

Abstract
Purpose
Mutations in PTEN-induced putative kinase 1 (Pink1), a mitochondrial serine/threonine kinase, cause a recessive inherited form of Parkinson’s disease (PD). Pink1 deletion in rats results in a progressive PD-like phenotype, characterized by significant motor deficits starting at 4 months of age. Despite the evidence of mitochondrial dysfunction, the pathogenic mechanism underlying disease due to Pink1-deficiency remains obscure.
Experimental design
Striatal synaptic mitochondria from 3-month-old Pink1-deficient rats were characterized using bioenergetic and mass spectroscopy (MS)-based proteomic analyses.
Results
Striatal synaptic mitochondria from Pink1-deficient rats exhibit decreased complex I-driven respiration and increased complex II-mediated respiration compared with wild-type rats. MS-based proteomics revealed 69 of the 811 quantified mitochondrial proteins were differentially expressed between Pink1-deficient rats and controls. Down-regulation of several electron carrier proteins, which shuttle electrons to reduce ubiquinone at complex III, in the Pink1-knockouts suggests disruption of the linkage between fatty acid, amino acid, and choline metabolism and the mitochondrial respiratory system.
Conclusions and clinical relevance
These results suggest that complex II activity is increased to compensate for loss of electron transfer mechanisms due to reduced complex I activity and loss of electron carriers within striatal nerve terminals early during disease progression. This may contribute to the pathogenesis of PD.
Keywords: bioenergetics, mitochondria, Pink1, proteomics, synapses
1 Introduction
Parkinson’s disease (PD) is the most common age-related human neurodegenerative movement disorder. The defining clinical characteristics of PD, known collectively as parkinsonism, include rigidity, resting tremor, bradykinesia, postural instability, and micrographia [1]. The selective, progressive loss of dopaminergic neurons of the substantia nigra pars compacta and the associated loss of dopaminergic inputs into the striatum lead to the observed impaired motor functions. This progressive loss of dopamine secreting neurons and the presence of ubiquitylated neuronal cytoplasmic inclusions termed Lewy bodies, represent the main neuropathological features of PD [2]. PD is predominantly sporadic; however, it is a complex multifactorial disease and familial monogenic forms have been linked to mutations in alpha-synuclein and leucine rich-repeat kinase 2 [3]. Additionally, loss-of-function mutations in the PTEN-induced kinase 1 (Pink1), DJ-1, and Parkin proteins promote neuronal death and lead to autosomal recessive parkinsonism [3]. Although these three proteins are implicated as key players in regulating mitochondrial health and quality [4–6], unraveling the link between the cause of mitochondrial dysfunction and the most prominent phenotypic changes observed in PD has proven complex.
Mitochondrial dysfunction and oxidant stress are widely viewed as major factors in PD pathogenesis [7, 8]. Further, a variety of pesticides and toxins that inhibit mitochondrial function including rotenone (Rot), paraquat, and 1-methyl-4-phenylpyridinium, all of which affect complex I of the electron transport chain (ETC), elicit parkinsonian symptoms [9]. Furthermore, deficiency in ETC complex I, mitochondrial ROS accumulation, and the presence of mitochondrial DNA (mtDNA) mutations are observed in PD patients [7, 8, 10]. While the loss-of-function mutations in Pink1, DJ-1, and Parkin lead to PD in humans, mice are surprisingly resistant to disease through transgenic knockout (KO) of these genes [11]. However, in rats, deletion of Pink1 and DJ-1 leads to phenotypic signs of PD, such as the development of significant motor symptoms starting at 4 months of age and dopaminergic cell loss (approximately 50% by 8 months of age) in the substantia nigra [12].
We previously revealed that in rats loss of the mitochondrial serine/threonine kinase Pink1 leads to significant mitochondrial proteomic alterations in the striatum prior to neuronal loss (at 4 months of age) that persist into the symptomatic stage (at 9 months of age) of PD-like pathological progression; however, significant alterations in striatal mitochondrial function were not observed at the early (4 month) time point, just the later (9 month) time point [13]. Here we hypothesized that synaptic mitochondria, of prime importance to neuronal function and likely vulnerable population to pathological change, would be particularly sensitive to the alterations due to loss of Pink1. The overall goal of this study was to uncover mitochondria-relevant biological pathways altered due to deletion of Pink1 that are instructive for PD progression. In this vein, we performed functional and mass spectrometry-based quantitative proteomic characterizations of striatal synaptic mitochondria from Pink1 KO rats and compared these to age (3 months) and sex (male) matched wild-type (WT) rats congenic to the KO rats to minimize variables other than inactivation of Pink1, to illuminate mitochondrial pathways that are altered prior to onset of phenotypic changes.
We found that loss of Pink1 leads to impaired complex I-driven respiration and increased complex II-mediated respiration in the striatal synaptic mitochondria at this early stage. Additionally, alterations in the striatal synaptic mitochondrial proteome due to Pink1 deficiency implicate defects in the ETC, fatty acid metabolism, oxidation reduction, and generation of precursor metabolites and energy early in PD progression. This combination of mitochondrial functional and proteomic analyses has allowed us to evaluate the role of striatal synaptic mitochondria in PD etiology, further characterize mitochondrial dysfunction observed with specific familial PD gene alterations (loss of Pink1 function), and expand our knowledge concerning the role of the PD-linked gene, Pink1, in PD pathogenesis.
2 Materials and methods
2.1 Materials
All chemicals used were purchased from Sigma-Aldrich with the following exceptions: Pierce BCA Protein Assay, formic acid (FA), Pierce 660 nm Protein Assay, SuperBlock Blocking Buffer were purchased from Thermo Fisher Scientific. Trypsin (V5111) was purchased from Promega.
2.2 Animals and brain tissue
Rats with targeted disruption of the Pink1 gene and the background inbred strain (Long Evans Hooded) were obtained from SAGE Labs. The generation and characterization of these Pink1-deficient rats has been described previously [12]. The rats were maintained on a 12-hour light/dark cycle in a temperature-controlled environment with free access to standard rat chow and water. The University of Nebraska Medical Center Institutional Animal Care and Use Committee approved all experimental procedures. Rats (male, 3 months of age) were rendered unconscious by isoflurane and sacrificed by decapitation. After euthanasia, the brain was quickly removed and placed on a pre-chilled petri dish. The petri dish was placed under the dissection microscope, and the striatum was dissected from the brain (the weight of each striatum was approximately 240 mg) and transferred to a microcentrifuge tube containing ice-cold mitochondrial isolation buffer (MSHE+BSA): 70 mM sucrose, 210 mM mannitol, 5 mM HEPES, 1 mM EGTA and 0.5% (w/v) fatty acid free (FAF)-BSA (pH 7.2). The striatum was then transferred to a Dounce homogenizer containing ice-cold MSHE+BSA and placed on ice for fresh tissue mitochondrial isolation.
2.3 Isolation of synaptic mitochondria
Synaptic mitochondria were isolated using our previously published method with slight modifications [14, 15]. All homogenization and centrifugation steps were performed on ice or at 4°C. The striata were homogenized in MSHE+BSA buffer using 10 strokes in a Dounce homogenizer. The homogenate was centrifuged at 1,300 × g for 3 min, the supernatant was collected, and the pellet was resuspended in MSHE+BSA and the centrifugation step was repeated. The supernatants were pooled and centrifuged at 21,000 × g for 10 min. The resulting pellet was resuspended in 15% Percoll and layered on top of a 24% and 40% Percoll gradient (prepared from 100% Percoll solution containing 70 mM sucrose, 210 mM mannitol, 5 mM HEPES, 1 mM EGTA, and 0.5% (w/v) FAF-BSA (pH 7.2). After ultracentrifugation for 8 min at 30,700 × g, the banding near the interface of the upper two layers of the gradient, containing isolated nerve terminals (synaptosomes), was collected and diluted in MSHE+BSA.
To isolate the synaptic mitochondria [14, 15], the synaptosomal fraction was transferred to a nitrogen cavitation vessel (Parr Instrument Co.) and the pressure was equilibrated to 900 psi for 15 min followed by depressurization to atmospheric pressure. The resultant released synaptic mitochondria suspension was added to the top of 24% Percoll and ultracentrifuged for 10 min at 30,700 × g. The pellet containing the synaptic mitochondria was resuspended in MSHE+BSA and centrifuged at 8,000 × g for 10 min. The pellets were washed twice with mitochondrial assay solution (MAS, 1×): 70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA and 0.2% (w/v) FAF-BSA (pH 7.2) and centrifuged at 8,000 × g for 10 min. The mitochondrial pellets were resuspended in a minimal volume of MAS. Total mitochondrial concentrations were determined using the BCA method (yielded approximately 180 μg per rat striatum) and the isolated mitochondria were used immediately for analysis. Independent isolations were performed for each experimental procedure (bioenergetics, mass spectrometry, and immunoblotting).
2.4 Mitochondrial bioenergetics assays
Oxygen consumption rates (OCR) were measured using 3 technical replicate wells for each independent biological replicate (n = 4 for WT and n = 5 for Pink1 KO) using the Seahorse XFe96 Extracellular Flux Analyzer (Seahorse Bioscience) coupling assay as described previously [16]. The isolated striatal synaptic mitochondrial amounts of 5 μg (complex I-mediated respiration) and 2.5 μg (complex II- and IV-mediated respiration) per well were found to be optimal and were used for subsequent experiments (Figure S1). The Seahorse Wave 2.2.0 software package was used for data calculation. Graphs were generated in Prism (GraphPad Software).
The isolated mitochondria were plated in a volume of 20 μl MAS with appropriate substrates and inhibitors for each complex studied (complex I, 10 mM pyruvate and 2 mM malate as substrate; complex II, 10 mM succinate as substrate in the presence of 2 μM Rot; complex IV, 10 mM ascorbate with 100 μM tetramethylphenylenediamine (TMPD) as substrate in the presence of 4 μM antimycin A (AA)) [17]. After attachment to the plate, 160 μl of MAS (containing inhibitors and/or substrates) was added to each well, and the plate was incubated at 37°C for 5 min to equilibrate temperature. The final concentrations for sequential injections were 4 mM ADP, 2.5 μg/ml oligomycin (Oligo), 4 μM FCCP, and 2 μM Rot (complex I assay), 4 μM AA (complex I and II assays) or 20 mM sodium azide (complex IV assay). After completion of the assays, the mitochondrial protein content within each well was quantified using the BCA method to confirm equal well loading and for normalization of the OCR data.
2.5 Mitochondrial mass spectrometry
2.5.1 Protein digestion
Rat striatal synaptic mitochondrial protein sample aliquots (35 μg) used for data-independent acquisition mass spectrometry were digested with trypsin using filter aided sample preparation [18]. The resultant peptides were desalted using Oasis mixed-mode weak cation exchange cartridges (Waters), dehydrated with a Savant ISS 110 SpeedVac concentrator (Thermo Fisher Scientific) and resuspended in 10 μl of 0.1% FA prior to quantification using a NanoDrop 2000 UV-vis Spectrophotometer (Thermo Fisher Scientific) in conjunction with the Scopes method for peptide quantification by absorbance at 205 nm [19].
2.5.2 SWATH-MS analysis
The samples of peptides (2 μg) from WT and Pink1 KO rat striatal synaptic mitochondrial lysates were analyzed in quadruplicate (four biological replicates per strain (WT and Pink1 KO), n = 4) by nano-LC-MS/MS in SWATH-MS mode on the 5600 TripleTOF instrument (SCIEX) and targeted data extraction was performed as previously described [13, 20, 21]. All fragment ion chromatograms were extracted and automatically integrated with PeakView software (Version 2.1, SCIEX). For peptide identification, our published reference spectral library was used [13, 20]. This library was generated in ProteinPilot (Version 4.5, SCIEX) using the Paragon algorithm and the default settings [22]. All searches were performed against the UniProt Rattus Norvegicus Proteome UP000002494 containing 7,942 reviewed proteins (Swiss-Prot). Combined results yielded a library of 3,241 proteins identified with high confidence (greater than 99%) that passed the global FDR from fit analysis using a critical FDR of 1%. In accordance with previously published work [23], we selected five peptides and five transitions option for quantitative analysis and performed targeted data extraction for each peptide. For each peptide the fragment ion chromatograms were extracted using the SWATH isolation window set to a width of 10 min and 50 ppm accuracy [23]. To calibrate retention times, synthetic peptides (Biognosys AG) were spiked-in to the samples in accordance with the manufacturer’s protocol. Data were normalized to the median peak ratios of common proteins in MarkerView software (Version 1.2.1, SCIEX). The mass spectrometry proteomics data have been deposited to the ProteomeXchange consortium via the PRIDE [24] partner repository with the dataset identifier PXD004558.
2.5.3. Bioinformatics
Heat maps of protein expression values were built with Multi Experiment Viewer (Version 4.9, http://www.tm4.org/mev.html). The Database for the Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov) Bioinformatics Resources 6.7 was used for functional annotation [25]. The SWATH-MS data was uploaded to the CyberT Web server (http://cybert.ics.uci.edu/) [26], which implements a t-test using a Bayesian regularization method for quantitative mass spectrometry analysis.
2.6 Immunoblotting
Isolated synaptic mitochondria obtained from eight biological replicates per rat strain (n = 8 for WT and Pink1 KO) were lysed using cell lysis buffer (4% SDS, 100 mM Tris, 0.1 M DTT). The total protein concentration was determined using the 660 nm Protein Assay. For each sample, 10 μg of protein was separated by SDS-PAGE on NuPAGE 4%-12% Bis-Tris gels (Thermo Fisher Scientific). After electrophoresis, the proteins were transferred onto nitrocellulose membranes using the iBlot Dry Blotting System (Thermo Fisher Scientific). After blocking with SuperBlock Blocking Buffer, membranes were probed overnight at 4°C with primary antibodies for Etfa (1:1000, Abcam, ab110316), Etfb (1:750, Abcam, ab104944), Ivd (1:2000, Proteintech, 10822-1-AP), Tomm20 (1:2000, Santa Cruz Biotechnology, sc-11415), and Atp5a1 (1:2000, Abcam, MS507). The IRDye 680RD goat anti-rabbit (LI-COR, 926-68071), IRDye 800CW goat anti-mouse (LI-COR, 926-32210) or IRDye 800CW donkey anti-goat (LI-COR, 925-32214) fluorescent secondary antibodies were used at a 1:20,000 dilution. Blots were imaged with an Odyssey Fc imaging system and quantified using LI-COR Image Studio software (Version 4.0, LI-COR). Bar graphs were generated in Prism.
2.7 Statistical Analysis
Statistical analyses were conducted in Prism using ANOVA on the complete Seahorse assay data set with Bonferroni multiple comparisons post-hoc testing [27] and unpaired two-tailed t-tests for immunoblotting. p < 0.05 was considered statistically significant. Data are expressed as mean ± SEM.
3 Results
3.1 Loss of Pink1 alters striatal synaptic mitochondrial respiration in rats
Previously, we have identified significant complex II-mediated respiration changes in 9-month-old but not 4-month-old Pink1-deficient rats, and this increase in state 3 (ADP-stimulated respiration) and state 3u (uncoupled respiration) respiration was found in the striatum but not the cortex [13]. These studies were performed on a mixed population of mitochondria, originating from all cells in the striatum. Here we focused on synaptic mitochondria, as a critical and vulnerable population, from 3-month-old Pink1 KO rats, an age prior to the symptomatic motor changes. We measured the mitochondrial respiratory states driven by complex I, complex II, and complex IV in synaptic mitochondria isolated from Pink1-deficient and WT rats. We assessed complex I respiration driven by malate/pyruvate (Figure 1A-C), complex II respiration driven by succinate (Figure 1D-F), and complex IV respiration driven by TMPD/ascorbate (Figure S2). Complex I-dependent respiration (state 3 (ADP-stimulated respiration) and state 3u (uncoupled respiration)) in Pink1 KO rat striatal synaptic mitochondria was significantly reduced compared with WT rats (Figure 1A and B). Additionally, complex I-mediated ATP production (state 3 – state 4o), maximal respiration (state 3u – Rot/AA), and spare respiratory capacity (state 3u – state 2) were significantly impaired (Figure 1C). In contrast, complex II-dependent respiration (state 2 (respiration in the presence of substrate but in the absence of ADP), state 3 (ADP-stimulated respiration), and state 3u (uncoupled respiration)) was significantly increased (Figure 1D and E), an effect we did not find until 9 months of age in total striatal mitochondria in Pink1 KO rats [13]. This increase in respiration corresponded with elevated complex II-driven basal mitochondrial respiration (state 2 – AA), ATP production (state 3 – state 4o), and maximal respiration (state 3u – AA) (Figure 1F). No significant complex IV respiratory changes were observed in Pink1-deficient rats compared to control (Figure S2). The respiratory control ratio (RCR), a measure of mitochondrial coupling, was assessed (RCR = state 3u/state 4o). No significant alteration was detected in RCR in the Pink1 KO rats for complex I- (RCR = 5.3 ± 0.7 (WT) and 5.3 ± 1.3 (Pink1 KO)), complex II- (RCR = 3.7 ± 0.3 (WT) and 3.1 ± 0.2 (Pink1 KO)), or complex IV- (RCR = 1.3 ± 0.04 (WT) and 1.3 ± 0.02 (Pink1 KO)) driven respiration. These findings suggest that loss of Pink1 leads to impaired complex I function within striatal nerve terminals and that complex II activity may be increased to compensate for reduced ATP levels.
Figure 1. Altered synaptic mitochondrial bioenergetics due to loss of Pink1.
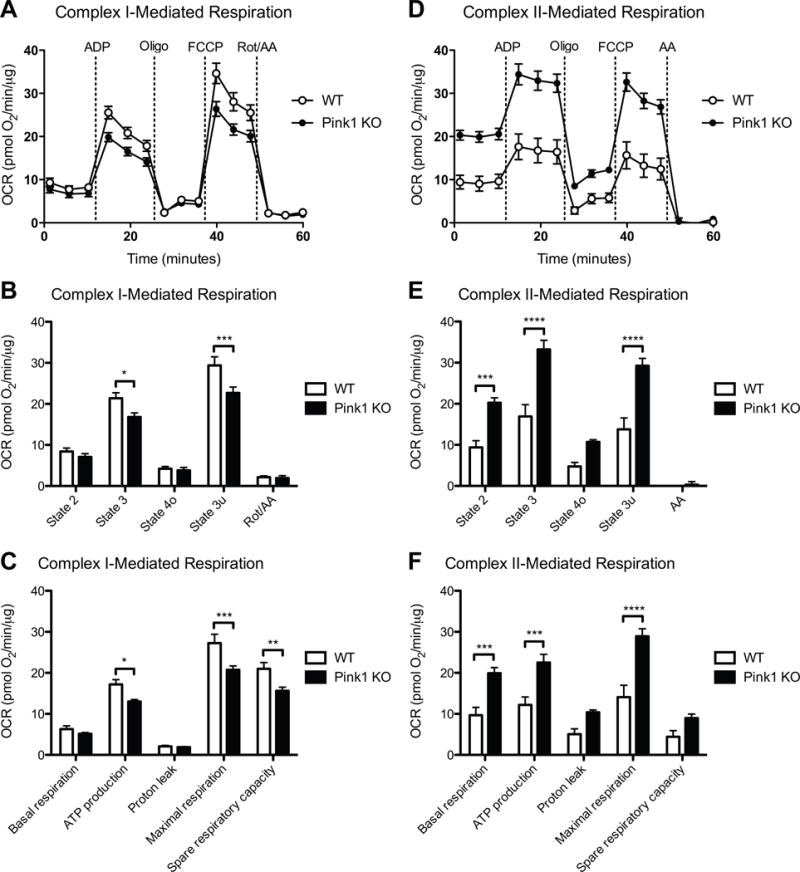
Striatal synaptic mitochondria from 3-month Pink1 KO and WT rats were isolated and assayed as described in the Materials and methods. (A) Graph output of coupling assay (complex I) of isolated synaptic mitochondria. Point-to-point OCR data are shown with pyruvate/malate as the substrate followed by addition of ADP, Oligo, FCCP, and Rot/AA. (B) Complex I-mediated state 2 (basal), state 3 (ADP-stimulated), state 4o (leak), and state 3u (uncoupled) respiration. *(p < 0.05), ***(p < 0.001), significantly lower in Pink1 (n = 5) versus WT rats (n = 4). (C) Basal respiration (state 2 – Rot/AA), ATP production (state 3 – state 4o), proton leak (state 4o – Rot/AA), maximal respiration (state 3u – Rot/AA), and spare respiratory capacity (state 3u – state 2) calculated from the coupling assay (A, B). *(p < 0.05), **(p < 0.01), ***(p < 0.001), significantly lower in Pink1 (n = 5) versus WT rats (n = 4). (D) Graph output of coupling assay (complex II) of isolated synaptic mitochondria. Point-to-point OCR data are shown with succinate as the substrate followed by addition of ADP, Oligo, FCCP, and AA. (E) Complex II-mediated state 2 (basal), state 3 (ADP-stimulated), state 4o (leak), and state 3u (uncoupled) respiration. ***(p < 0.001), ****(p < 0.0001), significantly higher in Pink1 (n = 5) versus WT rats (n = 4). (F) Basal respiration (state 2 – AA), ATP production (state 3 – state 4o), proton leak (state 4o – AA), maximal respiration (state 3u – AA), and spare respiratory capacity (state 3u – state 2) calculated from the coupling assay (D, E). ***(p < 0.001), ****(p < 0.0001), significantly higher in Pink1 (n = 5) versus WT rats (n = 4).
3.2 Proteomic alterations in striatal synaptic mitochondria from Pink1 deficient rats
Sequential window acquisition of all theoretical fragment-ion spectra (SWATH)-mass spectrometry (MS) is a proteomics strategy that systematically queries sample sets for the presence and quantity of essentially any protein of interest through mining of the complete fragment ion spectral libraries [23]. For our experiments, we have used our reference spectral library generated previously using mitochondria isolated from B35, H19-7/IGF-IR, PC12, and RN33B rat cell lines [20] and expanded through the addition of synaptic mitochondria isolated from rat brain [13]. The resulting library containing 3,241 proteins available for quantification by SWATH-MS was used to obtain quantitative levels of synaptic mitochondrial proteins from the striatum of 3-month-old Pink1 KO and WT rats.
Striatal synaptic mitochondria were isolated, protein lysates were trypsin digested, and the resultant peptides were quantified and used for shotgun proteomics, with four independent biological replicates for each strain (n = 4, WT and Pink1 KO). A total of 811 proteins were quantified in all samples from each experimental group. The complete list of the 811 identified proteins and quantification values for each biological replicate is provided in Table S1. We compared the reproducibility among the biological replicates in each group and found that the Pearson’s correlation coefficient (r2) range was from 0.95 to 0.98 in the WT group and from 0.97 to 0.99 in the Pink1 KO group (Figure S3), suggesting that the quantitative SWATH-MS data from replicates were highly reproducible. Consistent with published SWATH-MS analyses [28, 29], a ratio threshold (log2 Pink1 KO/WT) was determined in addition to a p-value threshold. The ratio threshold was determined from the normal distribution fit using 1 standard deviation (Figure 2A). Thus the absolute value of the z-score (normalized log2 ratios (Pink1 KO/WT)) had to be superior to 1.0. To optain p-values, the protein expression values for the biological replicates were analyzed using a Bayesian regularized t-test analysis [26], using p < 0.05 (uncorrected) for the threshold. Combining these thresholds, we obtained a list of 69 proteins differentially expressed in striatal synaptic mitochondria due to loss of Pink1 (Figure 2B; Table S2). The intensity changes of the differentially expressed proteins are shown as a heat map in Figure 2C. Bioinformatic analysis of these 69 proteins using DAVID functional annotation clustering tool [25] revealed several GO biological process terms that were altered according to our proteomics results (Table 1). The top term based on the list of differentially expressed proteins in striatal synaptic mitochondria from Pink1 KO rats was “electron transport chain”, which supports our functional findings of respiratory alterations. To further interrogate the proteins within the ETC, we generated heat maps of the protein expression changes identified by SWATH-MS for the ETC, oxidative phosphorylation (OXPHOS), and electron carriers important for transporting electrons to the respiratory chain (Figure 3A). Overall, the expression of the protein subunits of the ETC and OXPHOS complexes are not altered due to loss of Pink1; however, four subunits, Mtnd1, a mtDNA encoded core subunit of complex I, Ndufa10, which forms the ubiquinone binding site with Mtnd1 [30], Ndufs6, which is a component of the iron-sulfur fragment of complex I, and Uqcrq, which with cytochrome b, binds to ubiquinone (part of the Q-cycle within complex III) were found to be differentially expressed. Strikingly, the levels of several electron carrier proteins, particularly Etfa and Etfb, were found to be significantly down-regulated (Figure 3). These electron carriers are essential for shuttling electrons from at least nine distinct dehydrogenases that are involved in fatty acid β-oxidation, amino acid and choline metabolism to ubiquinone (part of the Q-cycle (series of reactions that describe how the sequential oxidation and reduction of Coenzyme Q10, between ubiquinol and ubiquinone forms results in pumping protons across the inner mitochondrial membrane) within complex III) [31, 32]. These findings suggest that the observed complex I functional impairment in striatal synaptic mitochondria from Pink1-deficient rats may in part be due to loss of core components of complex I. Further, the increased complex II activity could be compensatory for lower complex I activity as well as loss of electron transfer to the respiratory chain via electron carrier proteins.
Figure 2. Identification of differentially expressed striatal synaptic mitochondrial proteins induced by loss of Pink1.
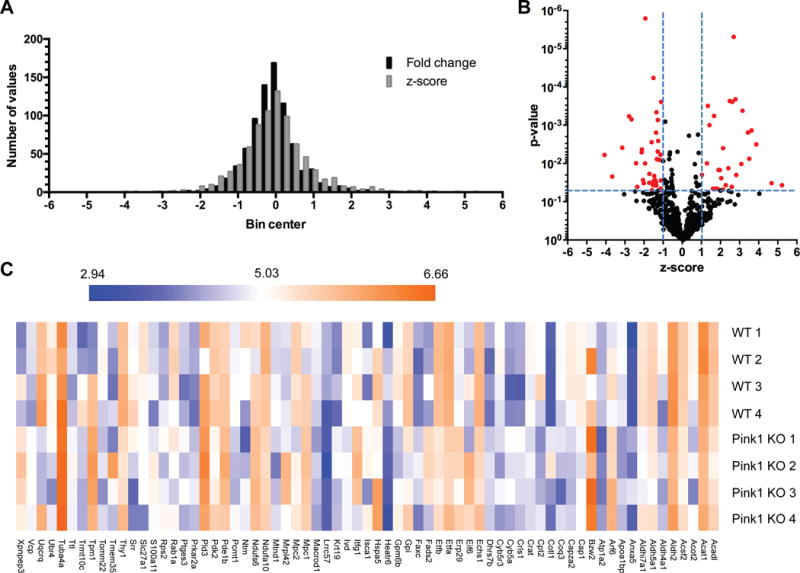
(A) Histogram of protein ratios (log2) in Pink1 KO versus WT. (B) The distribution of p-values (log10) and z-scores (log2) in 811 quantified proteins between the Pink1 KO and WT rats. A total of 69 proteins were selected as differentially expressed due to loss of Pink1, which exhibited a p-value < 0.05 and a z-score > 1 standard deviation (highlighted in red). (C) Heat map of the 69 proteins among four biological replicates between the WT and Pink1 KO rats. The log10 value of the SWATH-MS signal intensity is shown.
Table 1.
DAVID GO enrichment analysis for the 69 differentially expressed proteins.
| Biological Process Term | p-value | q-value | Genes |
|---|---|---|---|
| Electron transport chain | 1.9E-8 | 2.8E-5 | Aldh5a1, Cyb5a, Etfa, Etfb, Fads2, Mtnd1, Ndufa10, Ndufs6, Uqcrq |
| Oxidation reduction | 1.4E-7 | 2.1E-4 | Acadl, Aldh2, Aldh4a1, Aldh5a1, Aldh7a1, Cyb5a, Cyb5r3, Dhrs7b, Etfa, Etfb, Fads2, Ivd, Mtnd1, Ndufa10, Ndufs6, Uqcrq |
| Fatty acid metabolic process | 1.1E-6 | 1.6E-3 | Acot2, Acadl, Acsf2, Aldh5a1, Crat, Cpt2, Echs1, Fads2, Plges3, Slc27a1 |
| Generation of precursor metabolites and energy | 6.1E-6 | 9.1E-3 | Aldh5a1, Cyb5a, Etfa, Etfb, Fads2, Gpi, Mtnd1, Ndufa10, Ndufs6, Uqcrq |
Figure 3. Quantitative proteomics reveals altered expression of protein subunits in the ETC and electron carriers due to Pink1 deficiency.
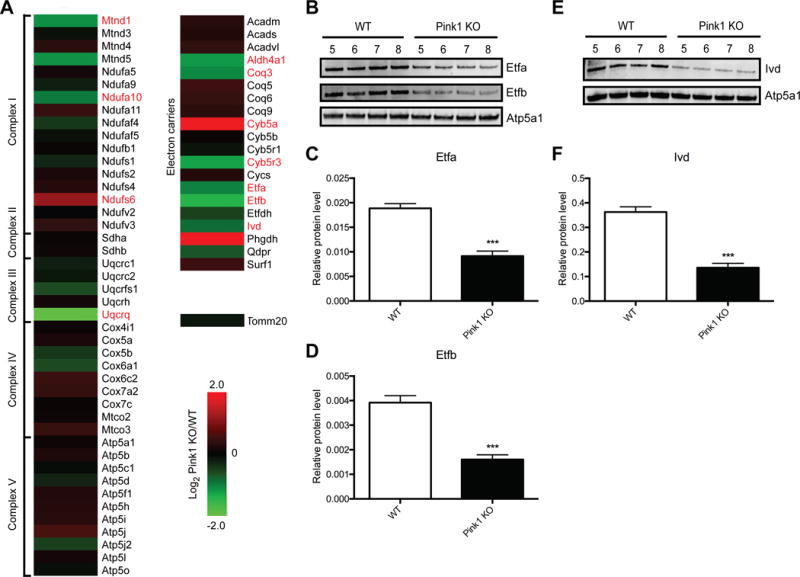
(A) Heat map of the protein expression changes in striatal synaptic mitochondria isolated from Pink1 KO compared to WT rats (Log2 Pink1 KO/WT). Proteins highlighted in red were identified as differentially expressed via proteomics. (B and E) Immunoblot validation of protein expression for selected proteins in (A). (B) Etfa, Etfb, and Atp5a1 antibodies were used for immuoblotting of striatal synaptic mitochondria isolated from WT and Pink1 KO rats (n = 4). Ivd and Atp5a1 antibodies were used in (E). (C, D, and F) Quantification of Western blot data in panels B and E. Values were normalized to Atp5a1. ***(p < 0.001), significantly lower in Pink1 versus WT rats.
3.3 Immunoblot validation experiments
To confirm the protein expression changes as determined by quantitative proteomics, striatal synaptic mitochondria were isolated from two independent groups of 3-month-old Pink1 KO and WT rats (n = 4 for each experiment per group). The results of the Western blot analyses confirmed our proteomics findings and revealed that Etfa and Etfb exhibit significantly reduced expression in striatal synaptic mitochondria from Pink1 KO rats when compared with WT (Figure 3B-D and S4A-C). The levels of Ivd, another electron carrier protein, were found to be significantly decreased via immunoblotting in the Pink1-deficient striatal synaptic mitochondria (Figure 3E-F and S4D-E) confirming differential expression as identified by our proteomics data. Further, the expression of Atp5a1 was shown to be unchanged due to loss of Pink1, consistent with the SWATH-MS results (Figure 3B, E and S4A). We also confirmed consistent levels of Tomm20 in striatal synaptic mitochondria from both WT and Pink1 KO rats as identified by our proteomics experiment (Figure S4D). These results validated our SWATH-MS data analysis.
4 Discussion
The selective, progressive loss of dopaminergic neurons of the substantia nigra pars compacta and the associated loss of nerve terminals within the striatum lead to the observed impaired motor functions in PD patients. Thus, this present study investigated the mitochondrial alterations in nerve terminals within the striatum rather than the substantia nigra to interrogate changes in the projections within the striatum that arise from the substantia nigra. We acknowledge that the striatal synaptosome preparation utilized in this study contains a heterogeneous population of nerve terminals within the striatum that arise from multiple brain regions (e.g. cortex, substantia nigra); however, as our data revealed significant striatal synaptic mitochondrial alterations, future studies aimed at studying specific populations of nerve terminals within the striatum (e.g. substantia nigra) are warranted to determine mechanisms that contribute to the development of PD symptoms due to loss of Pink1 [12].
Our data revealed that synaptic mitochondria from the striatum of 3-month-old presymptomatic Pink1-deficient rats exhibit reduced complex I-mediated respiration and elevated complex II-driven respiration. The functional alterations of the ETC complexes I and II were accompanied by intact complex IV-facilitated respiration. Additionally, the expression levels of 811 synaptic mitochondrial proteins were determined and of those 69 were differentially expressed in the Pink1 KO rats. The alterations in the striatal synaptic mitochondrial proteome due to loss of Pink1 implicate defects in the ETC, fatty acid metabolism, oxidation reduction, and generation of precursor metabolites and energy.
4.1 Synaptic Mitochondrial Bioenergetics
Rats with targeted disruption of Pink1 display progressive nigral neurodegeneration [12], suggesting that Pink1 may be essential for the survival of dopamingergic neurons in the substantia nigra pars compacta. Specifically, Pink1-deficient rats exhibit significant motor deficits starting at 4 months of age and approximately 50% dopaminergic neuron loss at 8 months of age [12]. An important aspect of PD-related changes in the brain due to loss of Pink1 is mitochondrial respiratory dysfunction. Our previous work examining the mitochondrial respiratory states of brain mitochondria from Pink1 KO rats revealed brain region and age specific effects of loss of Pink1 [13]. In particular, we observed increased complex II-mediated respiration (state 3 (ADP-stimulated respiration, controlled to similar extents by ATP turnover as well as substrate supply and oxidation) and state 3u (uncoupled respiration, controlled by substrate supply and oxidation)) in mitochondria from the striatum but not the cortex of Pink1-deficient rats [13]. Further, this functional change was observed only in striatal mitochondria from 9-month-old Pink1 KO rats (not in 4-month-old Pink1 KO rats) [13]. These previous studies were performed on a mixed population of mitochondria, originating from all cells in the striatum at 4 and 9 month of age, early and late during the symptomatic stage. Here we focused on synaptic mitochondria, as a critical and vulnerable population, from 3-month-old Pink1 KO rats, an age prior to the symptomatic changes. Our bioenergetic studies of synaptic mitochondria from the striatum of Pink1 KO rats demonstrate that similar complex II functional alterations (increased state 3 and state 3u) as well as increased state 2 respiration (steady state respiration, controlled largely by the activity of proton leak) exist within nerve terminals isolated from the striatum as were observed for the heterogeneous pool of mitochondria isolated from the striatum at 9 months; however, the changes identified in this study reach significance much earlier, at 3 months of age. Taken together, these results suggest that mitochondria located within striatal nerve terminals are functionally more sensitive to loss of Pink1 than the total heterogeneous pool of striatal mitochondria.
Striatal mitochondria from Pink1 KO mice show decreased mitochondrial respiration; however, the mice have a mild phenotype with no gross morphological changes [33]. While the mouse models fail to replicate many key features of PD, similar to Pink1 KO rats, Drosophila models of autosomal recessive PD exhibit age-dependent dopaminergic neuron loss [11, 34]. Previous studies have shown that mitochondria obtained from Pink1-deficient Drosophila display reduced mitochondrial respiration as a result of a decrease in ETC complex I and IV enzymatic activity [35]. Thus, in addition to interrogating complex II-driven respiration, we also examined complex I- and complex IV-mediated respiration. In the absence of complex IV functional alterations, we observed that striatal synaptic mitochondria from Pink1-deficient rats display impaired complex I-driven respiration (state 3 and state 3u). ADP-stimulated (state 3) respiration is mitochondrial respiration in the presence of ADP, allowing ATP synthase to function. State 3 is controlled by the activity of ATP turnover as well as substrate supply and oxidation, whereas state 3u (uncoupled respiration) is controlled by substrate supply and oxidation exclusively (ATP is not synthesized as ATP synthase is inhibited by Oligo).
These bioenergetics experiments revealed that mitochondrial dysfunction in Pink1 KO rats is due to impaired complex I respiration, specifically, lowered state 3 rates (dysfunction in ATP synthase) and reduced state 3u rates (dysfunction localized to substrate oxidation). Substrates that feed electrons to the ubiquinone pool, bypassing complex I, such as succinate (complex II), translocate fewer protons per electron pair than NADH-linked substrates (complex I), therefore the same proton cycling rate (to power ATP synthase) requires a higher respiration rate. During the process of OXPHOS, generation of ROS, by the ETC occurs. FAD-linked substrates (complex II) have been shown to support the highest rate of ROS production in the absence of inhibitors, whereas, ROS production supported by NADH-liked substrates (complex I) is not detectable in brain mitochondria unless complex I or complex III are inhibited [36]. Interestingly, loss of Pink1 in mice is associated with increased vulnerability to oxidative stress [33]. Further, because these substrates (i.e. succinate (complex II)) tend to generate a higher voltage (i.e. protonmotive force, Δp) across the membrane than NADH-liked substrates (complex I) and the inherent proton leak is highly voltage dependent, this increases the proton current through leak, even though the properties of the leak are not changed [37–39]. Our data is consistent with this, while complex I-mediated respiration rates are down 21.5% (state 3) and 22.8% (state 3u); complex II-driven respiration rates are up 49.1% (state 3) and 52.7% (state 3u). These findings suggest that the elevated complex II-driven respiration (state 3 and state 3u) may be compensatory to overcome the loss of complex I function, although perhaps at a cost (i.e. higher respiration to maintain ATP, elevated ROS generation, and increased proton current through leak).
In addition to altered respiration states, the striatal synaptic mitochondrial spare respiratory capacity (complex I) was significantly decreased in Pink1 KO rats. Failure of adequate mitochondrial spare respiratory capacity is a known mediator of neuronal death [40]. Since the striatum receives heavy innervation from the substantia nigral dopaminergic neurons, the degenerating population of cells during PD pathogenesis, synaptosomes isolated from the striatum would in part be comprised of nerve terminals derived from this vulnerable population of neurons. In fact, even non-manifesting patients with single heterozygous mutation in Pink1 display mild presynaptic dopaminergic striatal dysfunction [41]. Thus the observed reduced complex I spare respiratory capacity of synaptic mitochondria within the striatum might be a novel mechanism leading to neuron loss and would be expected to contribute to PD progression.
4.2 Synaptic Mitochondrial Protein Expression
Striatal synaptic mitochondrial proteomic alterations coincided with the observed bioenergetic alterations in the 3-month-old Pink1-deficient rats. A total of 811 proteins were quantified using SWATH-MS and of these 69 exhibited altered expression in striatal synaptic mitochondria from Pink1 KO compared to WT rats. Functional annotation of the 69 differentially expressed proteins using DAVID revealed enrichment of several biological processes important for mitochondrial function including “electron transport chain”, which supports our observation of respiratory alterations due to Pink1 deficiency. In order to characterize the proteomic changes associated with mitochondrial bioenergetics function, we considered the specific components of the ETC and OXPHOS machinery. While the majority of the ETC complex subunits remained unchanged in striatal synaptic mitochondria from Pink1 KO rats, levels of Ndufs6, a conserved subunit of the enzymatic core of complex I, was increased. Ndufs6 KO mice exhibit impairments in complex I as well as reduced levels of the fully assembled holoenzyme [42]. Further, patients with NDUFS6 mutations display a consistent reduction in complex I activity [42]. Thus, this subunit may be up-regulated in an attempt to salvage complex I activity in the Pink1 KO rat striatal synaptic mitochondria. Decreased expression of integral complex I subunits was observed including Mtnd1 and Ndufa10. Mtnd1 is encoded by mtDNA and pathogenic mutations within this gene are associated with multiple central nervous system (CNS) diseases, including PD [43]. Further, Mtnd1 is located close to Ndufa10 within complex I at a site important for the interaction of ubiquinone with complex I. In fact, Pink1 has been suggested to phosphorylate Ndufa10 and potentially regulate complex I ubiquinone reductase activity [30]. Our results support the idea that loss of Pink1 affects this important site of complex I and provides new evidence that this could be partially due to decreased expression of the subunits Mtnd1 and Ndufa10. In addition to altering ubiquinone coupling, altered expression of these subunits might lead to deficits in complex I assembly. Previous studies in Pink1 KO flies indicated that the observed reduced ETC complex activity was due to impaired ETC complex assembly [35]. Future studies utilizing Pink1 KO rats will be instrumental in interrogating which potential mechanism(s) is/are responsible for the altered bioenergetics.
Our proteomics data suggest that in addition to loss of complex I function, loss of other sources of electrons for input into complex III, may be driving the increase in complex II respiration to maintain ATP levels. In particular, similar to electrons from the complex II substrate succinate, electrons from fatty acid and amino acid metabolism enter the ETC by reducing the ubiquinone pool that links complexes I and III, thus bypassing complex I [31]. We identified significantly reduced expression of Etfa, Etfb, and Ivd (differential expression identified by proteomics and confirmed as significant by immunoblotting) in the striatal synaptic mitochondria from 3-month-old Pink1-deficient rats. The electron transfer protein Ivd has not been examined in PD; however, deficiency of Ivd activity in humans causes isovaleric acidemia, in which leucine cannot be degraded [44]. While the effect of reduced Ivd levels is not known in Pink1 deficiency, a build up of isovaleric acid, which can be neurotoxic, might exist. Furthermore, transfer of electrons to the respiratory chain via leucine degradation would be inhibited.
We have previously identified differential expression of Etfb in brain mitochondria from Pink1 KO rats at 10 days of age [45], suggesting this may be an early alteration. Further, down-regulation of these three proteins (Etfa, Etfb, and Ivd) was observed in the striatal mitochondria from 9-month-old Pink1 KO rats [13]. Although these electron carriers have not been directly linked to PD, it has been reported that neurotrophic effects of expression of Vegf-b and Gdnf in treatments studies using preclinical in vivo models of PD correlates with the expression of Etfb [46], potentially pointing to a pathway disrupted due to loss of Pink1. Deficiency of either electron carrier flavoprotein (Etfa or Etfb) or an electron-transfer flavoprotein dehydrogenase (Etfdh) causes the autosomal recessive disorder multiple acyl-CoA dehydrogenase deficiency (MADD) [47]. The metabolic defects associated with MADD (altered fatty acid oxidation and branched-chain amino acid catabolism) result in impaired ATP synthesis and excessive lipid accumulation. PD does involve deregulated lipid metabolism, a key event believed to contribute to CNS injury [48]. In fact, Pink1 has been shown to play a role in the regulation of mitochondrial fatty acid metabolism [49]. Further, mutations in Etfa and Etfb have been shown to result in ubiquinone deficiency [50]. Of note, ubiquinone deficiency is currently the only mitochondrial disease that can be successfully treated through dietary supplementation. Additionally, vitamin K2, which acts as an alternative electron carrier for ubiquinone due to similar structure has been shown to improve electron transfer in the context of Pink1 deficiency [51]. This demonstrates that targeting electron transfer mechanisms may be a viable therapeutic strategy for PD and warrants further investigation in the Pink1 KO rat model.
In summary, our combined functional and proteomic results highlight mitochondrial alterations due to loss of Pink1 and the role of striatal synaptic mitochondria early in familial PD pathogenesis. The observed significant decrease in striatal synaptic mitochondrial complex I-mediated respiration and increased complex II-driven respiration strongly suggests that complex II bioenergetics are elevated to maintain ATP levels and compensate for loss of complex I function (Figure 4). Additionally, our proteomics studies revealed 69 differentially expressed synaptic mitochondrial proteins in the striatum of Pink1 KO rats. The proteomic data further implicates ETC dysfunction and implies that loss of electron transfer to reduce ubiquinone contributes to altered respiratory chain function (Figure 4). These findings are significant because they indicate that mitochondria within striatal synapses are functionally sensitive to loss of Pink1 and indicate mitochondrial molecular processes that are altered prior to development of motor symptoms in Pink1 KO rats.
Figure 4. Schematic diagram of proposed model of striatal synaptic mitochondrial alterations due to Pink1 deficiency.
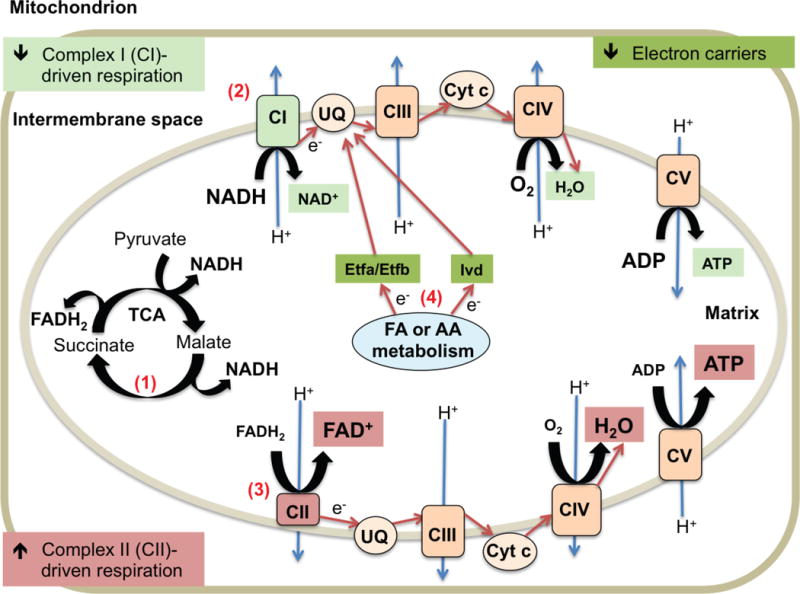
(1) Utilization of the provided complex I (CI) and complex II (CII) substrates (pyruvate/malate (CI) and succinate (CII)) during the Seahorse XF Analyzer assay yields NADH and FADH2, which transport electrons (e−) to the ETC (NADH to CI and FADH2 to CII). (2) Decreased CI-mediated respiration leads reduced ATP production. Highlighted in light green. (3) Increased CII-driven respiration results in increased ATP production to compensate. Highlighted in light red. (4) Decreased levels of electron carriers (Etfa, Etfb, and Ivd) impairs the transport of electrons from fatty acid (FA) and amino acid (AA) metabolism to ubiquinone (UQ) and complex III (CIII) of the ETC contributing to the increased CII function. Highlighted in dark green. Flow of e− indicated using red arrows. Proton (H+) pumping across the mitochondrial inner membrane indicated with blue arrows.
Supplementary Material
Statement of clinical relevance.
Parkinson’s disease (PD) is a common progressive neurodegenerative movement disorder. The etiology of PD remains unknown, although clinical and experimental evidence implicates the involvement of abnormal protein aggregation, oxidative stress, and mitochondrial dysfunction. The most convincing evidence for the involvement of mitochondrial dysfunction is the presence of mutations in the mitochondrial serine/threonine kinase Pink1 that cause recessive hereditary PD in several families. The clinical relevance of our observations is demonstrated by the fact that patients with Pink1 mutations display presynaptic dopaminergic striatal dysfunction and our data suggest that mitochondria located within striatal nerve terminals are functionally sensitive to loss of Pink1 and exhibit alterations prior to the development of motor symptoms in Pink1-deficient rats. Our combined functional and proteomic analyses reveals that striatal synaptic mitochondria from Pink1-deficient rats exhibit impaired complex I respiration and suggests that this impaired function is due to loss of integral complex I protein subunits. Further, an increase in complex II respiration was observed, which appears to be compensatory to the reduced complex I function. Our work suggests that complex I dysfunction and loss of electron transfer mechanisms to reduce ubiquinone within striatal synapses underlies, at least partially, the pathogenesis of this hereditary form of PD.
Acknowledgments
This study was supported by a Michael J. Fox Foundation (MJFF) grant #9524. We are grateful for technical support received from the UNMC Mass Spectrometry and Proteomics Core Facility, especially Dr. Pawel Ciborowski and Melinda Wojtkiewicz.
Abbreviations
- AA
antimycin A
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- ETC
electron transport chain
- FA
formic acid
- FAF
fatty acid free
- FCCP
carbonylcyanide-p-triflouromethoxyphenylhydrazone
- KO
knockout
- MAS
mitochondrial assay solution
- MSHE+BSA
mitochondrial isolation buffer
- mtDNA
mitochondrial DNA
- OCR
oxygen consumption rate
- Oligo
oligomycin
- OXPHOS
oxidative phosphorylation
- PD
Parkinson’s disease
- Pink1
PTEN-induced kinase 1
- Rot
rotenone
- SWATH-MS
sequential window acquisition of all theoretical fragment-ion spectra mass spectrometry
- TMPD
tetramethylphenylenediamine
- WT
wild-type
Footnotes
The authors have declared no conflict of interest.
References
- 1.Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- 2.Jellinger KA. A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim Biophys Acta. 2009;1792:730–740. doi: 10.1016/j.bbadis.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Shulman JM, De Jager PL, Feany MB. Parkinson’s disease: genetics pathogenesis. Annu Rev Pathol. 2011;6:193–222. doi: 10.1146/annurev-pathol-011110-130242. [DOI] [PubMed] [Google Scholar]
- 4.Bonifati V, Rizzu P, van Baren MJ, Schaap O, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 5.Bonifati V, Rohe CF, Breedveld GJ, Fabrizio E, et al. Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology. 2005;65:87–95. doi: 10.1212/01.wnl.0000167546.39375.82. [DOI] [PubMed] [Google Scholar]
- 6.Deng H, Dodson MW, Huang H, Guo M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schapira AH, Cooper JM, Dexter D, Jenner P, et al. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 8.Fariello RG. Experimental support for the implication of oxidative stress in the genesis of parkinsonian syndromes. Funct Neurol. 1988;3:407–412. [PubMed] [Google Scholar]
- 9.Martinez TN, Greenamyre JT. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid Redox Signal. 2012;16:920–934. doi: 10.1089/ars.2011.4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arthur CR, Morton SL, Dunham LD, Keeney PM, Bennett JP., Jr Parkinson’s disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Mol Neurodegener. 2009;4:37. doi: 10.1186/1750-1326-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dave KD, De Silva S, Sheth NP, Ramboz S, et al. Phenotypic characterization of recessive gene knockout rat models of Parkinson’s disease. Neurobiol Dis. 2014;70:190–203. doi: 10.1016/j.nbd.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Villeneuve LM, Purnell PR, Boska MD, Fox HS. Early Expression of Parkinson’s Disease-Related Mitochondrial Abnormalities in PINK1 Knockout Rats. Mol Neurobiol. 2016;53:171–186. doi: 10.1007/s12035-014-8927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stauch KL, Purnell PR, Fox HS. Quantitative proteomics of synaptic and nonsynaptic mitochondria: insights for synaptic mitochondrial vulnerability. J Proteome Res. 2014;13:2620–2636. doi: 10.1021/pr500295n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stauch KL, Purnell PR, Fox HS. Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging (Albany NY) 2014;6:320–334. doi: 10.18632/aging.100657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers GW, Brand MD, Petrosyan S, Ashok D, et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc. 2014;9:421–438. doi: 10.1038/nprot.2014.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 19.Scopes RK. Measurement of protein by spectrophotometry at 205 nm. Anal Biochem. 1974;59:277–282. doi: 10.1016/0003-2697(74)90034-7. [DOI] [PubMed] [Google Scholar]
- 20.Villeneuve LM, Stauch KL, Fox HS. Proteomic analysis of the mitochondria from embryonic and postnatal rat brains reveals response to developmental changes in energy demands. J Proteomics. 2014;109:228–239. doi: 10.1016/j.jprot.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haverland NA, Fox HS, Ciborowski P. Quantitative proteomics by SWATH-MS reveals altered expression of nucleic acid binding and regulatory proteins in HIV-1-infected macrophages. J Proteome Res. 2014;13:2109–2119. doi: 10.1021/pr4012602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shilov IV, Seymour SL, Patel AA, Loboda A, et al. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol Cell Proteomics. 2007;6:1638–1655. doi: 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Gillet LC, Navarro P, Tate S, Rost H, et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.O111.016717. O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vizcaino JA, Cote RG, Csordas A, Dianes JA, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang D, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 26.Kayala MA, Baldi P. Cyber-T web server: differential analysis of high-throughput data. Nucleic Acids Res. 2012;40:W553–559. doi: 10.1093/nar/gks420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerencser AA, Neilson A, Choi SW, Edman U, et al. Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem. 2009;81:6868–6878. doi: 10.1021/ac900881z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bourassa S, Fournier F, Nehme B, Kelly I, et al. Evaluation of iTRAQ and SWATH-MS for the Quantification of Proteins Associated with Insulin Resistance in Human Duodenal Biopsy Samples. PLoS One. 2015;10:e0125934. doi: 10.1371/journal.pone.0125934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang X, Meng Q, Gao J, Zhang S, et al. Label-free Quantitative Analysis of Changes in Broiler Liver Proteins under Heat Stress using SWATH-MS Technology. Sci Rep. 2015;5:15119. doi: 10.1038/srep15119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morais VA, Haddad D, Craessaerts K, De Bock PJ, et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science. 2014;344:203–207. doi: 10.1126/science.1249161. [DOI] [PubMed] [Google Scholar]
- 31.Schiff M, Froissart R, Olsen RK, Acquaviva C, Vianey-Saban C. Electron transfer flavoprotein deficiency: functional and molecular aspects. Mol Genet Metab. 2006;88:153–158. doi: 10.1016/j.ymgme.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 32.Rinaldo P, Matern D, Bennett MJ. Fatty acid oxidation disorders. Annu Rev Physiol. 2002;64:477–502. doi: 10.1146/annurev.physiol.64.082201.154705. [DOI] [PubMed] [Google Scholar]
- 33.Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008;105:11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gispert S, Ricciardi F, Kurz A, Azizov M, et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One. 2009;4:e5777. doi: 10.1371/journal.pone.0005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu W, Acin-Perez R, Geghman KD, Manfredi G, et al. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci U S A. 2011;108:12920–12924. doi: 10.1073/pnas.1107332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 37.Papa S, Lorusso M, Di Paola M. Cooperativity and flexibility of the protonmotive activity of mitochondrial respiratory chain. Biochim Biophys Acta. 2006;1757:428–436. doi: 10.1016/j.bbabio.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 38.Brand MD, Chien LF, Ainscow EK, Rolfe DF, Porter RK. The causes and functions of mitochondrial proton leak. Biochim Biophys Acta. 1994;1187:132–139. doi: 10.1016/0005-2728(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 39.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yadava N, Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neurosci. 2007;27:7310–7317. doi: 10.1523/JNEUROSCI.0212-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Madeo G, Schirinzi T, Martella G, Latagliata EC, et al. PINK1 heterozygous mutations induce subtle alterations in dopamine-dependent synaptic plasticity. Mov Disord. 2014;29:41–53. doi: 10.1002/mds.25724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Torraco A, Peralta S, Iommarini L, Diaz F. Mitochondrial Diseases Part I: mouse models of OXPHOS deficiencies caused by defects in respiratory complex subunits or assembly factors. Mitochondrion. 2015;21:76–91. doi: 10.1016/j.mito.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinto M, Moraes CT. Mitochondrial genome changes and neurodegenerative diseases. Biochim Biophys Acta. 2014;1842:1198–1207. doi: 10.1016/j.bbadis.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vockley J, Ensenauer R. Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity. Am J Med Genet C Semin Med Genet. 2006;142C:95–103. doi: 10.1002/ajmg.c.30089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Villeneuve LMP, P R, Stauch KL, Fox HS. Neonatal mitochondrial abnormalities due to PINK1 deficiency: Proteomics reveals early changes relevant to Parkinson’s disease. Data in Brief. 2001;6:428–432. doi: 10.1016/j.dib.2015.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yue X, Hariri DJ, Caballero B, Zhang S, et al. Comparative study of the neurotrophic effects elicited by VEGF-B and GDNF in preclinical in vivo models of Parkinson’s disease. Neuroscience. 2014;258:385–400. doi: 10.1016/j.neuroscience.2013.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olsen RK, Andresen BS, Christensen E, Bross P, et al. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum Mutat. 2003;22:12–23. doi: 10.1002/humu.10226. [DOI] [PubMed] [Google Scholar]
- 48.Adibhatla RM, Hatcher JF. Role of Lipids in Brain Injury Diseases. Future Lipidol. 2007;2:403–422. doi: 10.2217/17460875.2.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi J, Ravipati A, Nimmagadda V, Schubert M, et al. Potential roles of PINK1 for increased PGC-1alpha-mediated mitochondrial fatty acid oxidation and their associations with Alzheimer disease and diabetes. Mitochondrion. 2014;18:41–48. doi: 10.1016/j.mito.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bentinger M, Tekle M, Dallner G. Coenzyme Q–biosynthesis and functions. Biochem Biophys Res Commun. 2010;396:74–79. doi: 10.1016/j.bbrc.2010.02.147. [DOI] [PubMed] [Google Scholar]
- 51.Vos M, Esposito G, Edirisinghe JN, Vilain S, et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science. 2012;336:1306–1310. doi: 10.1126/science.1218632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.