Abstract
Age-related hearing loss (ARHL) is the most common sensory deficit in the elderly. The disease has a multifactorial etiology with both environmental and genetic factors involved being largely unknown. SLC7A8/SLC3A2 heterodimer is a neutral amino acid exchanger. Here, we demonstrated that SLC7A8 is expressed in the mouse inner ear and that its ablation resulted in ARHL, due to the damage of different cochlear structures. These findings make SLC7A8 transporter a strong candidate for ARHL in humans. Thus, a screening of a cohort of ARHL patients and controls was carried out revealing several variants in SLC7A8, whose role was further investigated by in vitro functional studies. Significant decreases in SLC7A8 transport activity was detected for patient’s variants (p.Val302Ile, p.Arg418His, p.Thr402Met and p.Val460Glu) further supporting a causative role for SLC7A8 in ARHL. Moreover, our preliminary data suggest that a relevant proportion of ARHL cases could be explained by SLC7A8 mutations.
Research organism: Human, Mouse
eLife digest
Age-related hearing loss affects about one in three individuals between the ages of 65 and 74. The first symptom is difficulty hearing high-pitched sounds like children’s voices. The disease starts gradually and worsens over time. Changes in the ear, the nerve that connects it to the brain, or the brain itself can cause hearing loss. Sometimes all three play a role. Genetics, exposure to noise, disease, and aging may all contribute. The condition is so complex it is difficult for scientists to pinpoint a primary suspect or develop treatments.
Now, Guarch, Font-Llitjós et al. show that errors in a protein called SLC7A8 cause age-related hearing loss in mice and humans. The SLC7A8 protein acts like a door that allows amino acids – the building blocks of proteins – to enter or leave a cell. This door is blocked in mice lacking SLC7A8 and damage occurs in the part of their inner ear responsible for hearing. As a result, the animals lose their hearing. Next, Guarch, Font-Llitjós et al. scanned the genomes of 147 people from isolated villages in Italy for mutations in the gene for SLC7A8. The people also underwent hearing tests.
Mutations in the gene for SLC7A8 that partially block the door and prevent the flow of amino acids were found in people with hearing loss. Some mutations in SLC7A8 that allow the door to stay open where found in people who could hear. The experiments suggest that certain mutations in the gene for SLC7A8 are likely an inherited cause of age-related hearing loss. It is possible that other proteins that control the flow of amino acids into or out of cells also may play a role in hearing. More studies are needed to see if it is possible to fix errors in the SLC7A8 protein to delay or restore the hearing loss.
Introduction
Age-related hearing loss (ARHL) or presbycusis is one of the most prevalent chronic medical conditions associated with aging. Indeed, more than 30% of people aged over 65 years suffer ARHL (Gates and Mills, 2005; Huang and Tang, 2010; Van Eyken et al., 2007). Clinically, ARHL is defined as a progressive bilateral sensorineural impairment of hearing in high sound frequencies mainly caused by a mixture of 3 pathological changes: loss of the hair cells of the organ of Corti (sensory), atrophy of the stria vascularis (metabolic) and degeneration of spiral ganglion neurons (SGN), as well as the central auditory pathway (neural) (Gates and Mills, 2005; Schuknecht, 1955; Yamasoba et al., 2013). ARHL has a complex multifactorial etiology with both genetic and environmental factors contributing (Cruickshanks et al., 2010; Christensen et al., 2001). Although most people lose hearing acuity with age, it has been demonstrated that genetic heritability affects the susceptibility, onset and severity of ARHL (Wingfield et al., 2007; Cruickshanks et al., 2001; Gates et al., 1999; Karlsson et al., 1997; Cruickshanks et al., 1998). Unfortunately, the complexity of the pathology coupled with highly variable nature of the environmental factors, which cause cumulative effects, increases the difficulty in identifying the genetic contributors underlying ARHL. Most of the findings from genome-wide association studies (GWAS) performed into adult hearing function could neither be replicated between populations, nor the functional validation of those candidates be confirmed (Dawes and Payton, 2016). Mouse models, including inbred strains, have been essential for the identification of several defined loci that contribute to ARHL (Bowl and Dawson, 2015).
SLC7A8/SLC3A2 is a Na+‐independent transporter of neutral amino acids that corresponds to system L also known as LAT2 (L-type Amino acid Transporter-2) (Pineda et al., 1999; Rossier et al., 1999; Oxender and Christensen, 1963). SLC7A8 is the catalytic subunit of the heterodimer and mediates obligatory exchange with 1:1 stoichiometry of all neutral amino acids, including the small ones (e.g. alanine, glycine, cysteine and serine), which are poor substrates for SLC7A5 (18), another exchanger with system L activity. Functional data indicate that the role of SLC7A8 is to equilibrate the relative concentrations of different amino acids across the plasma membrane instead of mediating their net uptake (Pineda et al., 1999; Meier et al., 2002; Verrey, 2003). The SLC7A8/SLC3A2 heterodimer is primarily expressed in renal proximal tubule, small intestine, blood-brain barrier and placenta, where it is thought to have a role in the flux of amino acids across cell barriers (Rossier et al., 1999; Bauch et al., 2003; Kanai and Endou, 2001; del Amo et al., 2008). So far, SLC7A8 research has been focused mainly on amino acid renal reabsorption. However, in vitro studies demonstrated that SLC7A8 could have a role in cystine efflux in epithelial cells and the in vivo deletion of Slc7a8 in a mouse model showed a moderate neutral aminoaciduria (Braun et al., 2011), suggesting compensation by other neutral amino acid transporters.
Therefore, in order to better understand the physiology of SLC7A8, we generated null Slc7a8 knockout mice (Slc7a8−/−) (Font-LLitjós, 2009) and (Figure 1—figure supplement 1A). Here, we describe the detection of a hypoacusic phenotype in the Slc7a8−/− mouse model and demonstrate that novel loss-of-function SLC7A8 mutations constitute a primary cause in the development of ARHL in a cohort of elderly people from two isolated villages in Italy.
Results
Slc7a8 ablation causes ARHL
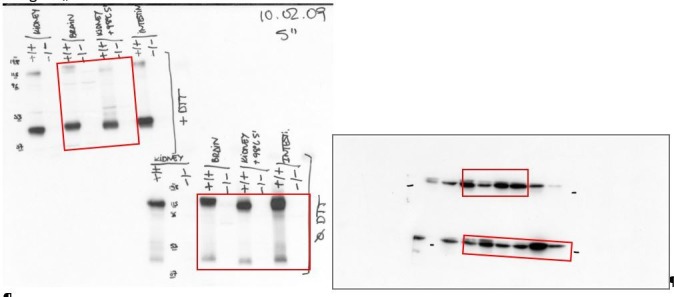
SLC7A8 is highly expressed in the kidney, intestine and brain, and neither full-length nor truncated SLC7A8 protein were detected in membrane samples of Slc7a8−/− mice (Figure 1A). The Allen Brain Atlas (Allen Institute for Brain Science, 2004) localizes mouse brain SLC7A8 to the cortical subplate, cerebellum, thalamus and olfactory bulb. Our results showed that SLC7A8 protein was localized to the plasma membrane of neuronal axons in different brain regions such as, the choroid plexus, subfornical organ, cerebral cortex and hypothalamus by immunohistochemistry (Figure 1—figure supplement 2A). This specific localization in the brain pointed to the possibility that the absence of the transporter could potentially lead to neurological disorders. Behavioral screening showed that absence of SLC7A8 in mice does not affect either learning or memory (Figure 1—figure supplement 3). In contrast, a significant reduction in latency was observed in the rotarod acceleration test indicating impairment in motor coordination in Slc7a8−/− mice (Figure 1—figure supplement 3G). Reaffirming poorer motor coordination performance in the Slc7a8−/− mice, an increased exposure to shock on the treadmill was also observed (Figure 1—figure supplement 3B). Interestingly, a marked impairment was observed in the pre-pulse inhibition of acoustic startle response, which assesses the response to a high intensity acoustic stimulus (pulse) and its inhibition by a weaker pre-pulse. The response to a 120 dB single-pulse was significantly reduced in Slc7a8−/− mice (Figure 1B). The higher threshold required for responding to the acoustic stimulus in the PPI tests in Slc7a8−/− animals could potentially be indicative of a hearing impairment or to a defect in the stress response signaling.
Figure 1. Hearing phenotype of C57BL6/J-129Sv Slc7a8 knockout mice.
(A) Representative image of western bloting of total membranes from kidney, brain and intestine of wild-type (+/+) and Slc7a8 knock out (-/-) mice in the absence (-) or presence (+) of 100 mM dithiothreitol reducing agent (DTT) of three independent biological samples for both sexes (male and female). Protein (50 μg) were loaded in 7% acrylamide SDS-PAGE gel. Molecular mass standard (KDa) are indicated. Red arrows point SLC7A8/CD98hc heterodimer band as well as the light subunit SLC7A8. Upper panel: Rabbit anti-SLC7A8. Bottom panel: Mouse anti-βactin. (B) Pre-Pulse Inhibition of the acoustic startle response (PPI). Mean and SEM are represented. Pulse: 120 dB single pulse. Pre-pulse inhibition test: six different pre-pulse intensities (70 to 90 dB) in pseudo random order with 15 s inter-trial intervals. Wild type (white circles, n = 19) and Slc7a8 −/− (blue circles, n = 15) from 4- to 7-month-old are represented. Significant differences were determined using paired Student’s t-test, ***p<0.001 (C–F) Hearing phenotype in wild-type (Slc7a8+/+, white, n = 11), heterozygous (Slc7a8+/−, green, n = 12) and knockout (Slc7a8−/−, blue, n = 14) mice, grouped by age (4–6 and 7–13 month old). (C,D) Auditory Brainstem Response (ABR) threshold in response to click, expressed as mean ±standard error (C), individual value (scatter plot, (D) and median (boxplot, (D). The significance of the differences was evaluated using ANOVA test, *p<0.05, **p<0.01 (Slc7a8−/− versus Slc7a8+/+) and # p<0.05 (Slc7a8−/− versus Slc7a8+/−). (E) Pie plot showing the percentage of normal hearing (all thresholds <45 dB SPL, white) mice and mice with mild (at least two tone burst threshold >45 dB SPL, orange) and severe (at least two tone burst threshold >60 dB SPL, red) hearing loss (HL), within each genotype and age group. (F) ABR thresholds in response to click and tone burst stimuli (8, 16, 24, 32 and 40 kHz) in mice from three genotypes separated by age group and hearing phenotype (normal hearing or hearing loss). Significant differences were determined using ANOVA test, *p<0.05, **p<0.01, ***p<0.001 (hearing impaired Slc7a8−/− versus normal hearing Slc7a8+/+) and # p<0.05 (hearing impaired Slc7a8−/− versus Slc7a8+/−).

Figure 1—figure supplement 1. Scheme of Slc7a8 knockout mouse generation.
Figure 1—figure supplement 2. SLC7A8 expression in mouse brain.

Figure 1—figure supplement 3. Behavior phenotype.

Figure 1—figure supplement 4. ABR latencies and amplitudes of C57BL6/J-129Sv Slc7a8 knockout mice.

Figure 1—figure supplement 5. Hearing phenotype of C57BL/6J Slc7a8 knockout mice.

Response to stress is modulated by the hypothalamic-pituitary-adrenal axis via the release of corticosterone from the adrenal cortex (Smith and Vale, 2006). As SLC7A8 is expressed in the murine pituitary gland (Figure 1—figure supplement 2A and S3H), plasma corticosterone levels under stressing conditions were analyzed. No differences were observed in corticosterone levels at either basal conditions, nor under restraint stress in the Slc7a8−/− group, indicating a normal stress response in the absence of SLC7A8 (Figure 1—figure supplement 3I). Thus, a hearing impairment in Slc7a8−/− animals was considered the most probable cause of the differences observed in the acoustic startle response test (Figure 1B). The impact of the ablation of SLC7A8 on the auditory system was tested initially on mice with a mixed C57BL6/J‐129Sv genetic background.
Auditory brainstem response (ABR) recording, which evaluates the functional integrity of the auditory system, was performed in Slc7a8−/− mice. Reinforcing our hypothesis, adult 4- to 6-month-old Slc7a8−/− mice showed significantly higher (p≤0.01) ABR thresholds in response to click stimulus, compared with age matched Slc7a8+/− and wild type mice, which maintain normal hearing thresholds (Figure 1C–E). The hearing loss observed in Slc7a8−/− mice affected the highest frequencies tested (20, 28 and 40 kHz) (Figure 1F). The analysis of latencies and amplitudes of the ABR waves in response to click stimuli, showed increased latency and decreased amplitude of wave I, but similar II-IV interpeak latency, in the Slc7a8−/− mice when compared with the other genotypes, pointing to a hypoacusis of peripheral origin without affectation of the central auditory pathway (Figure 1—figure supplement 4A to D).
Mice were grouped according to genotype, age and ABR threshold level and descriptive statistics calculated, showing that the penetrance of the hearing phenotype in the Slc7a8−/− mice is incomplete (Figure 1D and E). Therefore, mice were classified according to their hearing loss (HL) phenotype, defining normal hearing when ABR thresholds for all frequencies were <45 dB SPL, mild phenotype when at least two thresholds were between 45 and 60 dB SPL and severe hypoacusis when at least two thresholds were >60 dB SPL. At 4–6 months of age, Slc7a8−/− mice showed either severe (37.5%) or mild (25%) hearing loss, whilst mice from the other genotypic groups did not show hearing loss (Figure 1E). Next we studied 7–13 month-old mice, 50% of Slc7a8−/− mice presented severe hypoacusis and the hearing loss spread to lower frequencies with age. Slc7a8−/− mice with hearing loss showed statistically significant differences in ABR parameters when compared to the other genotypes (Figure 1F). Moreover, 43% of Slc7a8+/− mice developed mild hearing loss at 7–13 months, whereas the age-matched wild-type mice maintained intact hearing indicating a predisposition toward hearing loss in aged Slc7a8+/−mice (Figure 1E).
The onset and severity of ARHL is attributed to both environmental and genetic factors (Cruickshanks et al., 2010). As the environmental factors were well controlled in all the experiments, thus the phenotypic variability could be attributed as the consequence of individual genetic differences. Indeed, it has been described that several strains of inbred mice present a predisposition to suffer ARHL dependent on multiple genetic factors (Kane et al., 2012; Murillo-Cuesta et al., 2010). Here, the hearing loss phenotype was confirmed in a second mouse strain, the inbread C57BL6/J genetic background (Figure 1—figure supplement 5). Additionally, longitudinal study of Slc7a8−/− mice into the inbred C57BL6/J genetic background showed higher penetrance than the mixed background throughout the ages studied (Figure 2—figure supplement 2).
Localization and quantification of SLC7A8 in the inner ear
The presence of SLC7A8 has previously been reported in the mouse cochlea (Yang et al., 2011; Uetsuka et al., 2015; Sharlin et al., 2011), and specifically localized to the stria vascularis by liquid chromatography tandem mass spectrophotometry and by Western blotting (Uetsuka et al., 2015). Here, SLC7A8 was detected in wild-type mouse cochlea by immunofluorescence supporting its localization to the spiral ligament and spiral limbus from the basal to the apical regions of the cochlea (Figure 2A and B). SLC7A8 immunolabeling was not observed in the stria vascularis. We observed an intense expression of SLC7A8 in the spiral ligament surrounding the stria indicating that the SLC7A8 epitope (Figure 1—figure supplement 1B) is either hidden or absent in the stria vascularis. Quantification of SLC7A8 expression in the cochlea showed half a dose of the transporter in the Slc7a8+/−than in wild-type mice, and its ablation in Slc7a8−/− mice (Figure 2C). A closer study of SLC7A8 immunofluorescence showed that the transporter is also expressed in the spiral ganglia neurons area (SGN) (Figure 1—figure supplement 2B).
Figure 2. Immunolocalization of SLC7A8 in the mouse cochlea.
(A) Representative photomicrographs of cryosections of the base of the cochlea showing immunodetection for SLC7A8 (green) and s100 (red); and staining for DAPI (blue) or phalloidin (white) of wild type (upper row) and Slc7a8−/− mice (lower row). Scale bar, 100 µm. (B) On the left overlay image of a wild-type section indicating cochlea areas. Scale bar, 100 µm. On the right schematic drawing of the adult scala media adapted from Sanchez-Calderon et al. (2010). BC, border cells; CC, Claudius's cells; DC, Deiter's cells; HC, Hensen's cells; IC, intermediate cells; IHC, inner hair cells; IPC, inner phalangeal cells; Li, spiral limbus; MB, Basilar Membrane; OHC, outer hair cells; PC, pillar cells; RM, Reisner's membrane; SG, spiral ganglion; SL, spiral ligament; SV, stria vascularis; TM, tectorial membrane. (C) Quantification of SLC7A8 expression. Intensity of SLC7A8 immunofluorescence was normalized per mm2. Mean ±SEM from quadruplicates for each section, taken from apex, middle and basal cochlear turns of 4 wild-type (black), 3 Slc7a8+/− (green) and 4 Slc7a8−/− (blue) young (4- to 7-month-old) mice. Open and closed circles represent individual mice from C57BL6/J-129Sv or C57BL6/J backgrounds, respectevely. Unpaired Student’s t-test statistical analysis, p-values: *,≤0.05; **,≤0.01 and ***,≤0.001. (D) Quantification of SLC7A8 protein expression in the apex, middle and basal cochlear turns normalized per nuclei of young (2 month-old) (open bars) and old (12 month-old) (black bars) wild-type CBA mice. Data (mean ±SEM) were obtained from four cochlear sections obtained from three mice per group. Unpaired Student’s t-test statistical analysis, p-value: *,≤0.05.

Figure 2—figure supplement 1. Quantification of transcripts in the Slc7a8−/− mouse cochlea.
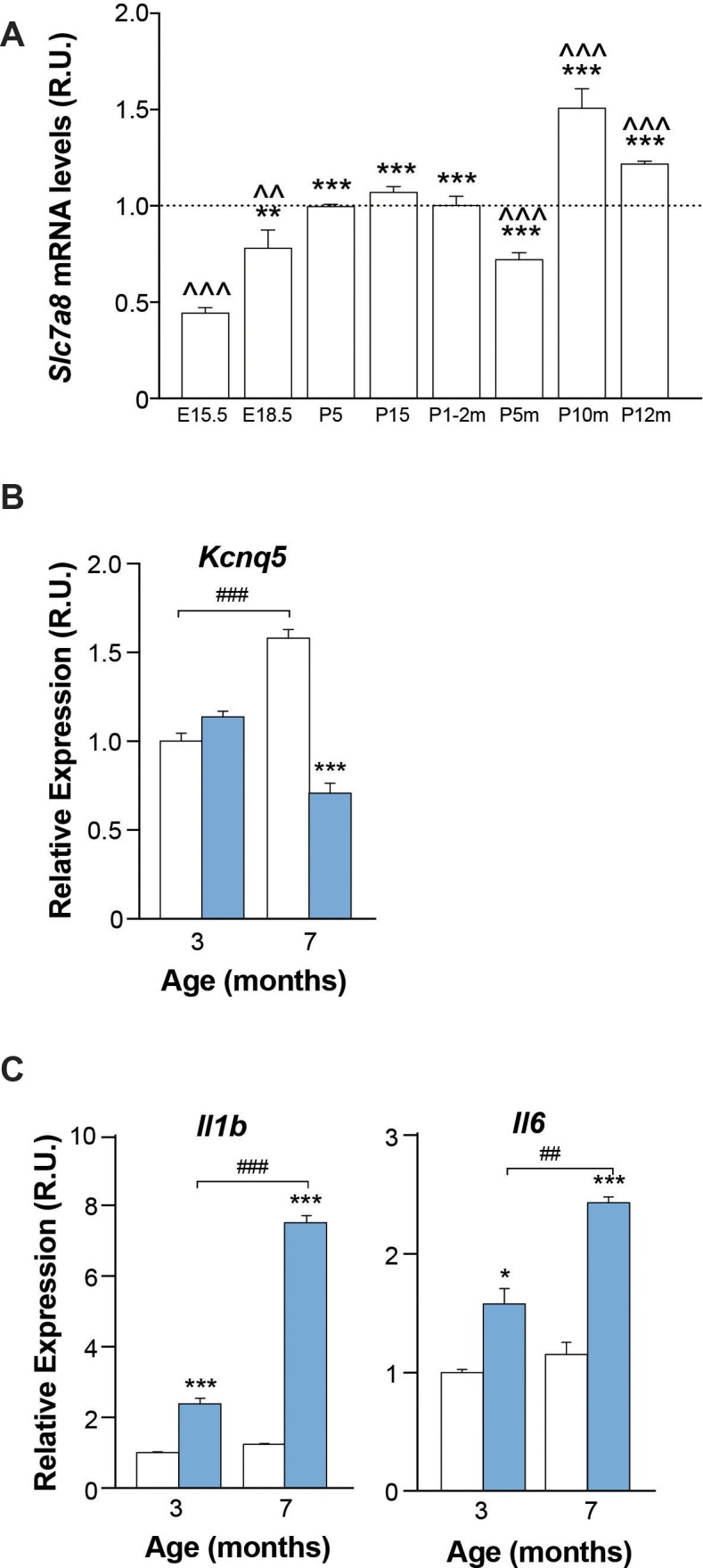
Figure 2—figure supplement 2. Progression of hearing phenotype of C57BL/6J Slc7a8 knockout mice.

The early HL onset and the progressive ARHL phenotype observed in Slc7a8−/− and Slc7a8+/− mice respectively, prompted us to compare the expression of SLC7A8 in wild-type cochlea at different ages (Figure 1D). Immunofluorescence quantification of SLC7A8 intensity at 2- and 12 months of age showed expression in the young mice and increased presence of the transporter in the older mice (Figure 2D). In the same line, Slc7a8 mRNA quantification from cochlea extracts showed a progressive increased expression throughout mouse life (Figure 2—figure supplement 1A).
Lack of Slc7a8 induced damage in the organ of Corti, spiral ganglion and stria vascularis
The cytoarchitecture of the inner ear was studied by hematoxylin/eosin staining (Figure 3), immunofluorescence (Figure 4 and Figure 4—figure supplement 1) and mRNA detection of several cochlear markers (Figure 3D and Figure 2—figure supplement 1). Most of the structures of the cochlear duct, including spiral ligament, spiral limbus, tectorial and basilar membranes showed a normal gross cytoarchitecture in the Slc7a8−/− mice. In contrast, in the basal turns of the cochlea we observed that 3 out of 6 Slc7a8−/− mice evaluated showed complete loss of hair cells and flat epithelia, while only one Slc7a8−/− mouse showed intact epithelia in the organ of Corti (Figure 3A). Likewise, loss of cells in the spiral ganglia, especially in the basal regions of the cochlea, was observed (Figure 3A). Slc7a8−/− mice at 4 to 7 months of age presented ~50% of cell loss in the spiral ganglion compared with wild type mice (Figure 3B). Decreased number of cells in the ganglia significantly correlates with ABR threshold and HL phenotype (Figure 3—figure supplement 1and B). Concomitantly with the loss of hair cells and spiral ganglion (SG) nuclei in Slc7a8−/− mice, the messenger levels of cell type specific biomarkers, such as the potassium voltage-gated channels Kcnq2, Kcnq3 and Kcnq5, and the transporter Slc26a5, which are expressed in the organ of Corti and SG were down-regulated respectively (Figure 3D and Figure 2—figure supplement 2B).
Figure 3. Cytoarchitecture of the Slc7a8−/− mouse cochlea.
(A) Hematoxylin and Eosin staining of the base of the cochlea. Representative photomicrographs taken from paraffin sections of wild-type and hipoacusic Slc7a8−/− mice. OC, Organ of Corti; SG, spiral ganglia region; and SL, spiral ligament. * Indicates loss of hair cells in the organ of Corti (first column), loss of neurons in the spiral ganglia (second column) and lower nuclei density in the spiral ligament (third column). Scale bar 100 μm. (B) Quantification of the number of neurons in the spiral ganglia (SG) in the basal turns of the cochlea. Y axis represents the mean nuclei quantification of 5 to 10 areas in SG. (C) Quantification of the number of nuclei in the spiral ligament (SL) of the basal turns of the cochlea by immunofluorescence using DAPI staining. For each sample, 12 overlaps of Z-stacks areas were used to quantify number of nuclei. Unpaired Student’s t-test statistical analysis: **, p≤0.01 (A to C) 4 wild-type (black), 3 Slc7a8+/− (green) and 4 Slc7a8−/− (blue) mice at 4 to 7-month-old are represented. Circles represent the average of the quadruplicate analysis performed in each mouse of C57BL6/J-129Sv (open) and C57BL6/J (filled) background. (D) Quantification of mRNA markers by RT-qPCR PCR. Cochlear gene expression of Slc26a5, Tbx18, Kcnq2 and Kcnq3 in the cochlea at 3-month-old and 7 months wild-type (white bars) and Slc7a8−/− (blue bars) C57BL6/J mice. Expression levels, normalized with Rplp0 gene expression, are represented as n-fold relative to control group. Values are presented as mean ±SEM of triplicates from pool samples of three mice per condition. Unpaired Student’s t-test statistical analysis, p-values: *p≤0.05; **p≤0.01; ***p≤0.001.

Figure 3—figure supplement 1. Correlations of the cell numbers and cell type biomarkers with HL phenotype.

Figure 4. Immunofluorescence of cochlear markers in the Slc7a8−/− mouse.
(A) Representative photomicrographs of cryosections (10 μm) from the basal turn of the cochlea from wild type (1 and 4), Slc7a8+/− (2 and 5) and Slc7a8−/− (3 and 6) mice labeled for Kir4.1 (green), phalloidin (red) and DAPI (blue) (1 to 3), or for s100 (red), phalloidin (cyan) and DAPI (blue) (4 to 6). Scale bar, 100 µm. (B, C and D) Graph representing the quantification of Kir4.1, s100 and phalloidin (Pha) labeling intensity in the basal turn of the cochlea. Means ± SEM, normalized per mm2 of 4 wild type (black bars), 3 Slc7a8+/− (green bars) and 4 Slc7a8−/− (blue bars) young (4- to 7-month-old) mice are represented. Individual circles represent the average of the quadruplicate analysis of sections from each mice of either C57BL6/J-129Sv (open) or C57BL6/J (filled) backgrounds. Unpaired Student’s t-test statistical analysis, p-value: *,≤0.05.

Figure 4—figure supplement 1. Quantification of the intensity of cell type biomarkers in apical and middle cochlear regions.

Less densely packed cells in the spiral ligament were observed in Slc7a8−/− than in wild-type mice (Figure 3A). Reinforcing this observation, the expression of Kir4.1, a potassium channel highly expressed in stria vascularis cells (Ando and Takeuchi, 1999), was also dramatically reduced by 50% in Slc7a8−/− (Figure 4B and Figure 4—figure supplement 1A). Likewise, decreased expression of Kir4.1 marker correlates with HL phenotype (Figure 3—figure supplement 1C). Phalloidin labeling of actin fibers in the basal cells of the stria vascularis was also decreased 50% in the base of the cochlea (Figure 4C and Figure 4—figure supplement 1).
SLC7A8 is abundantly expressed in fibrocytes of the spiral ligament and limbus (Figure 2), accordingly the number of fibrocytes in the spiral ligament decreased by 2/3 and 1/3 in the null and Slc7a8+/− mice, respectively (Figure 3C). Moreover, mice with severe HL phenotype showed 30% less number of fibrocytes in the spiral ligament (Figure 3—figure supplement 1D). The expression of the transcription factor Tbx18, essential for fibrocytes development and differentiation, was 50% less in Slc7a8−/− than in wild-type mouse cochleae (Figure 3D). In contrast, the expression of s100, fibrocyte types I and II marker, did not show significant differences (Figure 4D and Figure 4—figure supplement 1C).
Mutations in SLC7A8 are associated with ARHL
Once we associated mouse SLC7A8 transporter with deafness and identified it as a potential ARHL gene, screening for mutations in human populations was initiated. Whole genome sequencing (WGS) and audiogram test data obtained from 147 individuals from isolated villages in Italy were included in the study. The inclusion criteria were people 50 years old or older with an audiogram test done at high frequencies (Pure-tone audiometric PTA-H, 4 and 8 kHz). Individuals with pure-tone average for high frequencies (PTA-H) greater than or equal to 40 decibels hearing level (dB HL) were considered ARHL cases, whilst people with PTA-H less than 25 dB were considered as controls. A total of 66 cases suffering ARHL and 81 controls were selected. The gene-targeted studies conducted in this isolated cohort succeeded in detecting seven heterozygous missense variants (Table 1). Four of the variants: p.Val460Glu (V460E), p.Thr402Met (T402M), p.Val302Ile (V302I) and p.Arg418His (R418C) belong to ARHL cases (see Audiogram in Figure 5—figure supplement 1A) and other three: p.Arg8Pro (R8P), p.Ala94Thr (A94T) and p.Arg185Gln (R185L) to the control group (see Audiogram in Audiogram in Figure 5—figure supplement 1B).
Table 1. SLC7A8 Humans mutations found in ARHL and controls individuals.
| Phenotype | Age | Sex | Chr. 14 | Variant | Consequence | Code | Frequency | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Esp6500siv2 | 1000 g | Campion | ExAC | Studied cohort | |||||||
| ARHL | 75 | Female | 23597290 | 14:23597290 A / T | p.Val460Glu | V460E | NA | NA | 0.0013 | 0.00002475 | 0.015 |
| ARHL | 57 | Male | 23598917 | 14:23598917 G / A | p.Thr402Met | T402M | NA | NA | 0.0047 | 0.00002471 | 0.015 |
| ARHL | 75 | Male | 23608641 | 14:23608641 C / T (rs142951280) |
p.Val302Ile | V302I | 0.0005 | NA | 0.0047 | 0.0004613 | 0.015 |
| ARHL | 86 | Female | 23598870 | 14:23598870 G / A (rs146946494) |
p.Arg418Cys | R418C | 0.0005 | NA | 0.002 | 0.00002477 | 0.015 |
| control | 50 | Male | 23652101 | 14:23652101 C / G (rs141772308) |
p.Arg8Pro | R8P | 0.0008 | NA | 0.0013 | 0.0008156 | 0.012 |
| control | 50 | Male | 23635621 | 14:23635621 C / T (rs139927895) |
p.Ala94Thr | A94T | 0.0012 | 0.002 | 0.0013 | 0.00202 | 0.012 |
| control | 90 | Female | 23612368 | 14:23612368 C / A (rs149245114) |
p.Arg185Gln | R185L | NA | NA | 0.002 | 0.00002471 | 0.012 |
ARHL (age-related hearing loss). The age (years) of the subject when the Audiogram was performed is indicated. Variant [CHR: position reference/alternate (dbSNP135rsID)]. Consequence [HGUS annotation (protein change)]. Code [short description of the alternate variant]. Frequency of the mutations: Esp6500siv2 (NHLBI Exome Sequencing Project), 1000 g (1000 Genomes Project), Campion (The Allele Frequency Net Database) and ExAC (The Exome Aggregation Consortium).
All the mutations found in SLC7A8 cases and controls from isolated villages of Friuli Venezia Giulia exhibited different frequencies in comparison to public data bases, such as ExAC among others (see Table 1). According to ExAC database’s constrain metrics (Lek et al., 2016), the gene shows evidence of tolerance of both loss of function (pLi = 0) and missense variation (missense Z score = −0.14).
Functional studies of SLC7A8 mutations
A structural model of human SLC7A8 protein built using the homologous protein AdiC (Kowalczyk et al., 2011) in the outward-facing conformation (Rosell et al., 2014) (Figure 5—figure supplement 1C and D) was used to localize all the mutations identified here. Interestingly, three of the four mutations found in ARHL patients were located in very striking places: (i) V302 is a conserved amino acid located in the extracellular loop four which corresponds to the external lid that closes the substrate binding site when the transporter is open to the cytosol, (ii) T402 is located in transmembrane (TM) domain 10 facing to the substrate binding site, and (iii) V460 is located at the very end of TM domain 12, with potential interaction with the plasma membrane. In contrast, R418 is in the intracellular loop 5, between TM domain 10 and TM domain 11 and with no functional role described in transporters with the LeuT-fold (Krishnamurthy and Gouaux, 2012). Thus, three of these mutations were promising candidates to affect the transporter function due to their crucial location.
In vitro functional characterization of variants present in patients with ARHL and controls was performed by measuring amino acid uptake in HeLa cells co-transfected with the heavy subunit CD98hc and Strep tagged-SLC7A8 wild type and variants (Figure 5). Co-expression of the light (SLC7A8) and the heavy (CD98hc) subunits in the same cell increases the plasma membrane localization of the transporter (Rosell et al., 2014). All tested variants showed expression levels comparable to those of wild type, except for V460E that showed only 20% expression of wild -ype protein (Figure 5—source data 1), being the only variant that did not reach the plasma membrane as indicated by the lack of co-localization with wheat germ agglutinin staining (Figure 5A). Amino acid transport induced by SLC7A8 was analyzed for wild type and the identified variants (Figure 5B). All variants present in controls (R8P, R186L and A94T) conserved more than 80% of alanine transport compared with wild-type protein. Three variants found in patients with ARHL showed diminished alanine transport activity: T402M and V460E presented little residual transport activity (14.6 ± 2.6% and 3.6 ± 0.3% of wild-type activity, respectively) and R418C showed 50.7 ± 5.4% of wild-type alanine transport. Surprisingly, V302I presented similar alanine transport levels to wild type SLC7A8. Location of residue V302 within EL4 (within the external substrate lid (Figure 5—figure supplement 1D) led us to additionally measure a larger size SLC7A8 substrate, whose transport could potentially be more compromised than that of a small substrate (e.g. alanine). Interestingly, V302I transport activity of tyrosine was found to be only 40.0 ± 1.6% of wild-type SLC7A8. Because the V302I mutation showed a substrate-dependent impact, tyrosine transport in the other variants was also tested (Figure 5B). Other SLC7A8 variants found in patients with ARHL and controls showed similar decreased transport activity for alanine and tyrosine. Thus, the SLC7A8-induced tyrosine transport was clearly defective in the four variants found in patients with ARHL, whereas it was barely affected (>85% of wild-type transport activity) in the variants found in controls.
Figure 5. In vitro characterization of SLC7A8 mutants.
(A) Panel showing representative images of immunofluorescence of wild type and the indicated SLC7A8 mutants overexpressed in HeLa cells. Overlay of SLC7A8 (green), wheat germ agglutinin (WGA, membrane marker) (red) and the nuclear marker DAPI (blue) labeling. All SLC7A8 variants, except V460E, reached the plasma membrane. (B) Alanine (Ala) and tyrosine (Tyr) transport activity of human SLC7A8 wild type (WT) and mutants in transfected HeLa cells. SLC7A8 transport activity, corrected by SLC7A8-GFP expression, is presented as percentage of wild-type SLC7A8 transport activity. Data (mean ±SEM) corresponds to three independent experiments with quadruplicates. Mutants activity comparing with its, respectively, wild-type transport unpaired Student’s t-test statistical analysis is represented, p-values: *,≤0.05; **,≤0.01 and ***,≤0.001.

Figure 5—figure supplement 1. Audiogram of patients with ARHL and localization of the mutations in SLC7A8 protein.

Discussion
Here, we show that loss of function of the amino acid transporter SLC7A8 is associated with ARHL in both humans and mice. Full ablation of SLC7A8 transporter in mice produced a hearing loss defect with incomplete penetrance affecting mainly high-frequency sounds, a characteristic of ARHL (Figures 1C–F, S5 and S6). Interestingly, hearing loss severity increases with age in Slc7a8−/− mice (Figures 1C–F and S6). Similarly, Slc7a8 heterozygous mice showed increased hearing loss penetrance with age, as indicated by the late onset of the phenotype (starting from 7 months onwards) (Figures 1E, S5 and S6). In addition, SLC7A8 expression in wild type cochlea rises during ageing (Figure 2D and S7A). In patients with ARHL we identified four SLC7A8 variants that showed loss of function of transport of tyrosine (Figure 5B). Altogether, these results indicate that full SLC7A8 function is needed to keep an optimal hearing function throughout life, with half a dose of SLC7A8 being enough to accelerate ARHL phenotype in mice and humans.
The hearing loss (HL) phenotype in the Slc7a8−/− mice has been confirmed on two genetic backgrounds (mixed C57BL6/J-129Sv; Figure 1, and inbred C57BL6/J; Figure 1—figure supplement 5). Interestingly, onset and penetrance, but not severity, was increased in the hearing loss trait of Slc7a8−/− mice in the pure C57BL6/J background (Figure 1—figure supplement 5). It is well-known that the C57BL6/J background carry a mutation in the Cdh23 gene causing early onset of ARHL (Noben-Trauth et al., 2003; Mazelová et al., 2003). It is also worthwhile to mention that all the inbred C57BL6/J mice used to perform the experiments in this research were positive for the ARHL susceptibility allele A/A in Cadh23 (data not shown). Genetic linkage between both genes could be disregarded because both are located in different chromosomes (Slc7a8 in Chr:14 and Cdh23 in Chr:8). Therefore, non-additive severity of the hearing loss phenotype of Slc7a8 ablation and Cdh23 susceptibility allele suggests that both genes may share similar mechanisms of pathogenicity.
In line with the results observed in the mouse model, the four human mutations found in heterozygosis in ARHL patients showed a reduced SLC7A8 transporter activity meanwhile the mutations found in control group did not affect the transporter activity (Figure 5B). The predisposition of SLC7A8 to host deleterious variants, as shown by the in silico-patterns of missense and loss-of-function tolerance, could be explained because its aberration affects age-related hearing function, but its ablation is neither vital nor affects the reproduction of the mice (Slc7a8−/− showed same frequency of siblings as expected, data not shown). Furthermore, the presence of mutations in both ARHL cases and controls in our cohort with higher frequencies in respect to public databases could be explained as a result of isolation and inbreeding in our individuals; as isolation in a population could lead to an enrichment of deleterious variants due to relaxation of purifying selection (Xue et al., 2017). We also noted that in ExAC the mutations found in controls have a mean frequency that is seven times higher than the ones found in our cases, and we speculate that this could be an indirect hint of the higher deleteriousness of the variations found in our cases in respect to the controls. Thus, the present work points to SLC7A8 as a strong candidate gene involved in ARHL induction and the presented data suggest that a significant proportion (~3%) of ARHL cases could be explained by SLC7A8 mutations making it one of the major players so far described.
SLC7A8 was localized in key cochlear structures: the spiral ligament, spiral limbus and spiral ganglion (Figure 2A and S2B) likewise the three main pathological changes described in the ARHL were observed in the absence of SLC7A8: the hair cells of organ of Corti (sensory), the spiral ganglia (neural), and the spiral ligament and the stria vascularis (metabolic) (Figure 3).
The spiral ligament contributes to cochlear homeostasis and is crucial for normal hearing. Degradation of the spiral ligament can result in either one form of hereditary deafness through POU3F4 mutations at locus DFN3 (41) or in the loss of endocochlear potential (EP) in presbycusis mouse models (Wu and Marcus, 2003). In the spiral ligament, SLC7A8 expression was detected in fibrocytes, mostly in type I, close to the stria vascularis (Figure 2). In addition, a reduced number of total cells was observed in both Slc7a8−/− and Slc7a8+/− mice (Figure 3C). Type I fibrocytes are interconnected with the adjacent types II and V cells forming a gap junction-dependent cell system with a relevant role in ion homeostasis [for a review, see (Kikuchi et al., 2000)]. Deafness due to fibrocyte alterations has been described, which indicates the importance of their integrity for appropriate hearing (Minowa et al., 1999; Teubner et al., 2003; Boettger et al., 2003; Delprat et al., 2005; Trowe et al., 2008). Nonetheless, s100 expression (Figure 4C) appeared to be unaffected in the absence of SLC7A8. Interestingly, mutations in genes expressed in spiral ligament fibrocytes could affect stria vascularis function causing deafness, such as the ablation of the fibrocyte transcription factor POU3F4 that causes loss of fibrocytes IV and V in the spiral ligament, decreased cellular density in the stria vascularis and decreased expression of Kir4.1 (48). As the stria vascularis regulates nutrient transport and ion fluxes is responsible for the maintenance of the EP (Peter and Santi, 2001), which is the driving force required for neurotransmission after acoustic stimulus (Wangemann, 2006; Couloigner et al., 2006). We observed alterations in the stria vascularis, decreased expression of Kir4.1 and the basal cell marker phalloidin all correlating with HL phenotype in Slc7a8−/−, and similar traits in Slc7a8+/− mice (Figures 3A and 4B–C and S9). Moreover, is described that the ablation of the T-box transcription factor gene Tbx18, expressed in the spiral ligament, compromises fibrocytes differentiation (Trowe et al., 2008) and concomitant disruption of the architecture of the stria vascularis with almost complete absence of the basal cell layer, and down-regulation of Kir4.1 (52). Likewise, deletion of Pendrin (SLC26A4, PDS) (Cl-/I-/HCO3- anion exchanger expressed in mouse fibrocytes) showed pronounced signs of vestibular disease attributed to an altered EP (Everett et al., 2001). Concomitant with reported data, transcript levels of both Tbx18 and Slc26a4 are down-regulated in the Slc7a8−/− mouse (Figure 3D). Therefore, if we assume a defect in ion homeostasis in the absence of SLC7A8, we could expect an EP impairment that should also trigger vestibular damage. In line with this assumption, we observed impaired balance during gradual acceleration in rotarod test performance of Slc7a8−/− mouse (Figure 1—figure supplement 3G). Altogether, the data presented suggests that the absence of SLC7A8 in fibrocytes might contribute a metabolic component to the progression of hearing loss.
The reduction in the number of cells of the spiral ganglia in Slc7a8−/− mice to half of those in wild type (Figure 3A and B) and its correlation with ABR threshold at high frequencies (Figure 3—figure supplement 1) could be considered causative of neuronal hearing loss (Camarero et al., 2001), and the lack of expression of SLC7A8 in SG might directly contributed to this neurodegeneration (Figure 1—figure supplement 2B). SG axons are part of the auditory nerve and transmit signals from the organ of Corti to the brain. In addition, it has been described that SG degeneration may result in hair cells and sensory hearing loss (Stankovic et al., 2004; Sugawara et al., 2007; Zilberstein et al., 2012). SLC7A8 is expressed in the SG but not in the organ of Corti. However, Slc7a8−/− mice also showed loss of hair cells (Figure 3A) suggesting a potential negative feedback from the damaged SG similar to those described (Stankovic et al., 2004; Sugawara et al., 2007; Zilberstein et al., 2012).
SLC7A8/SLC3A2 exchanges all neutral amino acids except for proline (Pineda et al., 1999), and therefore either SLC7A8 ablation in mice or SLC7A8 loss-of-function mutations in humans can alter availability or concentration of a specific set of neutral amino acids in cells (especially fibrocytes and neurons) of the spiral ligament, spiral limbus and spiral ganglion. Three of the four ARHL mutations (T402M, R418C and V460E) showed similarly compromised transport of the amino acids tested (alanine and tyrosine), whereas V302I selectively showed a defect for the large amino acid tyrosine (Figure 5B). Mutation V302I, located within the external lid in the extracellular loop 4, might result in a steric hindrance with bulky substrates when closing the substrate cavity in the inward-facing conformation of the transporter. SLC7A8 loss-of-function might render alterations in the cell content of bulky neutral amino acids like branched chain amino acids or glutamine, which affect proteostasis and renewal of cell structures causing cell stress (Efeyan et al., 2015; Someya and Prolla, 2010). Caloric restriction, that involves both an increased branched amino content and protein degradation, showed an effective delay of age-related cochlear neuron degeneration (Someya et al., 2010; Bao and Ohlemiller, 2010). In any case, Slc7a8−/− cochlea presents signs of unresolved chronic inflammation with up-regulation of Il1b and Il6 mRNA (Figure 2—figure supplement 1C) and reduced activation of macrophages (down-regulation of Iba1 protein) (Suppl. Figure S8D). As SLC7A8 is also expressed in macrophages (BioGPS [Internet]. 2001), the role of the immune response in the hearing loss associated with Slc7a8−/− mice deserves further attention.
SLC7A8 also transports thyroid hormones (TH) (Zevenbergen et al., 2015; Hinz et al., 2017) as well as the dopamine precursor L-DOPA (Gomes and Soares-da-Silva, 2002; Pinho et al., 2004). Even though hypothyroidism causes hearing loss characterized by alterations in cochlear development (Peeters et al., 2015) and L-DOPA showed a protective role for cochlea during aging (Murillo-Cuesta et al., 2010), Slc7a8−/− mice showed neither hypothyroidism (Braun et al., 2011) nor alterations in L-DOPA plasma levels (data not shown). The lack of SLC7A8 might be compensated by other transporters like the main TH transporter MCT8 (Núñez et al., 2014). Moreover, we cannot disregard a local impact of a shortage of L-DOPA in the cochlea, which could influence its maintenance, altering the protective role of this metabolite. Therefore, in the absence of SLC7A8, three elements could play a role in the hearing loss phenotype: neutral amino acids, thyroid hormones and/or L-DOPA. Characterization of new SLC7A8 mutations with substrate-dependent transport activity will be necessary to draw a definitive conclusion as to the molecular mechanism of the SLC7A8 substrates involved in ARHL.
Conclusion
The present work provides evidence that the amino acid transporter SLC7A8/SLC3A2 has a direct role in age-related hearing-loss (ARHL). The ablation of SLC7A8 in a mouse model causes deafness with ARHL characteristics, defective audition at high-frequencies with early onset in homozygotes and progressive worsening in heterozygotes with age. Identification of rare variants in SLC7A8 gene together with amino acid transport loss-of-function in ARHL patients supports the concept that this gene has a role in the auditory system in association with other genetic and/or environmental factors.
This study highlights amino acid transporters as new targets to study in largely uncharacterized hearing disorders. The description of SLC7A8 as a novel gene involved in a complex trait such as ARHL demonstrates the importance of amino acid homeostasis in preserving auditory function and suggests that genetic screening should be extended to consider other amino acid transporters as potential new genes involved in cochlear dysfunction. Our results may enable the identification of individuals susceptible to developing ARHL, allowing for early treatment or prevention of the disease.
Materials and methods
All key research resources described in this section are summarized in Table 2.
Table 2. Key resources table.
| Reagent type (species) or resource |
Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Antibody | SLC7A8 antibody | Custom made | NA | Anti-Rabbit peptide sequence: PIFKPTPVKDPDSEEQP WB: 1:1000, IHC: 1/5000 and IF:1/200 |
| s100 | Sigma-Aldrich | Ref: S2532 | IF: 1/1000 | |
| Kir4.1 | Merck Millipore | Ref: AB5818 | IF: 1/200 | |
| BA1 | Abcam | Ref: ab5076 | IF: 1/200 | |
| Phalloidin | Thermo Fisher Scientific | Ref: A22287 | IF: 1/100 | |
| Donkey anti-Goat Alexa Fluor 546 |
Thermo Fisher Scientific | Ref: A-11056 | IF: 1/300 | |
| Donkey anti-Rabbit Alexa Fluor 488 |
Thermo Fisher Scientific | Ref: A-21206 | IF: 1/300 | |
| Goat anti-Mouse Alexa Fluor 546 |
Thermo Fisher Scientific | Ref: A-11030 | IF: 1/300 | |
| Goat anti-Rabbit Alexa Fluor 488 |
Thermo Fisher Scientific | Ref: A-11034 | IF: 1/300 | |
| WGA | Thermo Fisher Scientific | Ref: W21405 | labeled with Texas-Red IF: 1 mg/mL | |
| Anti-Strep Tag GT517 | Abcam | Ref: ab184224 | IF: 1/100 | |
| Goat-anti-mouse-FITC | Abcam | Ref: ab6785 | IF: 1/300 | |
| Behavior | Rotarod | Panlab | Ref:LE8500 | |
| Treadmill | Panlab | E8710MTS | ||
| Morris water maze | Panlab | SMART camera | circular tank (150 cm diameter, 100 cm high) | |
| PPI | Panlab | LE116 | ||
| Restrain stressor | Lab Research | Ref:G05 | ||
| ABR | Tucker Davis Technologies TDT |
System 3 Evoked | ||
| Mouse | C57BL6/J wild type | Harlam | Ref: 057 | C57BL/6JOlaHsd |
| C57BL6/J wild type | Jackson laboratory | Ref: 000664/Black | ||
| Slc7a8-/- chimera | Genoway | Customized Model Development |
Strategy Figure 1—figure supplement 1 | |
| Cell Line | HeLa | Sigma Aldrich | Ref: 93021013 | |
| Chemical compound, drug |
DTT dithiothreitol | SigmaAldrich | Ref:D9779 | |
| L- [3H]-labeled alanine | Perkin Elmer | Ref: NET348250UC | 1 μCi/ml | |
| [3 hr]-tyrosine | Perkin Elmer | Ref: NET127250UC | 1 μCi/ml | |
| Commercial assay or kit | Pierce BCA Protein Assay Kit |
Thermo Scientific | Ref:23225 | |
| ECL | GE Healthcare | Ref:RPN2232 | ||
| Corticosterone EIA kit | Enzo | Ref:ADI900097 | ||
| A + B conjugate | Vectastain | Ref: ABC kit | ||
| Rneasy | Qiagen | Ref: 74104 | ||
| High-capacity cDNA Reverse Transcription Kit |
Applied Biosystems | Ref: 4368813 | ||
| TaqMan Gene Expression Assay |
Applied Biosystems | potassium voltage-gated channel subfamily Q member 2 (Kcnq2) Mm00440080_m1; potassium voltage-gated channel subfamily Q member 3 (Kcnq3) Mm00548884_m1; potassium voltage-gated channel subfamily Q member 5 (Kcnq5) Mm01226041_m1; prestin (Slc26a5) Mm00446145_m1; T-box transcription factor TBX18 (Tbx18) Mm00470177_m1; interleukin one beta (Il1b) Mm00434228m1; interleukin 6 (Il6) Mm00446190m1; solute carrier family 7 (cationic amino acid transporter, y + system), member 8 (Slc7a8) Mm01318971m1 | ||
| QuikChange site-directed mutagenesis kit |
Stratagene | Ref: 200524 | ||
| Gene (human) | SLC7A8 | NCBI | NM_012244.3 | Protein NP_036376.2 (535AA) |
| Slc7a8 | NCBI | NM_016972.2 | Protein NP_058668.1 (531AA) | |
| Sequence-based reagent |
whole genome sequence |
Illumina | HiSeq 2000 | Data coverage was ranging from 4 to 10X |
| Sanger sequencing | Life Technologies | 3500 Dx Genetic Analyzer | ||
| BigDye | Life Technologies | ABI PRISM 3.1 Big Dye terminator |
||
| Software, algorithm | BioSig | Tucker Davis Technologies TDT |
NA | |
| Graph Pad Software | GraphPad Software, Inc |
Prism 4 | https://www.graphpad.com/scientific-software/prism/ | |
| SeqMan Pro software | DNAstar | https://www.dnastar.com/t-seqmanpro.aspx | sequencing assembly and analysis | |
| Annotations tools | ANNOVAR | http://annovar.openbioinformatics.org/en/latest/ | functional annotation of genetic variants DOI:10.1093 | |
| Genome Research | Bcftools | http://samtools.github.io/bcftools/ | ||
| SPSS 23.0 statistic software package |
IBM | NA | https://www.ibm.com/analytics/data-science/predictive-analytics/spss-statistical-software | |
| Transfected construct | Slc7a8 construct | Agilent | Catalog #212205 | Resistances: Neomycin and thymidine kinase |
| pcDNA3.1-StrepTag | ThermoFisher | Ref: V79020 | fused SLC7A8 or SLC3A2 | |
Mouse protocols
Animal experimentation complied with the ARRIVE guidelines and was conducted in accordance with Spanish (RD 53/2013) and European (Directive 2010/63/EU) legislations. All protocols used in this study were reviewed and approved by the Institutional Animal Care and Use Committee at IDIBELL in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAELAC accredited facility, B900010). Mice procedures were done according with scientific, humane, and ethical principles. The studied mouse model did not show phenotype differences comparing male and female. Thus, to ensure that our research represents both genders, the studies describes in this work were performed using both sexes equitably. The number of biological and experimental replicates is detailed in the legend of each figure.
Mouse model
Generation of the null Slc7a8 (Slc7a8−/−) was done by gene disruption. A coding region that includes exon 1 of the Slc7a8 gene was replaced for a neomycin resistance cassette by homologous recombination using a pBlueScript vector with two homologous arms (right: 6.1 kb and left: 2.3 kb) and two resistances (neomycin and thymidine kinase) in 5’ region of the gene (Figure 1—figure supplement 1A). ES cells transfection and microinjection experiments were done by GenOway (Lyon-France). Chimera mouse was outcrossed with a wild‐type C57BL6/J mouse to obtain first generation (F1) of Slc7a8 heterozygous (Slc7a8+/−) in a mixed C57BL6/J‐129Sv background. Intercross of F1 resulted in the analyzed F2 generation, which contemplates the three genotypes: wild type, Slc7a8+/− and Slc7a8−/− knockout mice. The pure inbred genetic background was generated backcrossing Slc7a8−/− F1 mice in the mixed C57BL6/J‐129Sv strain for 10 generations with pure C57BL6/J wild-type mice alternating male and females to avoid a genetic drift in the X and Y chromosomes.
Genotyping
Mice genotype was confirmed by triplex-PCR using DNA from the tail. Primers used were forward: 5’GGAGCGATCTGCGGAGTGA3’; reverse: 5’ACAGAGTGCGCTCCTACCCT3’ and reverse KO-specific: 5’CGGTGGGCTCTATGGGTCTA3’, and Standard DNA polymerase (Biotools Ref:10.002). The PCR products are 458 bp (wild type allele) and 180 bp (Slc7a8−/− allele) fragment.
Protein analysis
Protein analysis was done by western blotting using total membrane samples. Frozen tissues (50–100 mg) were homogenized in 5 mL of membrane buffer (25 mM HEPES – 4 mM EDTA – 250 mM sucrose – and protease inhibitors) and centrifuged at 10,000 rpm for 10 min at 4°C. Supernatant was centrifuged at 200,000xg for 1 hr at 4°C. The pellet was resuspended in 150 μL of membrane buffer using a 25G syringe. Pierce BCA Protein Assay Kit (Thermo Scientific Ref:23225) was used for protein quantification. Polyclonal rabbit antibody against mouse SLC7A8 protein was generated using an antigen against the C‐terminal region (peptide sequence: PIFKPTPVKDPDSEEQP) (Figure 1—figure supplement 1B). Serum extracts from inoculated rabbits were purified with protein G and used as primary antibody. Detection was by chemiluminescent reaction using ECL (GE Healthcare Ref:RPN2232) and autoradiography (Amersham Hyperfilm Ref:28906839). For specific SLC7A8 light subunit detection, samples were run in the presence of 100 mM of dithiothreitol (SigmaAldrich Ref:D9779).
Behavior tests
Rotarod (Panlab Ref:LE8500). The experimental design consisted of two training trials (TR) at the minimum speed (4 rpm) followed of two different tasks: (a) motor coordination and balance were assessed by measuring the latency to fall off the rod in consecutive trials with increasing fixed rotational speeds (FRS 4, 10, 14, 19, 24, and 34 rpm). The animals were allowed to stay on the rod for a maximum period of 1 min per trial and a resting period of 5 min was left between trials. (b) In the accelerating rod test, the rotation speed was increased from 4 to 40 rpm during two sessions of 1 min. For each trial, the elapsed time until the mouse fell off the rod was recorded. Treadmill (Panlab Ref:LE8710MTS): During two training trials (TR), the inclination of the treadmill was increased from 0° to 20° from the horizontal plane at different speeds (5, 10, 20, 30, 40 and 50 cm/s). Whenever an animal fell off the belt, foot shocks were applied for a maximal duration of 1 s. After the shock, mice were retrieved and placed back. Morris water maze (MWM): Mice were tested over 4 days (four trials/session, 10 min inter-trial intervals). The Morris Water Maze test consists of a circular tank (150 cm diameter, 100 cm high) filled with opaque water (with non-toxic white paint) and maintained at 21 ± 2°C. A removable circular platform (8 cm diameter) was located in a fixed position (NE quadrant) inside the pool. The pool was surrounded by white curtains, with cues affixed. The test was performed under low non-aversive lighting conditions (50 lux). An overhead camera connected to video-tracking software (SMART, Panlab SL., Spain) will be used to monitor the animal’s behavior. Latency to reach the platform, total distance travelled, speed and time in zones will be recorded for posterior data analysis. The maze was surrounded by white curtains with black patterns affixed, to provide an arrangement of spatial cues. A pre-training session was performed in which the platform was visible in the center (day 1), followed by five acquisition sessions during which the platform was submerged 2 cm below the water (days 2–6). In each trial, mice were introduced in the pool from one of the random starting locations. Mice failing to find the platform within 60 s. were placed on it for 10 s. At the end of every trial the mice were dried for 15 min in a heater. Escape latencies, length of the swimming paths and swimming speed for each animal and trial were monitored and computed by a tracking system connected to a video camera placed above the pool. Pre-pulse inhibition of acoustic startle response (PPI) (Panlab Ref:LE116): Training was 5 min of habituation time to the apparatus with a background noise level of 70 dB and then exposed to six blocks of 7 trial types in pseudo-random order with 15 s. inter-trial intervals. The trials: 1 s of a 120 dB, 8000 kHz sound preceded 100 ms. by a 40 ms pre-pulse (PP) sound of 74, 78, 82, 86 or 90 dB. The startle response was recorded for 65 ms, measuring every 1 ms. from the onset of the startle stimulus. Restrain stressor (LabResearch Ref:G05): Mice were habituated for 3 days prior the experiment collecting 10–15 μL of blood from tail. All sets were carried in the same room at the same time to minimize environmental variations and corticosterone fluctuations as a result of circadian rhythms. Mice were placed for 15 min in the conditioning unit and 75 μL of tail’s blood was collected. For recovery mice were placed into a clean cage for 90 min. Blood corticosterone were determined by Corticosterone EIA kit (Enzo Ref:ADI900097).
Auditory brainstem response test (ABR)
Hearing was evaluated by recording the auditory brainstem responses (ABR) with a System 3 TDT Evoked Potential Workstation (Tucker Davis Technologies TDT, Alachua, FL, USA) as previously described (Cediel et al., 2006; Riquelme et al., 2010). Briefly, mice were anesthetized with intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg), and placed inside a sound chamber. Broadband click (0.1 ms) and tone bursts (5 ms) at 8, 16, 20, 28 and 40 kHz were delivered with an open field speaker (MF1, TDT) at an intensity range from 90 to 10 dB sound pressure level (SPL) in 5–10 dB SPL steps. The electrical responses were amplified and averaged and the ANABR recordings analyzed with BioSig software (TDT) to determine hearing thresholds in response to each stimulus, peak and interpeak latencies and peak amplitudes. Animals were kept thermostatized and monitored during both anesthesia and the following recovery period.
Histology and immunohistochemistry
Mice were perfused through vascular system with 4% PFA and inner ear and brain samples were collected. The cochlea was dissected, post-fixed and decalcified in 0.3 M EDTA pH 6.5 (Sigma-Aldrich Ref:E1644) for seven days. Decalcified cochleae were embedded in OCT or paraffin as reported (Murillo-Cuesta et al., 2012). Deparaffinized cochlear sections were stained with hematoxylin and eosin for general cytoarchitecture evaluation. Immunohistochemistry: Floating brain tissue sections were incubated with 3% H2O2 in 10% methanol in PBS for 10 min. Blocking buffer with: 0.2% gelatine, 0.2% Triton x-100% and 10% FBS for 30 min. Primary antibody: anti-SLC7A8 1/500 in blocking buffer ON at 4°C with agitation. Secondary antibody: 1/200 biotinylated anti rabbit in blocking buffer for 1 hr at RT. Third antibody: 1/100 of A + B conjugate (Vectastain, Ref:ABC kit) in blocking buffer for 1 hr at RT. Develop staining: 0.03%DAB in PBS for 5 min. Reaction: incubate 0.03%DAB +1/10.000 H2O2 for 2–7 min with agitation. Reaction was stopped by rinsing with PBS. Sections were dried and dehydrated before mounting. Detection was using a bright-light microscope. Immunofluorescence: OCT tissue sections were permeabilized by incubating for 10 min with 0.1% Triton X-100 and incubated as reported (Sanchez-Calderon et al., 2010; de Iriarte Rodríguez et al., 2015b) with the following primary antibodies: anti-SLC7A8 (1/200), -s100 (1/1000, Sigma-Aldrich Ref:S2532), -Kir4.1 (1/200, Merck Millipore Ref:AB5818), -IBA1 (1/100, Abcam Ref:ab5076), or with Phalloidin (1/100, Thermo Fisher Scientific Ref:A22287), ON at 4°C. Sections were then incubated with secondary antibodies: (1:300, Thermo Fisher Scientific Ref:A-11034 Goat anti-Rabbit Alexa Fluor 488, Ref:A-11030 Goat anti-Mouse Alexa Fluor 546, Ref:A-21206 Donkey anti-Rabbit Alexa Fluor 488, Ref:A-11056 Donkey anti-Goat Alexa Fluor 546) for 2 hr at RT. Detection by confocal microscopy (Leica, Ref:LSM 780 Zeiss).
Fluorescence quantification
Four sections of apex, middle and basal turns of the cochlea were quantified using the same settings, including argon laser voltage, for the quantification. Using Fiji software, the sum of the intensity of all stacks (2.6 μm in the z axis along the 10 μm section) from the spiral ligaments + stria vascularis area was extracted. Data were analyzed with Prism 7 statistic software package (Graph Pad Software, Inc.). Statistical significance was determined by Student’s t test for unpaired samples. The number of biological and experimental replicates are detailed in the legend of each figure.
Quantitative RT-PCR
RNA was isolated using RNeasy (Qiagen) from 1 to 2 cochleae; its integrity and concentration were assessed using an Agilent Bioanalyzer 2100 (Agilent Technologies). At least, three mice per condition were used. cDNA was then generated by reverse transcription (High Capacity cDNA Reverse Transcription Kit; Applied Biosystems) and gene expression analyzed in triplicate by qPCR using TaqMan Gene Expression Assay kits (Applied Biosystems). The following probes were used: potassium voltage-gated channel subfamily Q member 2 (Kcnq2) Mm00440080_m1; potassium voltage-gated channel subfamily Q member 3 (Kcnq3) Mm00548884_m1; potassium voltage-gated channel subfamily Q member 5 (Kcnq5) Mm01226041_m1; prestin (Slc26a5) Mm00446145_m1; T-box transcription factor TBX18 (Tbx18) Mm00470177_m1; interleukin 1 beta (Il1b) Mm00434228m1; interleukin 6 (Il6) Mm00446190m1; solute carrier family 7 (cationic amino acid transporter, y + system), member 8 (Slc7a8) Mm01318971m1. PCR was performed on an Applied Biosystems 7900HT Real-Time PCR System using Hprt1 or RPLP0 as the endogenous housekeeping gene. Relative quantification values were calculated using the 2-ΔΔCt method. All procedures have been already reported (de Iriarte Rodríguez et al., 2015a).
ARHL cohort recruitment and clinical assessment
A total of 147 Subjects were recruited in North-Eastern Italy isolated villages (FVG Genetic Park) (Esko et al., 2013) and from one isolated village from Southern Italy (Carlantino). Subjects underwent a clinical evaluation to exclude any syndromic form of hearing loss or other systemic illnesses linked with sensorineural hearing loss. Audiometric tests using standard audiometers were carried out for each subject. Measurements have been obtained after any acoustically obstructing wax was removed. Thresholds for six different frequencies (0.25, 0.5, 1, 2, 4, 8 kHz) were measured and then a pure-tone average for high frequencies (P-TAH) was computed by taking the average of 4 and 8 kHz. To avoid non-genetic variations in the hearing phenotype (e.g. monolateral hearing loss), the best hearing ear was considered for each individual. Cases were defined as people older than 50 years old having PTAH ≥40, while controls were subjects more than 50 years old with PTAH ≤25.
All studies were approved by the Institutional Review Board of IRCCS Burlo Garofolo, Trieste, Italy and consent forms for clinical and genetic studies have been signed by each participant. All research was conducted according to the ethical standards as defined by the Helsinki Declaration.
Whole genome sequencing and mutation screening
Blood samples were collected and used to extract DNA using standard protocols. Low coverage whole genome sequence was generated using Illumina technology (Genome Analyzer and HiSeq 2000) at the Welcome Trust Sanger Institute and Beijing Genomics Institute. Data coverage was ranging from 4 to 10X. A multi-sample genotype calling was performed and standard quality filters were applied. The detailed pipeline has already been described elsewhere (Timpson et al., 2014). Variants belonging to SLC7A8 gene were extracted using bcftools [http://samtools.github.io/bcftools/] and annotated with ANNOVAR (Wang et al., 2010). Only the exonic variants were further considered. Finally, variants of interest were confirmed by direct Sanger sequencing on a 3500 Dx Genetic Analyzer (Life Technologies, CA), using ABI PRISM 3.1 Big Dye terminator chemistry (Life Technologies) per manufacturer’s instructions. Mutation frequencies were compared with public databases such as Esp6500siv2 (NHLBI Exome Sequencing Project), 1000 g (1000 Genomes Project), Campion (The Allele Frequency Net Database) and ExAC (The Exome Aggregation Consortium). For SLC7A8 we collected several statistics including the probability of loss of function intolerance (pLI), where the closer pLI value is to 1, the more LoF intolerant the gene could be considered. We also collected the missense Z score, a positive score indicates intolerance to missense variation whereas a negative Z score indicates that the gene had more missense variants than expected.
Site-directed mutagenesis
The QuikChange site-directed mutagenesis kit (Stratagene) was used to introduce point mutations in SLC7A8 sequence, according to the manufacturer's protocol. The pcDNA3.1-StrepTag fused SLC7A8 construct was used as template (Costa et al., 2013). Amino acid substitutions were introduced into SLC7A8 sequence using a compatible reverse primer and forward primers (Figure 5—source data 1). All primers annealed to the coding sequence, and the position of the mutated codon was underlined. All constructs were verified by DNA sequencing and then used for transient transfection.
Cell culture and transfection
HeLa cells (Sigma Aldrich, Ref: 93021013) were maintained at 37°C/5% CO2 in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% (v/v) fetal bovine serum, 50 units/ml penicillin, 50 μg/ml streptomycin, and 2 mM l-glutamine. HeLa cells were transiently transfected with plasmid constructions mentioned above with the use of Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. Amino acid transport and fluorescence microscopy analyses were carried out 48 hr after transfection.
Visualization of Strep-tagged amino acid transporters by fluorescence microscopy
To analyze the effect of the mutations on SLC7A8 protein expression and plasma membrane localization, fluorescence microscopy of Strep-tagged wild type and mutant transporters was performed on a semiconfluent monolayer of transfected HeLa cells cultured on glass coverslips. Glass coverslip-grown cells were incubated with 1 mg/ml wheat germ agglutinin (WGA) labeled with Texas-Red (Thermo Fisher Scientific) at 37°C for 10 min, rinsed three times with phosphate-buffered saline-Ca2+-Mg2+ and fixed for 15 min in 4% paraformaldehyde. Fixed cells were blocked in blocking buffer (10% FBS and 0.1% saponin in PBS) for 1 hr and then incubated for 1 hr with primary antibody (anti-Strep Tag GT517, 1/100; Abcam). Secondary goat-anti-mouse-FITC antibody (Life Technologies) was incubated for 2 hr protected from light and rinsed three times with phosphate-buffered saline. Nuclear staining was performed by incubating 1 µg/ml Hoechst (Thermo Fisher Scientific) for 10 min, rinsed three times with phosphate-buffered saline and then mounted with aqua-poly/mount coverslipping medium (Polysciences Inc.). Images were taken using a Nikon E1000 upright epifluorescence microscope. All images were captured during 200 ms except for those corresponding to V460E that were overexposed to 2 s to reveal the subcellular localization of this very low expressing variant. To quantify SLC7A8 wild type and mutated transporters expression levels in cells, a single in-focus plane was acquired. Using ImageJ (v1.48, NIH), an outline was drawn around each cell and area and mean fluorescence measured, along with several adjacent background readings. The total corrected cellular fluorescence (TCCF) = integrated density – (area of selected cell × mean fluorescence of background readings), was calculated.
Amino acid transport assay
Amino acid uptake was measured by exposing replicate cultures at room temperature to L- [3H]-labeled alanine or [3H]-tyrosine (1 μCi/ml; Perkin Elmer) in sodium-free transport buffer (137 mM choline chloride, 5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, and 10 mM HEPES, pH 7.4). Initial rates of transport were determined using an incubation period of 1 min and 50 µM of cold alanine or tyrosine. Assays were terminated by washing with an excess volume of chilled transport buffer. Cells were lysed using 0.1% SDS and 100 mM NaOH and radioactivity measured in a scintillation counter. Uptake values were corrected by their total corrected cellular fluorescence (TCCF) for all transporters except for V460E mutant, which does not reach the plasmatic membrane.
Statistical analysis
Behavior and ABR experiments using mice were not performed blind to genotype and treatment conditions, but as data acquisition was automated this will not affect data processing and analysis. The sample size was chosen according to the standard sample sizes used in the field and without applying any statistical method. The general criteria of exclusion were pre-established: (1) samples with a value that differed by more/less than two standard deviations from the mean value were excluded from the study. The statistical tests used in each experiment were appropriate to the type of groups, data and samples. Unpaired Student t-test was used for experiments with only two independent groups. Repeated measures two-way ANOVA was applied when we had to compare two independent groups (genotype as the between subjects factor) where repeated measurements of the dependent variable were obtained (Rotarod and PPI).
Data were analyzed with IBM SPSS 23.0 statistic software package (Chicago, IL). Statistical significance was determined by one-way analysis of variance (ANOVA) and Levene's F test to assess the equality of variances. When significant differences were obtained, post hoc comparisons were performed using Bonferroni or Tamhane tests to compare the three genotypes. Normal distribution of data and homogeneity of variances was assessed using Shapiro-Wilk and Levene tests, respectively. In most of the datasets these two assumptions were achieved. However, when not achieved and because we use comparable sample sizes and ANOVA is robust to normality violations, our results are still valid. Sphericity assumption was assessed using Mauchly’s test and when not achieved Greenhouse correction was taken. Posthoc tests were performed using Bonferroni correction for individual comparisons. Bonferroni p<0.05 was assumed as critical value for significance throughout the study. Statistical analyses were performed using SPSS package.
Acknowledgements
We thank to all volunteers that participate in this study, Professor Fernando Aguado for technical assistance in brain IHC, Professor Josep Chillarón for SLC7A8 antibody design, Dr. Miguel López de Heredia for critical reading of the manuscript, Saif Ahmed for the code to extract the images’ analysis data, the core-facilities: IRB Advanced Digital Microscopy Facility (Anna Lladó and Julien Colombelli), the Genomics facility at the IIBm (CSIC-UAM), Units F1, F3 and F6-SEFALer (CIBERER) for their technical support and GenOway for the help in the homologous recombination and transfection of the construct into ES cells to generate the Slc7a8−/− mice. IRB Barcelona is the recipient of a Severo Ochoa Award of Excellence from MINECO (Government of Spain). MD acknowledges support of the Spanish Ministry of Economy and Competitiveness, ‘Centro de Excelencia Severo Ochoa 2013–2017; MD: and V N acknowledge the support of the CERCA Programme/Generalitat de Catalunya.’ Isabel Varela-Nieto, Paolo Gasparini, Manuel Palacín and Virginia Nunes share leadership.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Meritxell Espino Guarch, Email: mespinoguarch@sidra.org.
Manuel Palacín, Email: manuel.palacin@irbbarcelona.org.
Virginia Nunes, Email: vnunes@idibell.cat.
Jeremy Nathans, Johns Hopkins University School of Medicine, United States.
Funding Information
This paper was supported by the following grants:
Qatar National Research Fund JSREP07-013-3-006 to Meritxell Espino Guarch.
CIBERER ACCI 2016 to Silvia Murillo-Cuesta, Manuel Palacín, Virginia Nunes.
CIBERER ACCI 2017 to Silvia Murillo-Cuesta, Manuel Palacín, Virginia Nunes.
Generalitat de Catalunya SGR 2014/1125 to Mara Dierssen.
Ministerio de Ciencia e Innovación SAF2016-79956-R to Mara Dierssen.
Seventh Framework Programme TARGEAR FP7-PEOPLE-2013-IAPP to Isabel Varela-Nieto.
Ministerio de Ciencia e Innovación SAF2014-53979-R-FEDER to Isabel Varela-Nieto.
Ministerio de Ciencia e Innovación SAF2015-64869-R-FEDER to Manuel Palacín.
Generalitat de Catalunya SGR2009-1355 to Manuel Palacín.
Spanish Health Institute Carlos III-FIS PI13/00121-R-FEDER to Virginia Nunes.
Generalitat de Catalunya SGR2009-1490 to Virginia Nunes.
Spanish Health Institute Carlos III-FIS PI16/00267-R-Feder to Virginia Nunes.
Additional information
Competing interests
No competing interests declared.
Author contributions
Conceptualization, Resources, Methodology, Writing—original draft, Writing—review and editing, Western Blot, Brain IHC, Stress Response, IF and H/E quantifications.
Conceptualization, Methodology, Writing—review and editing, Lat2-/- generation Behavior tests.
Formal analysis, Writing—review and editing, ABR tests Cochlear RT-qPCR.
Methodology, Writing—review and editing, In vitro uptake.
Methodology, Writing—review and editing, Cochlear sampling and cytoarchitecture Cochlear H/E and IF Cochlear RT-qPCR.
Methodology, Writing—review and editing, Subject samples.
Formal analysis, Writing—review and editing, Mutations screening.
Formal analysis, Writing—review and editing, Mutations screening.
Methodology, Writing—review and editing, Stress Response.
Methodology, Writing—review and editing, Western Blot.
Methodology, Writing—review and editing, Behavior tests.
Methodology, Mice maintenance and necropsy.
Methodology, Cochlear H/E and IF Mice maintenance and necropsy.
Conceptualization, Writing—review and editing.
Conceptualization, Resources, Methodology, Writing—review and editing, Behavior tests.
Conceptualization, Resources, Writing—review and editing.
Conceptualization, Resources, Writing—review and editing, Subject samples.
Conceptualization, Resources, Supervision, Writing—original draft, Writing—review and editing.
Conceptualization, Resources, Funding acquisition, Writing—original draft, Writing—review and editing.
Ethics
Human subjects: Ethical Consent Published in: Esko T, Mezzavilla M, Nelis M, Borel C, Debniak T, Jakkula E, et al. Genetic characterization of northeastern Italian population isolates in the context of broader European genetic diversity. Eur J Hum Genet. 2013;21(6):659-65.
Animal experimentation: Animal experimentation complied with the ARRIVE guidelines and was conducted in accordance with Spanish (RD 53/2013) and European (Directive 2010/63/EU) legislations. All protocols used in this study were reviewed and approved by the Institutional Animal Care and Use Committee at IDIBELL in a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAELAC accredited facility, B900010). Mice procedures were done according with scientific, humane, and ethical principles. The studied mouse model did not show phenotype differences comparing male and female. Thus, to ensure that our research represents both genders, the studies describes in this work were performed using both sexes equitably. The number of biological and experimental replicates is detailed in the legend of each figure.
Additional files
Major datasets
The following previously published dataset was used:
Esko T, author; Mezzavilla M, author; Nelis M, author; Borel C, author; Debniak T, author; Jakkula E, author; Julia A, author; Karachanak S, author; Khrunin A, author; Kisfali P, author; Krulisova V, author; Ausrele Kucinskiene Z, author; Rehnstrom K, author; Traglia M, author; Nikitina-Zake L, author; Zimprich F, author; Antonarakis SE, author; Estivill X, author; Glavac D, author; Gut I, author; Klovins J, author; Krawczak M, author; Kucinskas V, author; Lathrop M, author; Macek M, author; Marsal S, author; Meitinger T, author; Melegh B, author; Limborska S, author; Lubinski J, author; Paolotie A, author; Schreiber S, author; Toncheva D, author; Toniolo D, author; Wichmann HE, author; Zimprich A, author; Metspalu M, author; Gasparini P, author; Metspalu A, author. Genetic characterization of northeastern Italian population isolates in the context of broader European genetic diversity. 2013 https://www.ebi.ac.uk/biostudies/studies/S-EPMC3658181 Publicly available at EBI BioStudies (accession no: S-EPMC3658181)
References
- Allen Institute for Brain Science 2004. Slc7a8 - RP_050915_02_F01 - sagittal. Allen Mouse Brain Atlas. 72001998
- Ando M, Takeuchi S. Immunological identification of an inward rectifier K+ channel (Kir4.1) in the intermediate cell (melanocyte) of the cochlear stria vascularis of gerbils and rats. Cell and Tissue Research. 1999;298:179–183. doi: 10.1007/s004419900066. [DOI] [PubMed] [Google Scholar]
- Bao J, Ohlemiller KK. Age-related loss of spiral ganglion neurons. Hearing Research. 2010;264:93–97. doi: 10.1016/j.heares.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauch C, Forster N, Loffing-Cueni D, Summa V, Verrey F. Functional cooperation of epithelial heteromeric amino acid transporters expressed in madin-darby canine kidney cells. Journal of Biological Chemistry. 2003;278:1316–1322. doi: 10.1074/jbc.M210449200. [DOI] [PubMed] [Google Scholar]
- Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, Lemcke B, Horst J, Leuwer R, Pape HC, Völkl H, Hübner CA, Jentsch TJ. Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. The EMBO Journal. 2003;22:5422–5434. doi: 10.1093/emboj/cdg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowl MR, Dawson SJ. The mouse as a model for age-related hearing loss - a mini-review. Gerontology. 2015;61:149–157. doi: 10.1159/000368399. [DOI] [PubMed] [Google Scholar]
- Braun D, Wirth EK, Wohlgemuth F, Reix N, Klein MO, Grüters A, Köhrle J, Schweizer U. Aminoaciduria, but normal thyroid hormone levels and signalling, in mice lacking the amino acid and thyroid hormone transporter Slc7a8. Biochemical Journal. 2011;439:249–255. doi: 10.1042/BJ20110759. [DOI] [PubMed] [Google Scholar]
- Camarero G, Avendano C, Fernandez-Moreno C, Villar A, Contreras J, de Pablo F, Pichel JG, Varela-Nieto I. Delayed inner ear maturation and neuronal loss in postnatal Igf-1-deficient mice. Journal of Neuroscience. 2001;21:7630–7641. doi: 10.1523/JNEUROSCI.21-19-07630.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cediel R, Riquelme R, Contreras J, Díaz A, Varela-Nieto I. Sensorineural hearing loss in insulin-like growth factor I-null mice: a new model of human deafness. European Journal of Neuroscience. 2006;23:587–590. doi: 10.1111/j.1460-9568.2005.04584.x. [DOI] [PubMed] [Google Scholar]
- Christensen K, Frederiksen H, Hoffman HJ. Genetic and environmental influences on self-reported reduced hearing in the old and oldest old. Journal of the American Geriatrics Society. 2001;49:1512–1517. doi: 10.1046/j.1532-5415.2001.4911245.x. [DOI] [PubMed] [Google Scholar]
- Costa M, Rosell A, Álvarez-Marimon E, Zorzano A, Fotiadis D, Palacín M. Expression of human heteromeric amino acid transporters in the yeast Pichia pastoris. Protein Expression and Purification. 2013;87:35–40. doi: 10.1016/j.pep.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Couloigner V, Sterkers O, Ferrary E. What's new in ion transports in the cochlea? Pflügers Archiv - European Journal of Physiology. 2006;453:11–22. doi: 10.1007/s00424-006-0103-4. [DOI] [PubMed] [Google Scholar]
- Cruickshanks KJ, Klein R, Klein BE, Nondahl DM. Sunlight and the 5-year incidence of early age-related maculopathy: the beaver dam eye study. Archives of Ophthalmology. 2001;119:246–250. [PubMed] [Google Scholar]
- Cruickshanks KJ, Nondahl DM, Tweed TS, Wiley TL, Klein BE, Klein R, Chappell R, Dalton DS, Nash SD. Education, occupation, noise exposure history and the 10-yr cumulative incidence of hearing impairment in older adults. Hearing Research. 2010;264:3–9. doi: 10.1016/j.heares.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshanks KJ, Wiley TL, Tweed TS, Klein BE, Klein R, Mares-Perlman JA, Nondahl DM. Prevalence of hearing loss in older adults in beaver Dam, Wisconsin. The epidemiology of hearing loss study. American Journal of Epidemiology. 1998;148:879–886. doi: 10.1093/oxfordjournals.aje.a009713. [DOI] [PubMed] [Google Scholar]
- Dawes P, Payton A. Genetics of Age-Related Hearing Loss. Genetics of Deafness. 2016;20:84–96. doi: 10.1159/000444565. [DOI] [Google Scholar]
- de Iriarte Rodríguez R, Magariños M, Pfeiffer V, Rapp UR, Varela-Nieto I. C-Raf deficiency leads to hearing loss and increased noise susceptibility. Cellular and Molecular Life Sciences. 2015a;72:3983–3998. doi: 10.1007/s00018-015-1919-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Iriarte Rodríguez R, Pulido S, Rodríguez-de la Rosa L, Magariños M, Varela-Nieto I. Age-regulated function of autophagy in the mouse inner ear. Hearing Research. 2015b;330:39–50. doi: 10.1016/j.heares.2015.07.020. [DOI] [PubMed] [Google Scholar]
- del Amo EM, Urtti A, Yliperttula M. Pharmacokinetic role of L-type amino acid transporters LAT1 and LAT2. European Journal of Pharmaceutical Sciences. 2008;35:161–174. doi: 10.1016/j.ejps.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Delprat B, Ruel J, Guitton MJ, Hamard G, Lenoir M, Pujol R, Puel JL, Brabet P, Hamel CP. Deafness and cochlear fibrocyte alterations in mice deficient for the inner ear protein otospiralin. Molecular and Cellular Biology. 2005;25:847–853. doi: 10.1128/MCB.25.2.847-853.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. 2015;517:302–310. doi: 10.1038/nature14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko T, Mezzavilla M, Nelis M, Borel C, Debniak T, Jakkula E, Julia A, Karachanak S, Khrunin A, Kisfali P, Krulisova V, Aušrelé Kučinskiené Z, Rehnström K, Traglia M, Nikitina-Zake L, Zimprich F, Antonarakis SE, Estivill X, Glavač D, Gut I, Klovins J, Krawczak M, Kučinskas V, Lathrop M, Macek M, Marsal S, Meitinger T, Melegh B, Limborska S, Lubinski J, Paolotie A, Schreiber S, Toncheva D, Toniolo D, Wichmann HE, Zimprich A, Metspalu M, Gasparini P, Metspalu A, D'Adamo P. Genetic characterization of northeastern Italian population isolates in the context of broader European genetic diversity. European Journal of Human Genetics. 2013;21:659–665. doi: 10.1038/ejhg.2012.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Human Molecular Genetics. 2001;10:153–161. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- Font-LLitjós M. Reabsorció renal d'aminoàcids: anàlisi de mutacions de SLC7A9, el gen de cistinúria de tipus B, i generació d'un model murí knockout de Slc7a8. Barcelona: Barcelona University; 2009. [Google Scholar]
- Gates GA, Couropmitree NN, Myers RH. Genetic associations in age-related hearing thresholds. Archives of Otolaryngology–Head & Neck Surgery. 1999;125:654–659. doi: 10.1001/archotol.125.6.654. [DOI] [PubMed] [Google Scholar]
- Gates GA, Mills JH. Presbycusis. The Lancet. 2005;366:1111–1120. doi: 10.1016/S0140-6736(05)67423-5. [DOI] [PubMed] [Google Scholar]
- Gomes P, Soares-da-Silva P. Na+-independent transporters, LAT-2 and b0,+, exchange L-DOPA with neutral and basic amino acids in two clonal renal cell lines. Journal of Membrane Biology. 2002;186:63–80. doi: 10.1007/s00232-001-0136-8. [DOI] [PubMed] [Google Scholar]
- Hinz KM, Neef D, Rutz C, Furkert J, Köhrle J, Schülein R, Krause G. Molecular features of the L-type amino acid transporter 2 determine different import and export profiles for thyroid hormones and amino acids. Molecular and Cellular Endocrinology. 2017;443:163–174. doi: 10.1016/j.mce.2017.01.024. [DOI] [PubMed] [Google Scholar]
- Huang Q, Tang J. Age-related hearing loss or presbycusis. European Archives of Oto-Rhino-Laryngology. 2010;267:1179–1191. doi: 10.1007/s00405-010-1270-7. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Endou H. Heterodimeric amino acid transporters: molecular biology and pathological and pharmacological relevance. Current Drug Metabolism. 2001;2:339–354. doi: 10.2174/1389200013338324. [DOI] [PubMed] [Google Scholar]
- Kane KL, Longo-Guess CM, Gagnon LH, Ding D, Salvi RJ, Johnson KR. Genetic background effects on age-related hearing loss associated with Cdh23 variants in mice. Hearing Research. 2012;283:80–88. doi: 10.1016/j.heares.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson KK, Harris JR, Svartengren M. Description and primary results from an audiometric study of male twins. Ear and Hearing. 1997;18:114–120. doi: 10.1097/00003446-199704000-00003. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Kimura RS, Paul DL, Takasaka T, Adams JC. Gap junction systems in the mammalian cochlea. Brain Research Reviews. 2000;32:163–166. doi: 10.1016/S0165-0173(99)00076-4. [DOI] [PubMed] [Google Scholar]
- Kowalczyk L, Ratera M, Paladino A, Bartoccioni P, Errasti-Murugarren E, Valencia E, Portella G, Bial S, Zorzano A, Fita I, Orozco M, Carpena X, Vázquez-Ibar JL, Palacín M. Molecular basis of substrate-induced permeation by an amino acid antiporter. PNAS. 2011;108:3935–3940. doi: 10.1073/pnas.1018081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481:469–474. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroberardino L, Spindler B, Pfeiffer R, Skelly PJ, Loffing J, Shoemaker CB, Verrey F. Amino-acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nature. 1998;395:288–291. doi: 10.1038/26246. [DOI] [PubMed] [Google Scholar]
- Mazelová J, Popelar J, Syka J. Auditory function in presbycusis: peripheral vs. central changes. Experimental Gerontology. 2003;38:87–94. doi: 10.1016/S0531-5565(02)00155-9. [DOI] [PubMed] [Google Scholar]
- Meier C, Ristic Z, Klauser S, Verrey F. Activation of system L heterodimeric amino acid exchangers by intracellular substrates. The EMBO Journal. 2002;21:580–589. doi: 10.1093/emboj/21.4.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minowa O, Ikeda K, Sugitani Y, Oshima T, Nakai S, Katori Y, Suzuki M, Furukawa M, Kawase T, Zheng Y, Ogura M, Asada Y, Watanabe K, Yamanaka H, Gotoh S, Nishi-Takeshima M, Sugimoto T, Kikuchi T, Takasaka T, Noda T. Altered cochlear fibrocytes in a mouse model of DFN3 nonsyndromic deafness. Science. 1999;285:1408–1411. doi: 10.1126/science.285.5432.1408. [DOI] [PubMed] [Google Scholar]
- Murillo-Cuesta S, Camarero G, González-Rodríguez A, De La Rosa LR, Burks DJ, Avendaño C, Valverde AM, Varela-Nieto I. Insulin receptor substrate 2 (IRS2)-deficient mice show sensorineural hearing loss that is delayed by concomitant protein tyrosine phosphatase 1B (PTP1B) loss of function. Molecular Medicine. 2012;18:1–9. doi: 10.2119/molmed.2011.00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murillo-Cuesta S, Contreras J, Zurita E, Cediel R, Cantero M, Varela-Nieto I, Montoliu L. Melanin precursors prevent premature age-related and noise-induced hearing loss in albino mice. Pigment Cell & Melanoma Research. 2010;23:72–83. doi: 10.1111/j.1755-148X.2009.00646.x. [DOI] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nature Genetics. 2003;35:21–23. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez B, Martínez de Mena R, Obregon MJ, Font-Llitjós M, Nunes V, Palacín M, Dumitrescu AM, Morte B, Bernal J. Cerebral cortex hyperthyroidism of newborn mct8-deficient mice transiently suppressed by lat2 inactivation. PLoS One. 2014;9:e96915. doi: 10.1371/journal.pone.0096915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxender DL, Christensen HN. Distinct mediating systems for the transport of neutral amino acids by the ehrlich cell. The Journal of Biological Chemistry. 1963;238:3686–3699. [PubMed] [Google Scholar]
- Peeters RP, Ng L, Ma M, Forrest D. The timecourse of apoptotic cell death during postnatal remodeling of the mouse cochlea and its premature onset by triiodothyronine (T3) Molecular and Cellular Endocrinology. 2015;407:1–8. doi: 10.1016/j.mce.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter A, Santi PM. Cochlear Anatomy and Central Auditory Pathways. Otolaryngology: Head and Neck Surgery. 2001;Chapter 140 [Google Scholar]
- Pineda M, Fernández E, Torrents D, Estévez R, López C, Camps M, Lloberas J, Zorzano A, Palacín M. Identification of a membrane protein, LAT-2, that Co-expresses with 4F2 heavy chain, an L-type amino acid transport activity with broad specificity for small and large zwitterionic amino acids. Journal of Biological Chemistry. 1999;274:19738–19744. doi: 10.1074/jbc.274.28.19738. [DOI] [PubMed] [Google Scholar]
- Pinho MJ, Serrão MP, Gomes P, Hopfer U, Jose PA, Soares-da-Silva P. Over-expression of renal LAT1 and LAT2 and enhanced L-DOPA uptake in SHR immortalized renal proximal tubular cells. Kidney International. 2004;66:216–226. doi: 10.1111/j.1523-1755.2004.00722.x. [DOI] [PubMed] [Google Scholar]
- Riquelme R, Cediel R, Contreras J, la Rosa Lourdes RD, Murillo-Cuesta S, Hernandez-Sanchez C, Zubeldia JM, Cerdan S, Varela-Nieto I. A comparative study of age-related hearing loss in wild type and insulin-like growth factor I deficient mice. Frontiers in Neuroanatomy. 2010;4:27. doi: 10.3389/fnana.2010.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell A, Meury M, Álvarez-Marimon E, Costa M, Pérez-Cano L, Zorzano A, Fernández-Recio J, Palacín M, Fotiadis D. Structural bases for the interaction and stabilization of the human amino acid transporter LAT2 with its ancillary protein 4F2hc. PNAS. 2014;111:2966–2971. doi: 10.1073/pnas.1323779111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossier G, Meier C, Bauch C, Summa V, Sordat B, Verrey F, Kühn LC. LAT2, a new basolateral 4F2hc/CD98-associated amino acid transporter of kidney and intestine. Journal of Biological Chemistry. 1999;274:34948–34954. doi: 10.1074/jbc.274.49.34948. [DOI] [PubMed] [Google Scholar]
- Sanchez-Calderon H, Rodriguez-de la Rosa L, Milo M, Pichel JG, Holley M, Varela-Nieto I. RNA microarray analysis in prenatal mouse cochlea reveals novel IGF-I target genes: implication of MEF2 and FOXM1 transcription factors. PLoS One. 2010;5:e8699. doi: 10.1371/journal.pone.0008699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuknecht HF. Presbycusis. Transactions of the American Laryngological, Rhinological and Otological Society, Inc. 1955:401–418. [PubMed] [Google Scholar]
- Sharlin DS, Visser TJ, Forrest D. Developmental and cell-specific expression of thyroid hormone transporters in the mouse cochlea. Endocrinology. 2011;152:5053–5064. doi: 10.1210/en.2011-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues in Clinical Neuroscience. 2006;8:383–395. doi: 10.31887/DCNS.2006.8.4/ssmith. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Prolla TA. Mitochondrial oxidative damage and apoptosis in age-related hearing loss. Mechanisms of Ageing and Development. 2010;131:480–486. doi: 10.1016/j.mad.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya S, Tanokura M, Weindruch R, Prolla TA, Yamasoba T. Effects of caloric restriction on age-related hearing loss in rodents and rhesus monkeys. Current Aging Science. 2010;3:20–25. doi: 10.2174/1874609811003010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MH, Choi SY, Wu L, Oh SK, Lee HK, Lee DJ, Shim DB, Choi JY, Kim UK, Bok J. Pou3f4 deficiency causes defects in otic fibrocytes and stria vascularis by different mechanisms. Biochemical and Biophysical Research Communications. 2011;404:528–533. doi: 10.1016/j.bbrc.2010.12.019. [DOI] [PubMed] [Google Scholar]
- Stankovic K, Rio C, Xia A, Sugawara M, Adams JC, Liberman MC, Corfas G. Survival of adult spiral ganglion neurons requires erbB receptor signaling in the inner ear. Journal of Neuroscience. 2004;24:8651–8661. doi: 10.1523/JNEUROSCI.0733-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara M, Murtie JC, Stankovic KM, Liberman MC, Corfas G. Dynamic patterns of neurotrophin 3 expression in the postnatal mouse inner ear. The Journal of Comparative Neurology. 2007;501:30–37. doi: 10.1002/cne.21227. [DOI] [PubMed] [Google Scholar]
- Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Söhl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP, Petit C, Willecke K. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Human Molecular Genetics. 2003;12:13–21. doi: 10.1093/hmg/ddg001. [DOI] [PubMed] [Google Scholar]
- Timpson NJ, Walter K, Min JL, Tachmazidou I, Malerba G, Shin SY, Chen L, Futema M, Southam L, Iotchkova V, Cocca M, Huang J, Memari Y, McCarthy S, Danecek P, Muddyman D, Mangino M, Menni C, Perry JR, Ring SM, Gaye A, Dedoussis G, Farmaki AE, Burton P, Talmud PJ, Gambaro G, Spector TD, Smith GD, Durbin R, Richards JB, Humphries SE, Zeggini E, Soranzo N, UK1OK Consortium Members A rare variant in APOC3 is associated with plasma triglyceride and VLDL levels in Europeans. Nature Communications. 2014;5:4871. doi: 10.1038/ncomms5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowe MO, Maier H, Schweizer M, Kispert A. Deafness in mice lacking the T-box transcription factor Tbx18 in otic fibrocytes. Development. 2008;135:1725–1734. doi: 10.1242/dev.014043. [DOI] [PubMed] [Google Scholar]
- Uetsuka S, Ogata G, Nagamori S, Isozumi N, Nin F, Yoshida T, Komune S, Kitahara T, Kikkawa Y, Inohara H, Kanai Y, Hibino H. Molecular architecture of the stria vascularis membrane transport system, which is essential for physiological functions of the mammalian cochlea. European Journal of Neuroscience. 2015;42:1984–2002. doi: 10.1111/ejn.12973. [DOI] [PubMed] [Google Scholar]
- Van Eyken E, Van Camp G, Van Laer L. The complexity of age-related hearing impairment: contributing environmental and genetic factors. Audiology and Neurotology. 2007;12:345–358. doi: 10.1159/000106478. [DOI] [PubMed] [Google Scholar]
- Verrey F. System L: heteromeric exchangers of large, neutral amino acids involved in directional transport. Pflügers Archiv - European Journal of Physiology. 2003;445:529–533. doi: 10.1007/s00424-002-0973-z. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. The Journal of Physiology. 2006;576:11–21. doi: 10.1113/jphysiol.2006.112888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield A, Panizzon M, Grant MD, Toomey R, Kremen WS, Franz CE, Jacobson KC, Eisen SA, Lyons M. A twin-study of genetic contributions to hearing acuity in late middle age. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2007;62:1294–1299. doi: 10.1093/gerona/62.11.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Marcus DC. Age-related changes in cochlear endolymphatic potassium and potential in CD-1 and CBA/CaJ mice. JARO - Journal of the Association for Research in Otolaryngology. 2003;4:353–362. doi: 10.1007/s10162-002-3026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Mezzavilla M, Haber M, McCarthy S, Chen Y, Narasimhan V, Gilly A, Ayub Q, Colonna V, Southam L, Finan C, Massaia A, Chheda H, Palta P, Ritchie G, Asimit J, Dedoussis G, Gasparini P, Palotie A, Ripatti S, Soranzo N, Toniolo D, Wilson JF, Durbin R, Tyler-Smith C, Zeggini E. Enrichment of low-frequency functional variants revealed by whole-genome sequencing of multiple isolated European populations. Nature Communications. 2017;8:15927. doi: 10.1038/ncomms15927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasoba T, Lin FR, Someya S, Kashio A, Sakamoto T, Kondo K. Current concepts in age-related hearing loss: epidemiology and mechanistic pathways. Hearing Research. 2013;303:30–38. doi: 10.1016/j.heares.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Dai M, Wilson TM, Omelchenko I, Klimek JE, Wilmarth PA, David LL, Nuttall AL, Gillespie PG, Shi X. Na+/K+-ATPase α1 identified as an abundant protein in the blood-labyrinth barrier that plays an essential role in the barrier integrity. PLoS One. 2011;6:e16547. doi: 10.1371/journal.pone.0016547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zevenbergen C, Meima ME, Lima de Souza EC, Peeters RP, Kinne A, Krause G, Visser WE, Visser TJ. Transport of Iodothyronines by Human L-Type Amino Acid Transporters. Endocrinology. 2015;156:4345–4355. doi: 10.1210/en.2015-1140. [DOI] [PubMed] [Google Scholar]
- Zilberstein Y, Liberman MC, Corfas G. Inner hair cells are not required for survival of spiral ganglion neurons in the adult cochlea. Journal of Neuroscience. 2012;32:405–410. doi: 10.1523/JNEUROSCI.4678-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]