

Abstract
Background
Statins compete with DHEAS for influx through the SLCO2B1 transporter, which may prolong time to progression (TTP) on androgen deprivation therapy. Abiraterone acetate (AA) may also undergo SLCO-mediated transport. Based on preclinical findings showing antagonism, we hypothesized that statins may compete with AA for influx via SLCO2B1 and could negatively impact drug efficacy.
Methods
We queried two institutional clinical databases [Dana-Farber Cancer Institute (DFCI), Johns Hopkins University (JHU)] for CRPC patients treated with AA. Treatment duration was a surrogate for TTP. Associations between statin use and AA duration were estimated using the Kaplan-Meier method. Multivariable Cox regression modeling adjusted for known prognostic factors.
Results
Of the 224 DFCI and 270 JHU patients included, the majority (96%) had metastatic disease. Nearly half (41 and 45%) were statin users. In the DFCI cohort, there was a trend toward longer AA duration in statin users: 14.2 vs. 9.2 mo (HR 0.79, 95% CI: 0.57–1.09, p=0.14). There was no association between statin use and AA duration in the JHU cohort: 8.3 vs. 8.0 months (HR 0.89, 95% CI: 0.69–1.16, p=0.38) in the statin users vs. non-users, except for a trend in patients that had not previously received docetaxel or enzalutamide (HR 0.79; 95% CI: 0.57–1.10).
Conclusions
Contrary to our initial hypothesis, there was a trend towards longer (rather than shorter) AA duration in statin users in the entire DFCI cohort and in the enzalutamide- and docetaxel-naïve JHU patients. Together, these results do not support the hypothesis that statins interfere with AA efficacy.
Keywords: statins, prostate cancer, abiraterone acetate, duration, SLCO transport
Introduction
Statins are a frequently used class of agents to control serum lipid levels. A variety of anticancer properties have been attributed to them including anti-proliferative and pro-apoptotic effects as well as reduction in cholesterol and downstream androgens necessary for cell growth. The organic anionic transporter, SLCO2B1, enables a variety of anticancer compounds and hormones to enter cells including statins and the abundant adrenal androgen dehydroepiandrosterone sulfate (DHEAS). DHEAS is a precursor to more potent androgens, such as dihydroxytestosterone (DHT), which drive prostate cancer growth.
Prior work by our group has shown that in vitro, statins compete with DHEAS for influx by SLCO2B11, which may deplete the tumor’s available androgen pool. Clinically, this reduction may translate to improved cancer outcomes as we observed in our institutional cohort of men with hormone sensitive prostate cancer (HSPC) receiving initial androgen deprivation therapy (ADT). In our retrospective analysis of 926 patients, statin users had a significantly increased TTP on ADT compared to non-users (27.5 vs. 17.4 months, p<0.001, HR 0.83)1. Similarly, retrospective review of a large cohort of patients treated in a prospective trial evaluating ADT for HSPC found that statin use conferred a benefit in both overall and prostate cancer–specific survival2. Given the complementary laboratory and clinical data, we postulated that statins compete with DHEAS uptake leading to lower androgen availability and enhanced disease control on ADT. The widespread use of statins and their established safety profile make them attractive potential anticancer agents as adjuvants to androgen ablating therapies.
Suppression of the androgen pathway using agents such as luteinizing hormone-releasing hormone (LHRH) agonists is generally the first line of defense in metastatic prostate cancer. However, almost all men progress to a castration resistant state, which is largely lethal. In part, this resistance to initial androgen blockade may be due to increased utilization of weaker but abundant adrenal androgens such as dehydroepiandrosterone (DHEA) and its downstream more potent metabolite DHT3. In more recent years, agents that more fully block androgen synthesis such as the oral biosynthesis inhibitor, abiraterone acetate (AA), have been developed. AA blocks testicular, adrenal, and intratumoral CYP17A1 and the subsequent production of androgens such as DHEA, globally reducing the circulating androgen pool.
Given AA’s ability to induce clinically significant improvements in overall survival, TTP, pain control, and prostate-specific antigen (PSA) responses in both docetaxel refractory and naïve patients, it has become one of the most frequently used second line hormonal maneuvers4,5. Additionally, its tolerability and ease of oral administration have made it a frequent choice prior to chemotherapy for mCRPC. AA may also undergo SLCO-mediated transport given its steroidal structure. The latter is supported by findings from Mostaghel et al that genetic variation in SLCO genes was associated with intraprostatic AA levels and that LNCaP cells overexpressing SLCO had higher intracellular levels of AA6. More efficient transport of AA or of other substrates such as DHEAS may play a role in the acquired resistance to AA that almost all patients experience.
The objective of our study was to assess whether concurrent statin use influenced the clinical efficacy of AA in patients with advanced castration-resistant prostate cancer (CRPC). Based on initial in vitro work demonstrating a negative interaction, we hypothesized that concurrent statin use would counteract AA’s antitumor effects. After seeing a contrary trend to benefit in the initial Dana-Farber Cancer Institute (DFCI) cohort, we attempted to validate the findings using a second institutional cohort of patients from Johns Hopkins University (JHU).
Materials, Patients, and Methods
Preclinical studies
Cell lines and reagents
The hormone sensitive prostate cancer cell lines, LNCaP and 22RV1, were employed in the study. LNCaP and 22RV1 cells were maintained in RPMI 1640 and supplemented with 10% FBS and antibiotics. The stable inducible shSLCO2B1-expressing cell lines were used as previously described and maintained in medium containing puromycin1. All cell lines were regularly screened for mycoplasma (Sigma Venor GeM Mycoplasma Detection Kit). Atorvastatin was purchased from Santa Cruz Biotechnology. Abiraterone acetate, fluvastatin, pravastatin and simvastatin were purchased from Selleckchem.
Cell proliferation assay
Cell proliferation was determined using the WST-1 assay (Roche, Indianapolis, IN). Briefly, cells were inoculated in 96-well plates at a confluence of ~10% for 2 days followed by treatment with statins and/or AA. Cell proliferation assays were carried out on different days after treatment. Each experiment was performed in triplicate.
Clinical studies
We queried our IRB approved institutional clinical databases for patients who had received AA for M0 or M1 CRPC from 2008 to 2015 for the DFCI cohort and from 2009 to 2014 for the JHU cohort. Demographic and clinical outcomes data were retrieved. Data regarding statin use including on-therapy dates was retrospectively collected from the electronic medical record. Time on AA was used as a surrogate for time to progression (TTP). Duration of AA was defined as the time from AA initiation to the time when AA was discontinued or censored on the last known alive date. The duration of AA was estimated using the Kaplan-Meier method. The association of statin use and duration of AA was assessed using a Cox regression model adjusting for known prognostic factors (e.g., prior docetaxel or enzalutamide use; sites of metastases: bone or lymph node versus liver or other viscera). For the primary analysis, we used a Cox regression model to assess the association of statin use and AA duration after adjusting for prior use of enzalutamide and docetaxel for the 2 cohorts separately.
Results
Preclinical studies
Given that AA may also use SLCO2B1 as a transporter, we evaluated the effects of AA and statins alone and in combination in prostate cancer cell lines. The key finding was that AA’s effect on prostate cancer cell proliferation is SLCO2B1 dependent. Knockdown of SLCO2B1 (shSLCO2B1) in both 22RV1 and LNCaP cells attenuated AA’s inhibition of cell growth (Figure 1A). These findings suggest that AA’s efficacy depends on SLCO2B1 transport into cells.7 When evaluating for competition between AA and statins for uptake, we observed that atorvastatin attenuated AA’s inhibition of the prostate cancer cell proliferation and was SLCO2B1 dependent (Figure 1B). When SLCO2B1 is knocked down, AA’s inhibitory effects on cell growth are attenuated. The greatest degree of AA’s inhibition of cell growth occurs in the absence of statin but in the presence of intact SLCO2B1. In the presence of statin, inhibition of cell proliferation by AA is decreased and is concentration dependent (Figure 1B).
Figure 1.
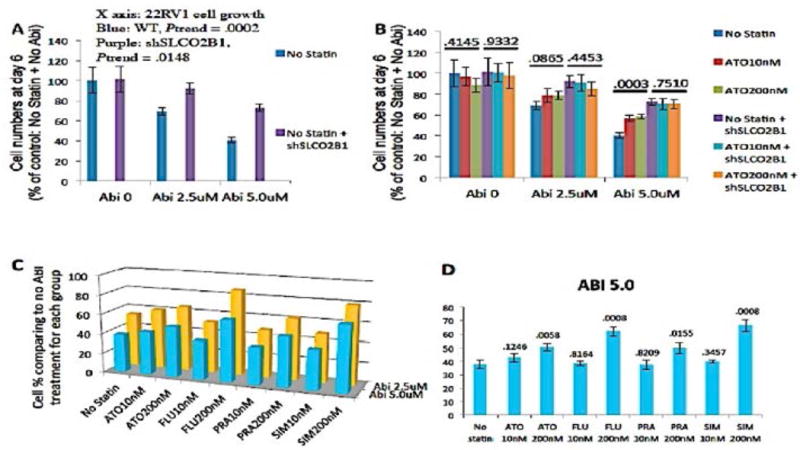
(A) AA (ABI) efficacy is dose and SLCO2B1 dependent. When SLC02B1 is knocked down (sh) in 22RV1 cells, ABI’s inhibition of cell growth is attenuated. (B) The greatest degree of AA inhibition occurs in the absence of statin but in the presence of intact SLCO2B1. In the presence of statin, AA’s inhibition of cell proliferation is decreased and is concentration dependent. When SLCO2B1 is knocked down, AA’s inhibitory effects on cell growth are attenuated. (C, D) LNCaP cells were treated with different concentration of AA indicated by “ABI” (no ABI treatment, 2.5 μM, 5.0 μM) and with or without various statins (ATO, atorvastatin; FLU, fluvastatin; PRA, pravastatin; SIM, simvastatin) at 10 nM and 200 nM. Relative cell numbers were examined by WST-1 assay at day 6. The value for each column represents the percentage of cell numbers comparing to the no ABI treatment condition within each statin treatment group. Fluvastatin induced the greatest attenuation of AA effects at the higher concentration (200 nM) whereas pravastatin showed the least inhibition of AA effects, especially when AA was used near the physiological concentration (2.5 μM).
To evaluate differences in inhibition among the different statins, we compared the effects of 4 common statins (Figure 1 C, D). Fluvastatin and simvastatin seemed to attenuate AA’s inhibition of cancer growth the most with pravastatin being less inhibitory.
Clinical observations
Based on the preclinical results showing antagonism, we hypothesized that statins may compete with AA for influx by SLCO2B1, which could negatively impact drug efficacy and treatment duration.
DFCI Cohort
We identified 224 patients treated with AA between 2008 and 2015 at DFCI who were eligible for analysis (Table 1). The majority of patients (96%) had metastatic disease at the time of AA initiation. Of these, 26% had received prior docetaxel and 7% had had prior enzalutamide. Nearly half were statin users (41%). Despite the lack of randomization, the two cohorts were well balanced in terms of baseline clinical factors such as Gleason score, prior local treatment, and site of metastases at AA start. There was a slightly higher incidence of M1 disease in non-users (20% vs. 11%). The median duration on AA was 10.7 months (range: <1 - 61 months). At the time of analysis, 160 (71%) patients had discontinued AA. Median follow-up from AA initiation was 27.8 months (range: 1.1–87.6).
Table 1.
Patient and clinical characteristics separated by institution
| DFCI | JHU | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All patients N=224 | Without statin N=132 | Statin user N=92 | All patients N=270 | Without statin N=148 | Statin user N=122 | |||||||
| N | %, median (q1, q3) | N | %, median (q1, q3) | N | %, median (q1, q3) | N | %, median (q1, q3) | N | %, median (q1, q3) | N | %, median (q1, q3) | |
| PSA at diagnosis | 212 | 9.4 (5.0,30.5) | 124 | 9.7 (5.1,30.9) | 87 | 8.7 (4.6, 28.5) | 234 | 11.9 (6.3, 54) | 133 | 12.8 (6.5, 83.6) | 101 | 11.3 (6.3, 34) |
| Gleason score | ||||||||||||
| 6 or less | 34 | 15% | 19 | 14% | 15 | 16% | 16 | 6% | 10 | 7% | 6 | 5% |
| 7 | 70 | 31% | 41 | 31% | 29 | 32 | 71 | 26% | 36 | 24% | 35 | 29% |
| 8 or more | 110 | 49% | 65 | 49% | 45 | 49% | 152 | 56% | 85 | 57% | 67 | 55% |
| Unknown | 10 | 4% | 7 | 5% | 3 | 3% | 31 | 11% | 17 | 11% | 14 | 11% |
| T stage | ||||||||||||
| T1 | 137 | 61% | 77 | 58% | 59 | 64% | 33 | 12% | 15 | 10% | 18 | 15% |
| T2 | 44 | 20% | 26 | 20% | 18 | 20% | 63 | 23% | 36 | 24% | 27 | 22% |
| T3 | 17 | 8% | 11 | 8% | 6 | 7% | 102 | 38% | 54 | 36% | 48 | 39% |
| T4 | 2 | <1% | 2 | 2% | 0 | 0% | 11 | 4% | 8 | 4% | 5 | 4% |
| Tx/Unknown | 24 | 11% | 16 | 12% | 9 | 10% | 61 | 23% | 37 | 25% | 14 | 20% |
| N stage | ||||||||||||
| N0 | 85 | 38% | 51 | 39% | 33 | 36% | 152 | 56% | 77 | 52% | 75 | 61% |
| N1 | 28 | 13% | 20 | 15% | 8 | 9% | 64 | 24% | 38 | 26% | 26 | 21% |
| Nx/Unknown | 111 | 50% | 61 | 46% | 51 | 55% | 54 | 20% | 33 | 22% | 21 | 17% |
| M stage | ||||||||||||
| M0 | 97 | 43% | 59 | 45% | 38 | 41% | 177 | 66% | 91 | 61% | 86 | 70% |
| M1 | 37 | 17% | 26 | 20% | 10 | 11% | 78 | 29% | 50 | 34% | 28 | 23% |
| Mx/Unknown | 90 | 40% | 47 | 36% | 44 | 48% | 15 | 6% | 7 | 5% | 8 | 7% |
| Primary local | ||||||||||||
| RP+/-RT | 105 | 47% | 61 | 46% | 44 | 48% | 114 | 42% | 62 | 42% | 52 | 43% |
| RT | 61 | 27% | 30 | 23% | 31 | 34% | 67 | 25% | 30 | 20% | 37 | 30% |
| None/Unknown | 58 | 26% | 41 | 31% | 17 | 18% | 89 | 33% | 56 | 38% | 31 | 27% |
| Age at start of AA | 224 | 68 (62, 75) | 132 | 68 (61, 74) | 92 | 70 (64, 76) | 279 | 70 (64, 75) | 148 | 68 (62, 73) | 122 | 72 (67, 77) |
| Year at start of AA | ||||||||||||
| Prior to 2011 | 6 | 3% | 4 | 3% | 2 | 2% | 3 | 1% | 3 | 2% | 1 | 1% |
| 2011 | 38 | 17% | 21 | 16% | 17 | 18% | 64 | 24% | 43 | 29% | 21 | 17% |
| 2012 | 83 | 37% | 53 | 40% | 30 | 33% | 92 | 34% | 48 | 32% | 43 | 35% |
| 2013 | 56 | 25% | 30 | 23% | 26 | 28% | 78 | 29% | 39 | 26% | 39 | 32% |
| 2014–2015 | 41 | 18% | 24 | 18% | 17 | 18% | 33 | 12% | 15 | 10% | 18 | 15% |
| Mets at AA start | 215 | 96% | 126 | 95% | 89 | 97% | 258 | 96% | 141 | 95% | 117 | 96% |
| PSA at AA start | 205 | 20.1 (5.4, 56.2) | 121 | 20.3 (6.6, 70.6) | 84 | 16.9 (4.5, 55.1) | 266 | 34.5 (11.3, 103) | 145 | 33.1 (10.6, 126) | 121 | 35.6 (13, 80.1) |
| Sites of mets | ||||||||||||
| Bone and LN | 192 | 86% | 114 | 86% | 78 | 85% | 206 | 76% | 115 | 78% | 91 | 75% |
| Soft tissue | 23 | 10% | 12 | 9% | 11 | 12% | 52 | 19% | 26 | 18% | 26 | 21% |
| Statin use at AA start | 92 | 41% | 92 | 100% | 122 | 45% | 122 | 100% | ||||
| Prior use of enza | 16 | 7% | 10 | 8% | 6 | 7% | 17 | 6% | 4 | 3% | 13 | 11% |
| Prior use of doce. | 59 | 26% | 35 | 27% | 24 | 26% | 79 | 29% | 49 | 33% | 30 | 25% |
AA: abiraterone acetate; Mets: metastases; LN: lymph node; Enza: enzalutamide; Doce: docetaxel
We observed a trend towards longer (rather than shorter) AA duration in statin users: 14.2 vs. 9.2 months (HR 0.79, 95% CI: 0.57–1.09, p=0.14, Figure 2A, Table 2). Similarly, in patients whose disease was naïve to docetaxel or enzalutamide, there was a trend towards longer AA use if patients were taking concurrent statins (HR 0.76; 95% CI: 0.50–1.15, Figure 2B). In univariate analysis, prior docetaxel (p=0.0001) or enzalutamide (p=0.0001) use was significantly associated with shorter AA duration (Table 2). Adjusting for prior use of docetaxel or enzalutamide and site of metastases did not alter the relationship between statin use and AA duration (HR 0.81, 95% CI: 0.58–1.11, p=0.18; Table 2). PSA at AA start was similar between the non-users and users (20.3 vs. 16.9 ng/dL, Table 1), and similar declines on AA therapy were achieved.
Figure 2.
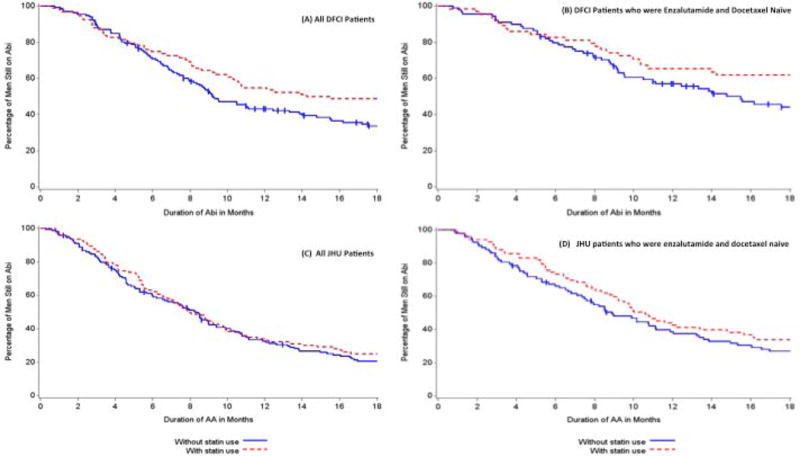
Kaplan-Meier analysis of association of statin use and AA duration (A) all DFCI patients, (B) DFCI patients who had not received prior enzalutamide or docetaxel, (C) all JHU patients, and (D) JHU patients who had not received prior enzalutamide or docetaxel.
Table 2.
Association of statin use at AA initiation and AA duration in DFCI and JHU cohorts
| N | N stopped | Median AA duration | HR and 95% CI (univariate) | P | HR and 95% CI (multivariable) | P | |
|---|---|---|---|---|---|---|---|
| DFCI Cohort | |||||||
| Statin use at AA initiation | 0.14 | * 0.18 ** 0.14 | |||||
| No statin use | 132 | 98 | 9.2 | 1 (reference) | 1 (reference) | ||
| With statin | 92 | 62 | 14.2 | 0.79 (0.57, 1.09) | * 0.81 (0.58, 1.11) ** 0.79 (0.57, 1.09) | ||
| JHU Cohort | |||||||
| Statin use at AA initiation | 0.38 | * 0.52 ** 0.49 | |||||
| No statin use | 143 | 128 | 8.3 | 1 (reference) | 1 (reference) | ||
| With statin | 122 | 104 | 8.0 | 0.89 (0.69, 1.15) | * 0.92 (0.70, 1.20) ** 0.91 (0.70, 1.19) |
Adjusting for prior use of enzalutamide, docetaxel, and sites of metastases
Adjusting for prior use of enzalutamide and docetaxel
JHU Cohort
From the JHU database, 270 patients with M0 or M1 CRPC, who had received AA between 2009 and 2014 and were eligible for analysis (Table 1). The majority of patients (96%) had radiographic metastatic disease at AA initiation. Similar to the DFCI cohort, approximately one-third of patients had received prior docetaxel (29%) and a minority, had received enzalutamide (6%). Almost half of the patients were statin users (45%). Despite the lack of randomization, the two JHU cohorts were well balanced in terms of baseline clinical factors such as Gleason score, history of prior local treatment, and site of metastases at AA initiation. There was a higher incidence of M1 disease at diagnosis in the JHU statin users (29%) compared to the DFCI cohort (17%). In the JHU cohort, the median duration on AA was 8.3 months (range: <1 to >48 months). At the time of data analysis, 236 (87%) had discontinued AA. Median follow-up from AA initiation was 30.5 months (range: <1 to 50.4 months).
There was no association between statin use and AA duration in the overall JHU cohort: 8.3 vs. 8.0 months (HR 0.89, 95% CI: 0.69–1.16, p=0.38, Table 2, Figure 2C) in the statin users vs. non-users. In patients whose disease was naïve to docetaxel and enzalutamide, the impact of statin use on duration of AA was similar to that observed in the DFCI cohort (HR 0.79; 95% CI: 0.57–1.10, Figure 2D). Adjusting for prior use of docetaxel, enzalutamide, and site of metastases confirmed the lack of association as in the univariate analysis (p=0.52, HR 0.92, 95% CI: 0.70, 1.20, Table 2). PSA at AA start was similar between the non-users and users (33.1 vs. 35.6 ng/dL, Table 1), and both cohorts achieved a similar degree of PSA decline.
In reviewing the clinico-demographic data (Table 1), we observed that the JHU patients appeared to have more advanced disease both at diagnosis and at AA initiation. They had higher Gleason grades, higher clinical T stages, and more nodal and metastatic disease at diagnosis. JHU patients also had a higher median PSA at time of AA initiation for CRPC (34.5 vs. 20.1 ng/dL).
Additionally, the proportion of JHU patients with poorer prognosis as defined by presence of visceral metastases was also higher at AA initiation (19% vs. 10%). Given these significant differences and potential heterogeneity between disease states across these two institutional cohorts, we summarized the clinical outcomes of these patients separately by center. Duration of AA therapy (8.3 months vs. 10.7 months) and overall survival (24.7 months vs. 36.6 months) were significantly shorter for JHU patients compared to DFCI patients (Figure 3, Table 3).
Figure 3. Duration of AA and Survival by Institute.
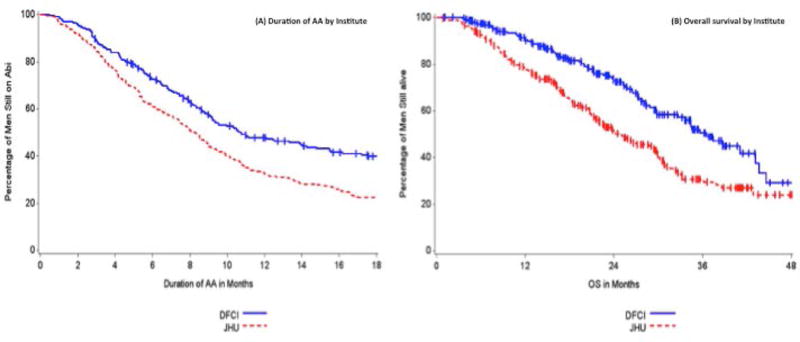
There was a significantly longer duration of AA and survival in patients at DFCI compared to JHU.
Table 3.
Outcomes of the two institutional cohorts by statin use
| DFCI All | DFCI Statin users | DFCI Non- users | JHU All | JHU Statin users | JHU Non-users | |
|---|---|---|---|---|---|---|
| AA duration (months) | 10.7 | 14.2 | 9.2 | 8.3 | 8.0 | 8.5 |
| Overall Survival (months) | 35.6 | 41.1 | 33.6 | 24.7 | 22.6 | 26.3 |
Discussion
Prior preclinical work and retrospective studies in patients with HSPC revealed how statins may compete with androgen uptake by the SLCO family of transporters. Similarly, in vitro studies showed a possible negative interaction between statins and AA, which we postulated might be due to competitive inhibition of SLCO2B1. Thus, we hypothesized that statins would compete with AA uptake by the prostate cancer cell and that patients on concurrent statins would have worse outcomes. However, contrary to our initial hypothesis, we observed a trend toward longer (rather than shorter) AA duration in statin users in our initial DFCI cohort at 14.2 vs. 9.2 months (HR 0.79, 95% CI: 0.57–1.09, p=0.14). The difference was even more pronounced in patients who were docetaxel or enzalutamide naïve (21.3 vs. 14.8 mo, HR 0.75, CI: 0.49–1.15). The differences could not be explained by imbalances in known baseline prognostic factors or by more advanced disease in the non-users.
Given the positive trend but small sample size of the DFCI cohort, we attempted to corroborate our findings in a similar cohort of JHU patients treated with AA for CRPC, who had matched, highly curated clinical and outcomes data. However, upon initial review of baseline clinical characteristics, we noticed that the JHU patients appeared to have more advanced prostate cancer as manifested by a higher percentage of patients with visceral metastases and a higher median PSA at the start of AA. The trend to benefit with statins in AA duration was not observed among the entire JHU cohort, and overall, the JHU patients had poorer outcomes with a median treatment duration of 8.3 months compared to 10.7 months in the DFCI cohort. However, in the JHU subset of patients, who were naïve to docetaxel and enzalutamide (and, thus, likely earlier in their disease course), the impact of statin use on duration of AA was similar to the DFCI patients (HR 0.79). Nonetheless, this difference did not reach statistical significance.
We considered several explanations for the disparate results between the cohorts. First, the positive trend observed in the DFCI cohort was not statistically significant and thus, there may be no interaction between statins and AA. A retrospective, small, single institution series of 108 patients with CRPC being treated with AA, did not find any difference in clinical benefit among the 19% of patients on statins compared to the non-users.8 Alternatively, both the German study and ours may be limited by sample sizes too small to definitively detect the benefit of concurrent statin use. However, the positive trend to benefit that we observed in the DFCI cohort is plausible for several reasons. In our previous study where we assessed the association of TTP on ADT, we found that statin users had a significantly longer TTP on ADT with a similar effect size (adjusted HR 0.83)1 and preclinical work by our group supplied a valid mechanism of in vitro inhibition of DHEAS uptake by statins. As both AA and DHEAS are substrates for SLCO2B1 mediated transport, it is conceivable that there could be an additive effect of both statins and AA in blocking DHEAS uptake by SLCO2B1. The antagonistic effect of statins on AA efficacy seen in vitro could be balanced by the decrease in DHEAS uptake and depleted intra-tumoral androgen pool. Additionally, Mostaghel et al have demonstrated significant variation in intra-prostatic levels of AA in patients with different SLCO2B1 genotypes6. If the mechanism is indeed mediated by SLCO transport, different SLCO SNPs may transport AA, DHEAS or statins with varying efficiency and potentially influence the clinical impact of concurrent AA and statin use.9 Adaptive increases in transport could result in more efficient use of residual non-gonadal androgens.10,11 Similarly, Mostaghel et al have observed that serum DHEAS levels remain abundant in men despite treatment with AA leaving a circulating reservoir for tumor use.10
The SLCO (OATP) transporters are expressed physiologically on multiple cells such as hepatocytes and mediate uptake of several hormones and drugs.12 SLCO1B1 and SLCO1B3 appear to be liver specific whereas SLCO2B1 appears to be more ubiquitously expressed throughout multiple organs of the body.12 One study observed that abiraterone and its metabolites inhibited the OATP1B1 (SLCO1B1) transporter in human hepatocytes in vitro but based on the calculated in vivo inhibition potential, these effects were thought to be negligible.13 Thus, one could postulate that competitive uptake between statins and AA by the various SLCO transporters in the liver could prolong drug exposure and influence disease control. Prospective pharmacokinetic evaluation could investigate this theory as well as whether patients with shorter durations or worse outcomes have lower concentrations of AA compared to responders.
The negative result in the entire JHU cohort but trend to benefit in the docetaxel- and enzalutamide-naïve patients suggests that disease state may matter. Our initial investigation of the anticancer effects of statins was in an earlier, hormone sensitive state. Among the AA patients, the DFCI cohort appeared to have less aggressive disease on average. Perhaps, the statin effect, if real, occurs in a less advanced, more hormone dependent state. As the disease progresses and becomes more reliant on non-androgen drivers, the use of statins as an anticancer therapy may not be as effective.
Of note, statin use was quite high at 41% to 45% in both institutional cohorts. This work highlights that commonly used medications can have potential interactions with our anticancer therapeutics and should be considered. Several other drugs such as metformin and aspirin have been considered in this light,14,15 some with ongoing prospective evaluation.
Additional limitations of this study include its retrospective nature and use of a surrogate endpoint. Missing data on statin use and small sample sizes may limit identification of smaller degrees of clinical impact. With respect to our primary endpoint, there are inherent limitations to using duration of treatment as a surrogate for true time to progression on AA and perhaps this could lead to variations in the differences observed, either positively or negatively. However, given that clinical practice outside of a controlled clinical trial setting encompasses much heterogeneity in the decision for drug cessation, e.g., PSA rise only vs. frank radiologic progression, it is reasonable to use AA duration as an indirect measure of clinical benefit.
In summary, contrary to our hypothesis derived from preclinical studies, there was a trend toward longer AA duration in statin users in the DFCI cohort. While this finding was not supported in the entire JHU cohort, who generally had more advanced disease, the subset of patients whose disease was naïve to enzalutamide and docetaxel appeared to have a similar trend and effect size with statin use. Thus, our findings do not support our initial hypothesis. Statins do not appear to adversely affect the efficacy of AA. Given our prior work demonstrating a median 10 month prolonged TTP on ADT in men concurrently taking statins (similar adjusted HR 0.83 vs. 0.79 on AA), in vitro findings of inhibition of DHEAS uptake by statins, and the trend toward a beneficial effect of statin on AA efficacy in the DFCI cohort and docetaxel- and enzalutamide-naive JHU patients, we postulate that there could be an additive effect of statins blocking DHEAS uptake and by AA inhibiting androgen synthesis in better risk patients. Ideally, future work will be geared towards prospective evaluation of the clinical efficacy of a combined AA and statin approach integrated with preclinical studies designed to demonstrate how statins may influence SLCO-mediated transport of DHEAS and AA and tumor control in vitro and in vivo using both patient sera and tissue samples.
Acknowledgments
Funding & Research Support:
This work was supported by a Dana-Farber Prostate Cancer SPORE P50CA090381; Department of Defense (DOD - W81XWH-14-1-0515).
Lauren C. Harshman was partially funded by a 2013 Prostate Cancer Foundation Young Investigator Award.
Benjamin L. Maughan was partially funded by a Conquer Cancer Foundation YIA.
Emmanuel S. Antonarakis was partially funded by grant P30 CA006973.
Footnotes
Disclosures:
LC Harshman: Advisory: Genentech, Dendreon, Pfizer, Medivation/Astellas, Kew Group, Theragene; Research to the institution: Bayer, Sotio, BMS, Merck, Takeda, Dendreon/Valient, Jannsen, Medivation/Astellas, Genentech, Pfizer, Takeda, Jannsen; Travel accommodations: Sanofi
L Werner: None
A Tripathi: None
X Wang: none
BL Maughan: none
ES Antonarakis: Honoraria: Sanofi, Dendreon, Medivation, Janssen Biotech, ESSA, Astellas Pharma; Consultant/adviser: Sanofi, Dendreon, Medivation, Janssen Biotech, ESSA, Astellas Pharma; Research funding to institution: Janssen Biotech, Johnson & Johnson, Sanofi, Dendreon, Aragon Pharmaceuticals, Exelixis, Millenium Pharm, Genentech, Novartis, Astellas Pharmaceuticals, and Tokai Pharma; Patents: co-inventor of a biomarker technology that has been licensed to Qiagen and Tokai; Travel/Accommodations/Expenses: Sanofi, Dendreon, Medivation
M Nakabayashi: none
R McKay: Research funding to institution: Bayer, Pfizer
M Pomerantz: none
LA Mucci: none
ME Taplin: Research funding and advisory board: Janssen
CJ Sweeney: Consulting and research support: Janssen, Astellas, Medivation, Bayer, Sanofi; Consulting; Pfizer
GSM Lee: none
PW Kantoff: Consultant: Astellas, Bayer, Bellicum, BIND Biosciences Inc., BN Immunotherapeutics, DRGT, Genetech/Roche, Ipsen Pharmaceuticals, Janssen, Metamark, Merck, MTG Therapeutics, Omnitura, OncoCellMDX, OncoGenex, Progenity, Sanofi, Tarveda Therapeutics, Thermo Fisher, Bavarian Nordic; Investments: Bellicum, DRGT, Placon, Tarveda Therapeutics; Editorial services: UpToDate; Advisory board: Memorial Sloan kettering Cancer Center, University of Pennsylvania, Thomas Jefferson University, MD Anderson Cancer Center, Northwestern University
References
- 1.Harshman LC, Wang X, Nakabayashi M, et al. Statin Use at the Time of Initiation of Androgen Deprivation Therapy and Time to Progression in Patients With Hormone-Sensitive Prostate Cancer. JAMA oncology. 2015;1(4):495–504. doi: 10.1001/jamaoncol.2015.0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamilton RJDK, Crook JM, O’Callaghan CJ, Higano CS, Dearnaley D, Horwitz E, Goldenberg L, Gospodarowicz MK, Klotz L. The association between statin use and outcomes in patients initiating androgen deprivation therapy. J Clin Oncol. 2015;33(suppl 7) doi: 10.1016/j.eururo.2020.12.031. abstr 145. [DOI] [PubMed] [Google Scholar]
- 3.Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer research. 2008;68(11):4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368(2):138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mostaghel EA, Cho E, Zhang A, et al. Association of Tissue Abiraterone Levels and SLCO Genotype with Intraprostatic Steroids and Pathologic Response in Men with High-Risk Localized Prostate Cancer. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harshman LCWL, Nakabayashi M, Tripathi A, McKay RR, Wang X, Pomerantz M, Mucci LA, Taplin ME, Sweeney C, Lee GSM, Kantoff PW. The impact of statin use on abiraterone acetate (AA) treatment duration in patients with castration-resistant prostate cancer (CRPC) J Clin Oncol. 2016;34(suppl 2S) abstr 196. [Google Scholar]
- 8.Boegemann M, Schlack K, Fischer AK, et al. Influence of Statins on Survival Outcome in Patients with Metastatic Castration Resistant Prostate Cancer Treated with Abiraterone Acetate. PLoS One. 2016;11(9):e0161959. doi: 10.1371/journal.pone.0161959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Harshman LC, Xie W, et al. Association of SLCO2B1 Genotypes With Time to Progression and Overall Survival in Patients Receiving Androgen-Deprivation Therapy for Prostate Cancer. J Clin Oncol. 2016;34(4):352–359. doi: 10.1200/JCO.2015.62.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tamae D, Mostaghel E, Montgomery B, et al. The DHEA-sulfate depot following P450c17 inhibition supports the case for AKR1C3 inhibition in high risk localized and advanced castration resistant prostate cancer. Chemico-biological interactions. 2015;234:332–338. doi: 10.1016/j.cbi.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho E, Montgomery RB, Mostaghel EA. Minireview: SLCO and ABC transporters: a role for steroid transport in prostate cancer progression. Endocrinology. 2014;155(11):4124–4132. doi: 10.1210/en.2014-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagenbuch B, Gui C. Xenobiotic transporters of the human organic anion transporting polypeptides (OATP) family. Xenobiotica. 2008;38(7–8):778–801. doi: 10.1080/00498250801986951. [DOI] [PubMed] [Google Scholar]
- 13.Monbaliu J, Gonzalez M, Bernard A, et al. In Vitro in Vivo Drug-Drug Interaction Studies to Assess the Effect of Abiraterone Acetate, Abiraterone, and Metabolites of Abiraterone on CYP2C8 Activity. Drug Metab Dispos. 2016 doi: 10.1124/dmd.116.070672. [DOI] [PubMed] [Google Scholar]
- 14.Chang SL, Harshman LC, Presti JC., Jr Impact of common medications on serum total prostate-specific antigen levels: analysis of the National Health and Nutrition Examination Survey. J Clin Oncol. 2010;28(25):3951–3957. doi: 10.1200/JCO.2009.27.9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Preston MA, Riis AH, Ehrenstein V, et al. Metformin use and prostate cancer risk. Eur Urol. 2014;66(6):1012–1020. doi: 10.1016/j.eururo.2014.04.027. [DOI] [PubMed] [Google Scholar]