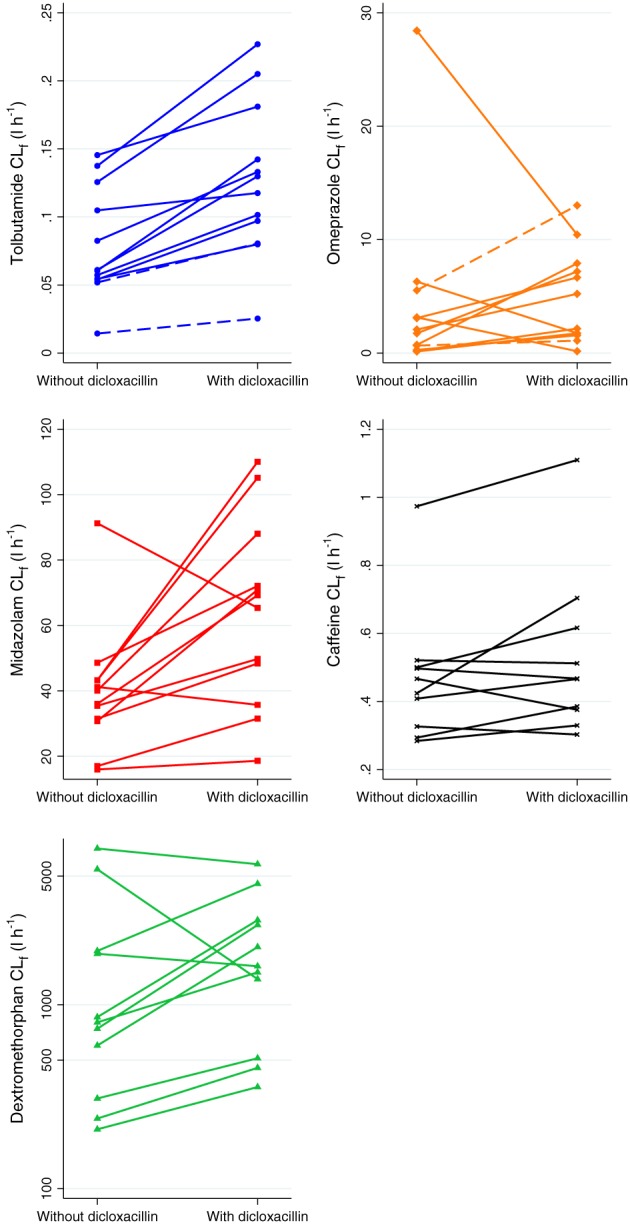
Formation clearance (CLf) for tolbutamide and midazolam with and without dicloxacillin. CLf was calculated as: Amount of metabolite in urine0–t/AUC of substrate0–t, where AUC is the area under the plasma concentration–time curve and t is time. One cytochrome P450 m(CYP) 2D6 poor metabolizer was excluded from the dextromethorphan graph, to avoid clustering of the remaining individuals. Two individuals were excluded from the caffeine analysis owing to ingestion of caffeine during the study. Dashed lines represent CYP2C9 poor metabolizers for tolbutamide and CYP2C19 ultrarapid metabolizers for omeprazole
With concomitant dicloxacillin treatment, the AUC0–24 h of all probe drugs was significantly reduced, without changes in CLR (Table 1, Table S3, Figure 3). Tolbutamide AUC0–24 h and Cmax were reduced by 27% and 7%, respectively, driven by increased formation of the 4‐hydroxytolbutamide metabolite, as reflected in a 64% increase in the CLf of the metabolite (Figure 4). Omeprazole AUC0–24 h and Cmax were 67% and 60% lower, respectively, with dicloxacillin exposure. Although the CLf of the CYP2C19‐mediated 5‐hydroxyomeprazole metabolite was twice as high with dicloxacillin exposure, this was not statistically significant {P = 0.18; geometric mean ratio [GMR] [95% confidence interval (CI): 2.01 (0.69, 5.89]}. Dicloxacillin treatment also led to a 46% reduction in midazolam AUC0–24 h, attributed to increased CYP3A4 metabolism, as reflected by a 59% increase in the CLf of the α‐hydroxymidazolam metabolite. The ratio of the midazolam‐to‐metabolite AUC0–24 h increased slightly, with significant imprecision [GMR (95% CI): 1.27 (0.89, 1.80)]. Dicloxacillin also caused minor changes in caffeine pharmacokinetics; caffeine AUC0–24 h was 19% lower with dicloxacillin with no change in CLf, suggesting that the changes in AUC0–24 h were not caused by increased CYP1A2‐mediated paraxanthine metabolism. Dicloxacillin also caused a marked 48% decrease in dextromethorphan AUC0–24 h.
Human hepatocytes
The effect of dicloxacillin on the expression and activity of CYP3A4, CYP2C9, CYP2B6 and CYP1A2 was assessed in cryopreserved hepatocytes from four donors (two male and two female). Data from all donors are shown in Figure 5, and individual data from the four donors are shown in Figures S1–S4. All data are shown relative to the 0.1% DMSO control, and all mentioned results were statistically significant. Dicloxacillin led to dose‐dependent upregulation of CYP3A4, CYP2C9 and CYP2B6, but not CYP1A2 expression, in all hepatocyte samples (Figure 5A). CYP3A4 mRNA expression increased 11.0‐ to 42.0‐fold, while CYP2C9 and CYP2B6 mRNA expression increased by 1.7‐ to 2.7‐fold and 2.5‐ to 2.9‐fold, respectively (Figure 5A). Corresponding increases in enzyme activity were 4.0‐ to 16.0‐fold for CYP3A4, 1.7‐ to 3.2‐fold for CYP2C9, and 3.0‐ to 5.0‐fold for CYB2B6. Surprisingly, CYP1A2 enzyme activity also increased (2.6‐ to 6.3‐fold) (Figure 5A). Luciferase assays revealed that dicloxacillin activated PXR, but not CAR or AhR, in a dose‐dependent manner (Figure 6). Similar trends were seen for the chemically related isoxazolyl penicillin, flucloxacillin (Figure 1), with increases in the expression and activity of CYP3A4, CYP2C9, CYP2B6 and CYP1A2 (Figures S5–S8). The induction of CYP enzymes by flucloxacillin was less pronounced than with dicloxacillin, which may have been caused by a decrease in PXR activation by flucloxacillin (Figure 6).
Figure 5.
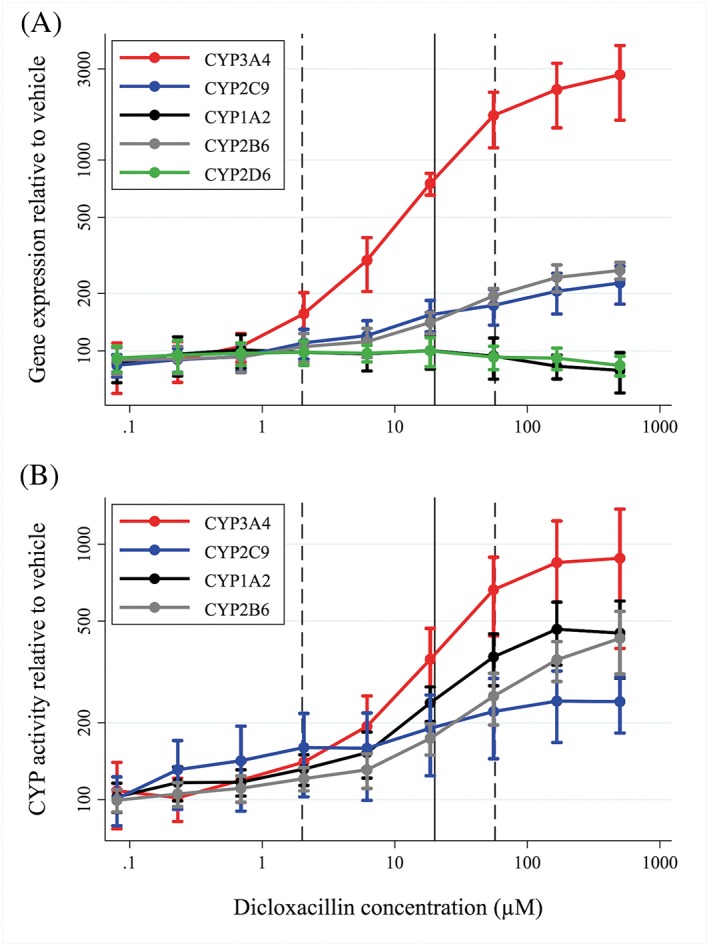
Increased gene expression (A) and activity (B) of cytochrome P450 (CYP) 3A4, CYP2C9 and CYP2B6 caused by dicloxacillin treatment of human hepatocytes (mean of four donors). Data are shown as mean fold increase relative to vehicle (0.1% dimethyl sulfoxide) with standard deviations. Solid and dashed lines represent average (Css, avg), minimum (Css, min) and maximum concentration of dicloxacillin (Css, max) at steady state at 500 mg dicloxacillin four times daily in healthy volunteers 4
Figure 6.
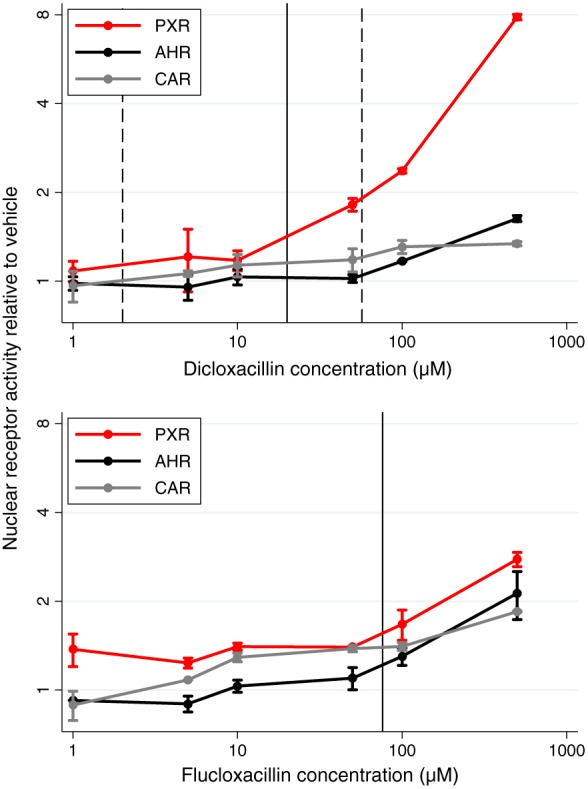
Nuclear receptor activity relative to vehicle induced by dicloxacillin (A) and flucloxacillin (B). AhR, aryl hydrocarbon receptor; CAR, constitutive androstane receptor; PXR, pregnane X receptor. Data are shown as mean fold‐increase relative to vehicle (0.1% dimethyl sulfoxide) with standard deviations. Solid and dashed lines for dicloxacillin represent average (Css, avg), minimum (Css, min) and maximum concentration of dicloxacillin (Css, max) at steady state at 500 mg dicloxacillin four times daily in healthy volunteers 4, while the solid line for flucloxacillin represents the maximum plasma concentration after a 750 mg single dose of flucloxacillin in healthy volunteers 3
Discussion
The present study confirmed the inductive potential of the isoxazolyl penicillin, dicloxacillin, on the expression and activity of drug‐metabolizing CYP enzymes. CYP3A4‐, CYP2C9‐ and CYP2C19‐mediated metabolism were induced by 10 days of treatment with 1 g dicloxacillin three times daily in a five‐drug clinical pharmacokinetic study in healthy men. From experiments in human hepatocytes, we showed that the mechanism for this induction is likely to be PXR activation leading to increased expression and activity of CYP enzymes, and we hypothesize that this extends to flucloxacillin.
The primary weakness of the study was the use of midazolam as a buccal administration. This may lead to larger variability in midazolam pharmacokinetics than intravenous administration but the randomization procedure served to minimize the impact of the within‐subject variability of the midazolam absorption profile. While absolute bioavailability following buccal administration in adults is about 75%, it is not known how much undergoes intestinal absorption 35. As CYP3A4 is highly expressed in both the liver and the intestines, the increased midazolam metabolism observed in the present study is likely to have been caused by a combination of induction of hepatic enzymes and intestinal first‐pass metabolism. In our study, we are unable to discriminate between intestinal and hepatic CYP3A4 induction. As we were likely to have captured less effect on intestinal CYP3A4 activity, our results may reflect some conservative quantitative bias. While not statistically significant, the AUC ratio of midazolam to metabolite did suggest a change but the precision of the estimate was wide. AUC estimation of metabolite is subject to several sources of bias, specifically with respect to the proportion, variation and rate of renal elimination of the glucuronidated metabolite, all of which affects metabolite AUC. Quantification of the amount of metabolite in the urine following deglucuronidation is therefore more robust and provides a better estimate of CLf and enzyme activity. A minor weakness was the lack of protein analysis from the in vitro study. However, as both mRNA expression and activity of CYP enzymes were increased following dicloxacillin exposure, it is likely that protein levels also increased.
The main strength of the study was the full pharmacokinetic profiles in the plasma and urine of five probe drugs and their main metabolites. This allows pharmacokinetic phenotyping of several CYP enzymes simultaneously, and these extensive data provide an excellent overview of the different metabolic pathways induced by dicloxacillin. The clinical data from the present study are supported by in vitro assessment in human hepatocytes, which is considered the gold standard for in vitro assessment of alterations in drug metabolism. Collectively, the in vivo and in vitro data strongly support dicloxacillin being a clinically relevant inducer of CYP3A4 and CYP2C metabolism.
The current results provide a mechanistic explanation for our previous observation that dicloxacillin decreased INR levels in patients receiving warfarin 14. Dicloxacillin activates PXR, causing increased expression of CYP2C9, leading to increased CYP2C9‐mediated metabolism of warfarin and lower clinical efficacy. CYP3A4 induction may be of significance as well, as the metabolism of the warfarin R‐enantiomer, a less potent inhibitor of vitamin K epoxide reductase 36, is catalysed by CYP3A4 37. These results are also in line with previous in vitro data indicating that dicloxacillin is a CYP3A4 inducer 17. We showed a similar extent of induction of probe substrate metabolism for CYP3A4, and also CYP2C9.
Both omeprazole AUC0–24 h and Cmax were significantly reduced after dicloxacillin exposure, although CLf for the 5‐hydroxy metabolite was unchanged. This was likely to have been caused by one outlier with a large reduction in CLf (threefold) after dicloxacillin exposure (Figure 4). This is in stark contrast to the increased CLf observed for the majority of other subjects (two of the other 11 individuals had minor reductions in CLf after dicloxacillin exposure; Figure 4). Interestingly, we showed that dicloxacillin also decreased the AUC0–24 h of dextromethorphan. Besides CYP2D6, CYP3A4 also catalyses the metabolism of dextromethorphan 38. As CYP2D6 is not thought to be significantly inducible and the CLf of dextrorphan is unchanged, the observed effect on dextromethorphan pharmacokinetics is likely to have been caused by induction of 3‐methoxymorphinan formation by CYP3A4. Caffeine AUC0–24 h is also affected but no change in CLf was observed. CYP1A2 is responsible for around 95% of the primary metabolism of caffeine but CYP3A4, CYP2C8 and CYP2C9 may also be involved in the metabolism 39. It is plausible that induction of these enzymes may lead to the marginally increased caffeine metabolism that we observed. The present study did not assess the length of the observed induction after conclusion of dicloxacillin treatment. A previous study showed that 7 days of treatment with rifampicin produced enzyme induction, but the induced activities were no longer detectable 8 days after conclusion of rifampicin treatment 40. Another clinical study showed that the inductive effect of rifampicin on CYP3A4 persisted for about 4 weeks following 28 days of treatment 41.
Owing to the brevity of the exposure to dicloxacillin (the standard treatment duration is 7–10 days), CYP induction is not expected to be of substantial clinical significance for a large proportion of widely used drugs. The clinical efficacy of many drugs, such as cholesterol‐lowering agents, is largely a result of long‐term treatment, and 10 days of induction should not have a substantial impact on the overall therapeutic efficacy. However, the clinical efficacy of drugs with a narrow therapeutic range, such as antiepileptic agents or immunosuppressants, may be affected by concomitant dicloxacillin treatment. Penicillins were previously suspected of causing therapeutic failure of oral contraceptives 42 but a potential mechanism for this observation was never identified, and no solid evidence has substantiated the existence of these putative drug–drug interactions. Owing to the substantial induction of CYP3A4 by dicloxacillin, it is plausible that concomitant use of dicloxacillin and oral contraceptives increases the metabolism of the latter, and may lead to a risk of therapeutic failure and unwanted pregnancy. This putative drug–drug interaction warrants properly designed pharmacokinetic studies to determine its magnitude and clinical relevance.
Our in vitro results lend credence to a hypothesis that treatment with the chemically related drug, flucloxacillin (Figure 1 ), more widely used in the UK 43, 44, may induce CYP enzymes, although possibly to a lesser extent than dicloxacillin. Induction of CYPs by flucloxacillin should be quantified by an adequately designed in vivo pharmacokinetic drug–drug interaction study to guide recommendations for the use of these isoxazolyl penicillins. We also showed that dicloxacillin induces CYP2B6 expression and activity. Whether this translates to changes in the clinical pharmacokinetics of CYP2B6 substrates needs to be elucidated, and is especially relevant for antiretroviral drugs such as efavirenz.
In conclusion, dicloxacillin is an inducer of CYP3A4‐ and CYP2C‐mediated metabolism in healthy volunteers and in human hepatocytes. The effect is clinically relevant, and we recommend caution when prescribing dicloxacillin to users of drugs with a narrow therapeutic window that are metabolized by CYP2C9, CYP2C19 or CYP3A4.
Competing Interests
There are no competing interests to declare.
The authors would like to thank Birgitte Damby Sørensen for her thorough and timely analytical work, Anders Højgaard Hansen for supplying Figure 1 and Søren Feddersen for assisting with genotyping. This study was supported by the Odense University Hospital Free Research Fund (Grant #29‐A1534), the Danish Research Council of Independent Research | Medical Sciences (Grant #DFF‐5053‐00042) and the A.P. Møller Foundation for the Advancement of Medical Science.
Supporting information
Figure S1 Effects of dicloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor HU1765
Figure S2 Effects of dicloxacillin affects expression and activity of cytochrome P450 CYP enzymes in cryopreserved hepatocytes from donor TRL4034
Figure S3 Effects of dicloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor 1057
Figure S4 Effects of dicloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor OII
Figure S5 Effects of flucloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor HU1765
Figure S6 Effects of flucloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor TRL4034
Figure S7 Effects of flucloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor 1057
Figure S8 Effects of flucloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor OII
Table S1 Genotype analysis identified two cytochrome P450 (CYP) 2C9 (CYP2C9*2/*3 and CYP2C9*3/*3) and one CYP2D6 (CYP2D6*4/*4) poor metabolizers and two CYP2C19 ultrarapid metabolizers (CYP2C19*17/*17)
Table S2 Sensitivity analysis of the noncompartmental analysis, excluding poor‐ and ultrarapid metabolizers of cytochrome P450 (CYP) 2C9, CYP2C19 and CYP2D6
Table S3 Results from noncompartmental analysis of all included individuals for all five probe drugs and their primary metabolites
Stage, T. B. , Graff, M. , Wong, S. , Rasmussen, L. L. , Nielsen, F. , Pottegård, A. , Brøsen, K. , Kroetz, D. L. , Khojasteh, S. C. , and Damkier, P. (2018) Dicloxacillin induces CYP2C19, CYP2C9 and CYP3A4 in vivo and in vitro . Br J Clin Pharmacol, 84: 510–519. doi: 10.1111/bcp.13467.
References
- 1. Rayner C, Munckhof WJ. Antibiotics currently used in the treatment of infections caused by Staphylococcus aureus . Intern Med J 2005; 35(Suppl. 2): S3–16. [DOI] [PubMed] [Google Scholar]
- 2. Sandberg A, Jensen KS, Baudoux P, Van Bambeke F, Tulkens PM, Frimodt‐Møller N. Intra‐ and extracellular activities of dicloxacillin against Staphylococcus aureus in vivo and in vitro . Antimicrob Agents Chemother 2010; 54: 2391–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Røder BL, Frimodt‐Møller N, Espersen F, Rasmussen SN. Dicloxacillin and flucloxacillin: pharmacokinetics, protein binding and serum bactericidal titers in healthy subjects after oral administration. Infection 1995; 23: 107–112. [DOI] [PubMed] [Google Scholar]
- 4. Wu G, Zheng Y, Zhou H, Hu X, Liu J, Zhai Y, et al Safety and pharmacokinetics of dicloxacillin in healthy Chinese volunteers following single and multiple oral doses. Drug Des Devel Ther 2015; 9: 5687–5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goossens H, Ferech M, Vander Stichele R, Elseviers M. ESAC Project Group. Outpatient antibiotic use in Europe and association with resistance: a cross‐national database study. Lancet 2005; 365: 579–587. [DOI] [PubMed] [Google Scholar]
- 6. Schmidt M, Hallas J, Laursen M, Friis S. Data resource profile: Danish online drug use statistics (MEDSTAT). Int J Epidemiol 2016; 45: 1401–142g. [DOI] [PubMed] [Google Scholar]
- 7. Murai Y, Nakagawa T, Yamaoka K, Uno T. Comparative pharmacokinetics of metabolism and urinary excretion of isoxazolylpenicillins in man. Chem Pharm Bull 1983; 31: 3292–3301. [DOI] [PubMed] [Google Scholar]
- 8. Susanto M, Benet LZ. Can the enhanced renal clearance of antibiotics in cystic fibrosis patients be explained by P‐glycoprotein transport? Pharm Res 2002; 19: 457–462. [DOI] [PubMed] [Google Scholar]
- 9. Putnam WS, Woo JM, Huang Y, Benet LZ. Effect of the MDR1 C3435T variant and P‐glycoprotein induction on dicloxacillin pharmacokinetics. J Clin Pharmacol 2005; 45: 411–421. [DOI] [PubMed] [Google Scholar]
- 10. Krstenansky PM, Jones WN, Garewal HS. Effect of dicloxacillin sodium on the hypoprothrombinemic response to warfarin sodium. Clin Pharm 1987; 6: 804–806. [PubMed] [Google Scholar]
- 11. Mailloux AT, Gidal BE, Sorkness CA. Potential interaction between warfarin and dicloxacillin. Ann Pharmacother 1996; 30: 1402–1407. [DOI] [PubMed] [Google Scholar]
- 12. Lacey CS. Interaction of dicloxacillin with warfarin. Ann Pharmacother 2004; 38: 898. [DOI] [PubMed] [Google Scholar]
- 13. Khalili H, Nikvarz N, Najmeddin F, Dashti‐Khavidaki S. A probable clinically significant interaction between warfarin and cloxacillin: three case reports. Eur J Clin Pharmacol 2013; 69: 721–724. [DOI] [PubMed] [Google Scholar]
- 14. Pottegård A, Henriksen DP, Madsen KG, Hellfritzsch M, Damkier P, Stage TB. Change in international normalized ratio among patients treated with dicloxacillin and vitamin K antagonists. JAMA 2015; 314: 296–297. [DOI] [PubMed] [Google Scholar]
- 15. Kennedy B, Larcombe R, Chaptini C, Gordon DL. Interaction between voriconazole and flucloxacillin during treatment of disseminated Scedosporium apiospermum infection. J Antimicrob Chemother 2015; 70: 2171–2173. [DOI] [PubMed] [Google Scholar]
- 16. Comuth WJ, Comuth JA, Hauer RNW, Malingré MM. Interaction of flucloxacillin and quinidine. Eur J Clin Pharmacol 2011; 68: 891–893. [DOI] [PubMed] [Google Scholar]
- 17. Yasuda K, Ranade A, Venkataramanan R, Strom S, Chupka J, Ekins S, et al A comprehensive in vitro and in silico analysis of antibiotics that activate pregnane X receptor and induce CYP3A4 in liver and intestine. Drug Metab Dispos 2008; 36: 1689–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen Y, Ferguson SS, Negishi M, Goldstein JA. Induction of human CYP2C9 by rifampicin, hyperforin, and phenobarbital is mediated by the pregnane X receptor. J Pharmacol Exp Ther 2004; 308: 495–501. [DOI] [PubMed] [Google Scholar]
- 19. Wohlfarth A, Naue J, Lutz‐Bonengel S, Dresen S, Auwärter V. Cocktail approach for in vivo phenotyping of 5 major CYP450 isoenzymes: development of an effective sampling, extraction, and analytical procedure and pilot study with comparative genotyping. J Clin Pharmacol 2012; 52: 1200–1214. [DOI] [PubMed] [Google Scholar]
- 20. Zadoyan G, Rokitta D, Klement S, Dienel A, Hoerr R, Gramatté T, et al Effect of Ginkgo biloba special extract EGb 761® on human cytochrome P450 activity: a cocktail interaction study in healthy volunteers. Eur J Clin Pharmacol 2012; 68: 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doroshyenko O, Rokitta D, Zadoyan G, Klement S, Schläfke S, Dienel A, et al Drug cocktail interaction study on the effect of the orally administered lavender oil preparation silexan on cytochrome P450 enzymes in healthy volunteers. Drug Metab Dispos 2013; 41: 987–993. [DOI] [PubMed] [Google Scholar]
- 22. Streetman DS, Bleakley JF, Kim JS, Nafziger AN, Leeder JS, Gaedigk A, et al Combined phenotypic assessment of CYP1A2, CYP2C19, CYP2D6, CYP3A, N‐acetyltransferase‐2, and xanthine oxidase with the ‘Cooperstown cocktail’. Clin Pharmacol Ther 2000; 68: 375–383. [DOI] [PubMed] [Google Scholar]
- 23. Jetter A, Kinzig‐Schippers M, Skott A, Lazar A, Tomalik‐Scharte D, Kirchheiner J, et al Cytochrome P450 2C9 phenotyping using low‐dose tolbutamide. Eur J Clin Pharmacol 2004; 60: 165–171. [DOI] [PubMed] [Google Scholar]
- 24. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap) – a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009; 42: 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Acharya C, Hooker AC, Türkyılmaz GY, Jönsson S, Karlsson MO. A diagnostic tool for population models using non‐compartmental analysis: the ncappc package for R. Comput Methods Programs Biomed 2016; 127: 83–93. [DOI] [PubMed] [Google Scholar]
- 26. Halladay JS, Wong S, Khojasteh SC, Grepper S. An ‘all‐inclusive’ 96‐well cytochrome P450 induction method: measuring enzyme activity, mRNA levels, protein levels, and cytotoxicity from one well using cryopreserved human hepatocytes. J Pharmacol Toxicol Methods 2012; 66: 270–275. [DOI] [PubMed] [Google Scholar]
- 27. Halladay JS, Wong S, Jaffer SM, Sinhababu AK, Khojasteh‐Bakht SC. Metabolic stability screen for drug discovery using cassette analysis and column switching. Drug Metab Lett 2007; 1: 67–72. [DOI] [PubMed] [Google Scholar]
- 28. Yueh M‐F, Kawahara M, Raucy J. Cell‐based high‐throughput bioassays to assess induction and inhibition of CYP1A enzymes. Toxicol In Vitro 2005; 19: 275–287. [DOI] [PubMed] [Google Scholar]
- 29. Yueh M‐F, Kawahara M, Raucy J. High volume bioassays to assess Cyp3a4‐mediated drug interactions: induction and inhibition in a single cell line. Drug Metab Dispos 2005; 33: 38–48. [DOI] [PubMed] [Google Scholar]
- 30. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE, et al The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 2015; 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 2015; 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dicloxacillin summary of product characteristics [online]. Available at http://mri.cts‐mrp.eu/download/DK_H_2321_002_FinalSPC.pdf (last accessed 25 May 2017)
- 35. Buccolam summary of product characteristics [online]. Available at https://www.medicines.org.uk/emc/medicine/25538#PHARMACOKINETIC_PROPS (last accessed 16 May 2017).
- 36. Padrini R, Quintieri L. R‐warfarin anticoagulant effect. Br J Clin Pharmacol 2017; 83: 2305–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther 1997; 73: 67–74. [DOI] [PubMed] [Google Scholar]
- 38. Yu A, Haining RL. Comparative contribution to dextromethorphan metabolism by cytochrome P450 isoforms in vitro: can dextromethorphan be used as a dual probe for both CYP2D6 and CYP3A activities? Drug Metab Dispos 2001; 29: 1514–1520. [PubMed] [Google Scholar]
- 39. Kot M, Daniel WA. The relative contribution of human cytochrome P450 isoforms to the four caffeine oxidation pathways: an in vitro comparative study with cDNA‐expressed P450s including CYP2C isoforms. Biochem Pharmacol 2008; 76: 543–551. [DOI] [PubMed] [Google Scholar]
- 40. Inui N, Akamatsu T, Uchida S, Tanaka S, Namiki N, Karayama M, et al Chronological effects of rifampicin discontinuation on cytochrome P450 activity in healthy Japanese volunteers, using the cocktail method. Clin Pharmacol Ther 2013; 94: 702–708. [DOI] [PubMed] [Google Scholar]
- 41. Reitman ML, Chu X, Cai X, Yabut J, Venkatasubramanian R, Zajic S, et al Rifampin's acute inhibitory and chronic inductive drug interactions: experimental and model‐based approaches to drug‐drug interaction trial design. Clin Pharmacol Ther 2011; 89: 234–242. [DOI] [PubMed] [Google Scholar]
- 42. Weaver K, Glasier A. Interaction between broad‐spectrum antibiotics and the combined oral contraceptive pill: a literature review. Contraception 1999; 59: 71–78. [DOI] [PubMed] [Google Scholar]
- 43. Aujla RS, Bryson DJ, Gulihar A, Taylor GJ. Trends in orthopaedic antimicrobial prophylaxis in the UK between 2005 and 2011. Ann R Coll Surg Engl 2013; 95: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Francis NA, Hood K, Lyons R, Butler CC. Understanding flucloxacillin prescribing trends and treatment non‐response in UK primary care: a clinical practice research datalink (CPRD) study. J Antimicrob Chemother 2016; 71: 2037–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of dicloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor HU1765
Figure S2 Effects of dicloxacillin affects expression and activity of cytochrome P450 CYP enzymes in cryopreserved hepatocytes from donor TRL4034
Figure S3 Effects of dicloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor 1057
Figure S4 Effects of dicloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor OII
Figure S5 Effects of flucloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor HU1765
Figure S6 Effects of flucloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor TRL4034
Figure S7 Effects of flucloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor 1057
Figure S8 Effects of flucloxacillin on the expression and activity of cytochrome P450 (CYP) enzymes in cryopreserved hepatocytes from donor OII
Table S1 Genotype analysis identified two cytochrome P450 (CYP) 2C9 (CYP2C9*2/*3 and CYP2C9*3/*3) and one CYP2D6 (CYP2D6*4/*4) poor metabolizers and two CYP2C19 ultrarapid metabolizers (CYP2C19*17/*17)
Table S2 Sensitivity analysis of the noncompartmental analysis, excluding poor‐ and ultrarapid metabolizers of cytochrome P450 (CYP) 2C9, CYP2C19 and CYP2D6
Table S3 Results from noncompartmental analysis of all included individuals for all five probe drugs and their primary metabolites