

Abstract
Perosamine (4-amino-4,6-dideoxy-D-mannose), or its N-acetylated form, is one of several dideoxy sugars found in the O-antigens of such infamous Gram-negative bacteria as Vibrio cholerae O1 and Escherichia coli O157:H7. It is added to the bacterial O-antigen via a nucleotide-linked version, namely GDP-perosamine. Three enzymes are required for the biosynthesis of GDP-perosamine starting from mannose-1-phosphate. The focus of this investigation is on GDP-perosamine synthase from Caulobacter cresentus, which catalyzes the final step in GDP-perosamine synthesis, the conversion of GDP-4-keto-6-deoxymannose to GDP-perosamine. The enzyme is PLP-dependent and belongs to the aspartate aminotransferase superfamily. It contains the typically conserved active site lysine residue, which forms a Schiff base with the PLP cofactor. Two crystal structures were determined for this investigation: a site-directed mutant protein (K186A) complexed with GDP-perosamine and the wild-type enzyme complexed with an unnatural ligand, GDP-3-deoxyperosamine. These structures, determined to 1.6 Å and 1.7 Å resolution respectively, revealed the manner in which products, and presumably substrates, are accommodated within the active site pocket of GDP-perosamine synthase. Additional kinetic analyses using both the natural and unnatural substrates revealed that the Km for the unnatural substrate was unperturbed relative to the natural substrate, but the kcat was lowered by a factor of approximately 200. Taken together these studies shed light on why GDP-perosamine synthase functions as an aminotransferase whereas another very similar PLP-dependent enzyme, namely GDP-4-keto-6-deoxy-D-mannose 3-dehydratase or ColD, catalyzes a dehydration reaction using the same substrate.
Graphical Abstract
The lipopolysaccharide or LPS is a major component of the outer membrane of Gram-negative bacteria. It is a complex entity composed of three parts: the outermost O-specific polysaccharide, the middle core polysaccharide, and the innermost portion referred to as Lipid A (1, 2). The O-specific polysaccharide, or O-antigen, typically contains repeating units of three to five sugars. It varies among bacterial strains in its sugar composition, linkage, and sequence. Often the LPS contains unusual dideoxy sugars such as D-perosamine, D-tyvelose, or L-colitose, among others (3). The building blocks for the synthesis of the O-antigen are the nucleotide-linked versions of these sugars, namely GDP-perosamine, CDP-tyvelose, and GDP-colitose.
In recent years, the biochemical pathways for the production of the 3(4),6-dideoxyhexoses have become the focus of significant research attention (4). Most of these pathways begin with the attachment of α-D-hexose-1-phosphate to an NMP moiety via a nucleotidyltransferase. Subsequently, the C-6′ hydroxyl group is removed, and the C-4′ hydroxyl group is oxidized to a keto-functionality yielding NDP-4-keto-6-deoxyhexose. This reaction is catalyzed by NDP-hexose 4,6-dehydratase. Both the nucleotidyltransferases and the dehydratases have been well characterized with respect to structure and/or function (5, 6). Importantly, the NDP-4-keto-6-deoxyhexose intermediate represents the branching point for all of the subsequent enzymatic reactions that ultimately lead to the production of such unusual sugars as perosamine.
The focus of this investigation is GDP-perosamine synthase (Scheme 1), which catalyzes the formation of GDP-D-perosamine from GDP-4-keto-6-deoxymannose (7). Interest in this enzyme grew from our previous structural investigation of GDP-4-keto-6-deoxy-D-mannose-3-dehydratase, an enzyme involved in colitose production and hereafter referred to as ColD (8–10). Like GDP-perosamine synthase, ColD utilizes GDP-4-keto-6-deoxymannose as a substrate (Scheme 1). However, rather than catalyzing the amination of the sugar C-4′, ColD functions to remove the hydroxyl group at C-3′. ColD and GDP-perosamine synthase display an amino acid sequence identity of ~23%, and both of these enzymes require pyridoxal 5′-phosphate (PLP) and L-glutamate for activity. Whereas GDP-perosamine synthase contains the typically conserved lysine residue responsible for anchoring the PLP cofactor to the protein, ColD contains a histidine residue thereby precluding covalent bond formation between the protein and the cofactor.
Scheme 1.

We recently solved the structure of GDP-perosamine synthase from Caulobacter crescentus CB15 with crystals grown in the presence of α-ketoglutarate (11). From this initial investigation of the C. crescentus GDP-perosamine synthase, we were able to produce a novel GDP-linked sugar, GDP-4-amino-3,4,6-trideoxy-D-mannose, hereafter referred to as GDP-3-deoxyperosamine (11). This was accomplished by reacting the ColD product with GDP-perosamine synthase and in the presence of L-glutamate (Scheme 1). This study also revealed that GDP-perosamine synthase is a dimer with two regions, delineated by Arg 19 to Ser 33 and Tyr 221 to Gln 236, providing extensive subunit:subunit contacts. These two regions from one subunit project towards the active site of the second subunit (and vice versa). The individual subunits of GDP-perosamine synthase are characterized by a seven-stranded mixed β-sheet, a two-stranded antiparallel β-sheet, and 12 α-helices as shown in Figure 1a. The overall fold of the enzyme places it into the well-characterized aspartate aminotransferase superfamily (12–14). Shown in Figure 1b is a close-up view of the interactions between the protein and the α-ketoglutarate ligand. A key electrostatic interaction between Arg 231 from the second subunit of the dimer and α-ketoglutarate serves to anchor the ligand into the active site region.
Figure 1.

Molecular architecture of C. crescentus GDP-perosamine synthase. (a) A ribbon representation of the dimer. The initial structure solved contained PLP attached to Lys 186 via a Schiff base (indicated in sphere representation). The local twofold rotational axis relating one subunit to the other lies in the plane of the figure as indicated by the black arrow. (b) Close-up view of the active site. Crystals used for the initial structural analysis of GDP-perosamine synthase were grown in the presence of α-ketoglutarate, and the structure was determined to 1.8 Å resolution. The Lys 186/PLP internal aldimine and α-ketoglutarate are highlighted in slate and green bonds respectively. Possible hydrogen bonding interactions are depicted by dashed lines. All figures were prepared with the software package PyMOL (21).
Perhaps one of the most intriguing biochemical questions regarding ColD and GDP-perosamine synthase is why one functions as a dehydratase whereas the other acts as an aminotransferase, respectively. Is it simply a result of ColD containing a histidine rather than the lysine residue normally found in PLP-dependent enzymes, or are there other factors involved? To further address this fascinating issue, we report here the structural analysis of GDP-perosamine synthase solved in the presence of either GDP-perosamine, its natural product or GDP-3-deoxyperosamine. Taken together, the high resolution structures described herein provide a more detailed understanding of the active site geometry of GDP-perosamine synthase, and allow for its comparison with the substrate binding pocket of ColD.
Materials and Methods
Cloning, Site-Directed Mutagenesis, Expression, and Purification of GDP-Perosamine Synthase
The gene encoding GDP-perosamine synthase was cloned from C. crescentus as previously described (11) and used to produce the pET28t-Per plasmid required for this investigation. Lys 186 was changed to an alanine through site-directed mutagenesis using the QuikChange Mutagenesis Kit (Stratagene). Mutagenesis was confirmed by DNA sequence analysis. The protein was expressed and purified as previously described (11), and subsequently dialyzed against 25 mM Tris-HCl (pH 8.0) and 100 mM NaCl. For crystallization experiments, the protein was concentrated to 25 mg/mL as estimated by the absorbance at 280 nm using an extinction coefficient of 1.40 (mg/mL)−1cm−1. Wild-type enzyme required for the structural and functional studies was prepared as previously reported (11).
Crystallization of GDP-Perosamine Synthase
Crystallization conditions were first surveyed by the hanging drop method of vapor diffusion using a sparse matrix screen developed in the laboratory. Large single crystals of his-tagged GDP-perosamine synthase K186A mutant protein were subsequently grown via batch methods by mixing 20 μL of the protein solution (at 25 mg/mL) with 20 μL of a precipitant solution containing 100 mM MES (pH 6.5), 20% poly(ethylene glycol) 8000, 2 mM PLP, and 2 mM glutamate. The crystals grew to maximum dimensions of about 0.1 × 0.3 × 0.6 mm in 1 week. They belonged to space group P21 with two dimers in the asymmetric unit. Crystals of wild-type GDP-perosamine synthase were grown as previously reported (11).
Structural Analysis of GDP-Perosamine Synthase
GDP-perosamine and GDP-3-deoxyperosamine were produced as described elsewhere (11). Crystals of the wild-type GDP-perosamine synthase were transferred to a synthetic mother liquor consisting of 100 mM MES (pH 6.5), 50 mM NaCl, 26% poly(ethylene glycol) 8000, 2 mM PLP, 2 mM α-ketoglutarate, and 20 mM GDP-3-deoxyperosamine. The K186A GDP-perosamine synthase crystals were transferred to the same synthetic solution, but with 20 mM GDP-perosamine replacing the GDP-3-deoxyperosamine. Crystals were incubated at room temperature in these solutions for over 24 hours. α-ketoglutarate was used in the soaking solutions to ensure that the cofactor remained in the PLP state thereby allowing the amino sugar to react with the cofactor.
Single crystals were subsequently transferred to cryoprotectant solutions containing 100 mM MES (pH 6.5), 150 mM NaCl, 26% poly(ethylene glycol) 8000, 15% ethylene glycol, 2 mM PLP, and 2 mM α-ketoglutarate, with the addition of either 30 mM GDP-perosamine (for the K186A crystals) or 30 mM GDP-3-deoxyperosamine (for the wild-type crystals). X-ray data were collected from the K186A crystal at the SBC Beamline 19-BM (Advanced Photon Source, Argonne National Laboratory, Argonne, IL). The data set was processed with HKL2000 (15) and scaled with SCALEPACK (15). X-ray data were collected from the wild-type crystal on a Bruker Proteum CCD detector system. The x-ray source was CuKα radiation from a Rigaku RU200 x-ray generator operated at 50 kV and 90 mA. These x-ray data were processed with SAINT version 7.06A (Bruker AXS Inc.) and internally scaled with SADABS version 2005/1 (Bruker AXS Inc). Relevant x-ray data collection statistics are presented in Table 1. The structure of the K186A GDP-perosamine synthase containing GDP-perosamine was solved via molecular replacement with the program EPMR (16), employing the wild-type GDP-perosamine synthase determined in this laboratory as a search model. All solvent molecules, Lys 186, and the coordinates for the PLP cofactor were omitted from the search probe. The resulting model had an initial R-factor of 38%. Alternate cycles of manual model building using the program Coot (17) and least-squares refinement of the model with TNT (18) reduced the overall R-factor to 16.9% for all measured x-ray data to 1.6 Å resolution. Ramachandran statistics indicate that 89.0% of the residues adopt ϕ, ψ angles in the “most favored,” 10.9% in the “additionally allowed,” and 0.1% in the “generously allowed” regions.
Table 1.
X-ray Data Collection Statistics.
| Enzyme Complexed with GDP-perosamine | Enzyme Complexed with GDP-3-deoxyperosamine | |
|---|---|---|
| space group | P21 | P21 |
| unit cell dimensions (Å) | a = 50.1, b = 151.9, c = 105.7, β = 102.1° | a = 50.4, b = 152.2, c = 105.8, β = 101.8° |
| resolution limits | 50 – 1.6 (1.66 – 1.6)b | 100 – 1.7 (1.8 – 1.7)b |
| number of independent reflections | 186452 (16930) | 161560 (22419) |
| completeness (%) | 91.9 (83.8) | 94.6 (83.2) |
| redundancy | 6.3 (4.6) | 3.7 (1.7) |
| avg I/avg σ(I) | 11.5 (7.6) | 10.7 (2.3) |
| Rsym (%)a | 4.1 (21.4) | 8.6 (42.4) |
Rsym = (Σ|Σ I − Ī|/ΣI) × 100.
Statistics for the highest resolution bin.
The structure of the wild-type enzyme containing GDP-3-deoxyperosamine was solved via difference Fourier methods, utilizing an initial rigid body refinement with TNT. Alternate cycles of manual model building and least squares refinement reduced the R-factor to 19.9% for all measured x-ray data to 1.7 Å resolution. Relevant refinement statistics are presented in Table 2. Ramachandran statistics indicate that 87.8% of the residues adopt ϕ, ψ angles in the “most favored,” whereas 12.2% lie in the “additionally allowed” regions.
Table 2.
Least-Squares Refinement Statistics.
| Enzyme Complexed with GDP-perosamine | Enzyme Complexed with GDP-3-deoxyperosamine | |
|---|---|---|
| resolution limits (Å) | 30 - 1.6 | 30 - 1.7 |
| R-factora (overall) %/no. reflections | 16.9/186447 | 19.9/161554 |
| R-factor (working) %/no. reflections | 16.6/167517 | 19.7/153306 |
| R-factor (free) %/no. reflections | 23.8/18930 | 26.3/16248 |
| number of protein atomsb | 11299 | 11408 |
| number of heteroatomsc | 1479 | 1195 |
| Average B values | ||
| protein atoms (Å2) | 28.2 | 21.5 |
| ligands (Å2) | 28.7 | 42.8 |
| solvent (Å2) | 39.1 | 26.8 |
| weighted RMS deviations from ideality | ||
| bond lengths (Å) | 0.013 | 0.014 |
| bond angles (°) | 2.01 | 2.10 |
| trigonal planes (Å) | 0.009 | 0.009 |
| general planes (Å) | 0.018 | 0.018 |
| torsional anglesd (°) | 16.4 | 16.7 |
R-factor = (Σ|Fo − Fc|/Σ|Fo|) × 100 where Fo is the observed structure-factor amplitude and Fc is the calculated structure-factor amplitude.
These include multiple conformations for E145, V162, R231, Q236 and E313 in Subunit2, K43 and I188 in Subunit 3 and I188 in Subunit 4 in the enzyme/GDP-perosamine complex; and S32, and V39 in Subunit 1, and S32, V39, V162, I225, R231, and I312 in Subunit 2 of the enzyme/GDP-3-deoxyperosamine complex.
Heteroatoms include 1259 water molecules, four PLP-linked sugar ligands, and three ethylene glycols for the enzyme/GDP-perosamine complex and 1039 water molecules, four PLPs, four GDP-3-deoxyperosamine ligands, and two ethylene glycols.
The torsional angles were not restrained during the refinement.
Enzymatic Assays
GDP-mannose-4,6-dehydratase was purified as previously described (10). GDP-4-keto-6-deoxymannose was produced by reacting 5 mM GDP-mannose (Sigma), 0.1 mM NADP+, and 3 μM GDP-mannose-4,6-dehydratase in Buffer A (20 mM HEPES, pH 7.5, 50 mM NaCl, 10 mM MgCl2) for 2 hours at room temperature. The reaction was filtered through a 30-kDa cut-off filter to remove the protein. The flow-through was purified on an ÄKTA™ Purifier HPLC (GE Healthcare) equipped with a Resource-Q 6 mL anion exchange column (GE Healthcare). The column was equilibrated with Buffer A, after which the reaction was loaded onto the column, washed and eluted with a linear gradient to 30% Buffer B (20 mM HEPES, pH 7.5, 1 M NaCl, and 10 mM MgCl2). GDP-4-keto-6-deoxymannose eluted in 110 mM NaCl.
The activity of wild-type GDP-perosamine synthase was assessed using an HPLC-based assay whereby the decrease of GDP-4-keto-6-deoxymannose was measured over time. Reactions were carried out at room temperature and consisted of varying concentrations of GDP-4-keto-6-deoxymannose (0.010–0.200 mM) or GDP-4-keto-3,6-dideoxymannose (0.006–0.080 mM), L-glutamate (0.005–1.50 mM), 100 μM PLP, and 1.9 μg/mL GDP-perosamine synthase (for the GDP-4-keto-6-deoxymannose reaction) or 19 μg/mL GDP-perosamine synthase (for the GDP-4-keto-3,6-dideoxymannose reaction) in Buffer C (20 mM HEPES, pH 7.5, 110 mM NaCl, and 10 mM MgCl2). At appropriate time intervals, samples of the reaction were removed and treated with HCl to 80 mM. Carbon tetrachloride was added to the sample, which was subsequently mixed. The sample was then centrifuged, and the aqueous fraction was removed and diluted tenfold with Buffer A. The diluted sample was loaded onto the HPLC equipped with a 1 mL Resource-Q anion exchange column equilibrated with Buffer A. The sample was eluted with Buffer B, and the concentration of GDP-4-keto-6-deoxymannose in the sample was determined by comparing the observed GDP-4-keto-6-deoxymannose peak integration to that of a standard sample.
Reaction rates determined from the assay using the natural substrate were then fitted to Equation 1, whereas the rates from the assay using the unnatural substrate were fitted to equation 2.
| (Eq.1) |
| (Eq.2) |
The aminotransferase activity of K186A GDP-perosamine synthase was determined by monitoring the production of GDP-perosamine as described elsewhere (11). The mutant enzyme was incapable of producing GDP-perosamine, further supporting the role of Lys 186 as the catalytic acid/base.
Amino Donor Assay
Solutions containing 0.020 mM GDP-4-keto-6-deoxymannose, 0.100 mM PLP, 2 μg/mL GDP-perosamine synthase, and 10 mM of an amino donor (L-glutamate, L-glutamine, L-serine, L-aspartate, L-asparagine, L-alanine, L-glycine, or L-arginine) were incubated in Buffer C at room temperature. The reactions were terminated after 30 minutes and analyzed by HPLC as described above.
Results and Discussion
Amino Donor Assays
Previous studies on the GDP-perosamine synthases from E. coli O157:H7 (19) and Vibrio cholerae O1 (20) demonstrated differences in their amino donor requirements. E. coli GDP-perosamine synthase can only utilize L-glutamate as its amino donor, whereas the V. cholerae enzyme can utilize L-glutamate or L-glutamine, (albeit at a reduced rate). To determine the amino donor requirement for the C. crescentus GDP-perosamine synthase, reactions were set up in the presence of L-glutamate, L-glutamine, L-serine, L-aspartate, L-asparagine, L-alanine, L-glycine, or L-arginine. Of these, only the reaction with L-glutamate produced detectable levels of GDP-perosamine formation (data not shown). As noted in the introduction, we recently determined the molecular architecture of wild-type GDP-perosamine synthase in its internal aldimine form and complexed with α-ketoglutarate (11). The positioning of L-glutamate during the course of the catalytic reaction can thus be inferred from this structure. In particular, the guanidinium group of Arg 231 (from the second subunit) is clearly positioned to interact with the γ-carboxylate group of α-ketoglutarate (Fig. 1b). The fact that C. crescentus GDP-perosamine synthase can only use L-glutamate as its amino donor is not surprising. It is likely that L-glutamine and the other amino acids tested do not form strong enough interactions (if any at all) with Arg 231 to properly align the α-amino group for transfer to the PLP cofactor. Note that Arg 231 is also conserved in the E. coli and V. cholerae enzymes.
Steady-State Kinetics
As discussed below, we observed GDP-3-deoxyperosamine in the electron density maps for the wild-type enzyme, but were unable to observe a sugar ligand when wild-type crystals were soaked with the natural substrate, GDP-perosamine. As such, we suspected that the ability of GDP-perosamine synthase to utilize the alternative substrate was impaired relative to that of the natural substrate. To test this hypothesis, the steady-state kinetic parameters of GDP-perosamine synthase for both the natural (GDP-4-keto-6-deoxymannose) and unnatural (GDP-4-keto-3,6-dideoxymannose) substrates were determined.
An HPLC-based assay was used to measure the decrease of substrate over time. When the reaction rates at varying natural substrate concentrations were plotted, it was apparent that at high concentrations it was inhibiting enzymatic activity. Therefore, these data were fitted to Equation 1 that describes a ping-pong mechanism in which the sugar substrate is a competitive inhibitor versus glutamate. The effect of substrate inhibition was negligible in the reaction with the unnatural substrate, however. Thus, the data were fitted to Equation 2, which describes a standard ping-pong mechanism. The derived steady-state kinetic parameters are presented in Table 3.
Table 3.
Kinetic Parameters for C. crescentus GDP-perosamine Synthase.
| Km sugar (mM) | Km glutamate (mM) | Ki sugar (mM) | kcat (s−1) | kcat/Km sugar (M−1s−1) |
|---|---|---|---|---|
| GDP-4-keto-6-deoxymannose reaction | ||||
| 0.013 ± 0.006 | 4.6 ± 1.4 | 0.089 ± 0.034 | 2.7 ± 0.6 | (2.1 ± 1.1) × 105 |
| GDP-4-keto-3,6-dideoxymannose reaction | ||||
| 0.016 ± 0.003 | 0.13 ± 0.02 | N/A | 0.015 ± 0.001 | (9.4 ± 1.8) × 102 |
Although the Km for the unnatural substrate is unperturbed relative to the natural substrate, the kcat is lower by a factor of approximately 200. These parameters suggest that the unnatural substrate is accommodated in the active site nearly as well as the natural substrate, but the absence of the 3′-OH group affects the alignment of the sugar moiety, and thus it is not properly positioned for efficient catalysis. As a result of the decrease in kcat, less glutamate is required to saturate the enzyme, which leads to a drop in the Km for glutamate. It is this decrease in the glutamate Km that eliminates the substrate inhibition phenomenon observed with the natural substrate.
Active Site of K186A GDP-Perosamine Synthase in Complex with GDP-Perosamine
The crystals used in this investigation contained two dimers in the asymmetric unit. Given that the α-carbon positions for all four subunits in the asymmetric unit are very similar with root-mean-square deviations between the individual chains ranging from 0.17 Å to 0.26 Å, the following discussion refers only to the second dimer in the x-ray coordinate file.
Initially, crystals of the wild-type enzyme were soaked in GDP-perosamine, but the corresponding electron density maps never demonstrated convincing density for the nucleotide-linked sugar. Thus, in order to trap GDP-perosamine into the catalytic cleft of the enzyme, a site-directed mutant protein was constructed whereby the active site Lys 186 was changed to an alanine residue. Note that the α-carbons for the subunits of the wild-type enzyme and the K186A protein superimpose with a root-mean-square deviation of ~0.15 Å, thereby indicating no major structural perturbations resulting from the site-directed mutation.
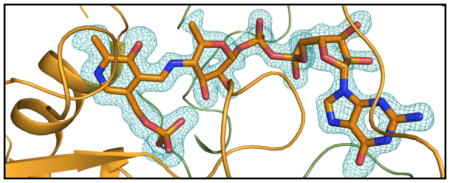
Shown in Figure 2a is the electron density for the PLP cofactor and GDP-perosamine. As can be seen, a Schiff base between the PLP cofactor and the nucleotide-linked sugar has been trapped in the active site region. This is referred to as the external aldimine. The pyranosyl moiety of GDP-perosamine adopts the B2,5 boat rather than the more typical chair conformation. Those amino acid residues located within ~3.5 Å of the external aldimine are shown in Figure 2b. The phosphate of the PLP moiety is anchored to the protein via the side chains of Thr 61 and Ser 181 from Subunit 3 and Asn 229 from Subunit 4. Additional hydrogen bonds to the phosphoryl group are provided by the backbone amide nitrogens of Gly 60 and Thr 61 and two water molecules. The pyranosyl group of GDP-perosamine is positioned into the active site of the enzyme via a hydrogen bond between the side chain of Asn 185 (Subunit 3) and the ring oxygen and between the 2′-OH group and a water. The side chain of Phe 183 (Subunit 3) abuts the pyranosyl ring. Arg 315 from Subunit 3 and Arg 220 and Tyr 221 from Subunit 4 provide most of the hydrogen bonding/electrostatic interactions between the protein and the pyrophosphoryl group of GDP-perosamine. The nucleotide ribose adopts the C2′-endo pucker, and its 3-OH group hydrogen bonds to the side chain of Glu 313. Finally, the guanine base interacts extensively with the protein via the side chains of Thr 29 and Ser 32, the backbone amide nitrogens of Ile 31 and Ser 32, and the carbonyl group of Thr 29, all of which are contributed by Subunit 4. In addition to these hydrogen bonds, the indole side chain of Trp 30 from Subunit 4 forms a parallel stacking interaction with the guanine base.
Figure 2.

The structure of K186A GDP-perosamine synthase in complex with GDP-perosamine. (a) Electron density corresponding to the bound nucleotide-linked sugar. The map was calculated with coefficients of the form (Fo - Fc), where Fo was the native structure factor amplitude and Fc was the calculated structure factor amplitude. Atoms corresponding to the PLP cofactor and the GDP-perosamine ligand were excluded from the coordinate file. The map was contoured at 3σ. (b) Close-up view of the active site with bound GDP-perosamine. Amino acids lying with ~3.5 Å of the PLP/GDP-perosamine complex are shown. Those residues highlighted in slate correspond to Subunit 3 in the x-ray coordinate file, whereas those displayed in green belong to Subunit 4. Residue labels ending with an asterisk correspond to Subunit 4. The PLP/GDP-perosamine complex is depicted in gold bonds. Potential hydrogen bonds are indicated by the dashed lines.
Active Site of Wild-type GDP-Perosamine Synthase in Complex with GDP-3-deoxyperosamine
Like that for the enzyme/GDP-perosamine complex, the crystals used to examine the enzyme/GDP-3-deoxyperosamine structure contained two dimers in the asymmetric unit. Given that the α-carbons for the four subunits superimpose with root-mean-square deviations between 0.22 Å to 0.30 Å, the following discussion refers only to the first dimer in the x-ray coordinate file. For this complex structure, it was not necessary to employ the K186A mutant form to trap the ligand in the active site. Shown in Figure 3a is the electron density observed in the active site cleft, which is consistent with GDP-3-deoxyperosamine. Given the resolution of the x-ray data (1.7 Å), however, it is not possible to completely rule out that a small fraction of the density corresponds to GDP-4-keto-3-deoxyperosamine or possibly the external aldimine. Again, the pyranosyl moiety of GDP-3-deoxyperosamine adopts the B2,5 boat conformation. A closeup view of the active site is presented in Figure 3b. As can be seen, the hydrogen bonding interactions that serve to position GDP-3-deoxyperosamine into the active site are virtually identical to those observed for the enzyme/GDP-perosamine complex.
Figure 3.

The structure of wild-type GDP-perosamine synthase in complex with GDP-3-deoxyperosamine. (a) Electron density corresponding to the bound nucleotide-linked sugar. The map was calculated with coefficients of the form (Fo - Fc), where Fo was the native structure factor amplitude and Fc was the calculated structure factor amplitude. Atoms corresponding to the GDP-3-deoxyperosamine ligand were excluded from the coordinate file. The map was contoured at 3σ. (b) Close-up view of the active site with bound GDP-3-deoxyperosamine. Amino acids lying with ~3.5 Å of the PLP/GDP-3-deoxyperosamine complex are shown. Those residues highlighted in slate correspond to Subunit 2 in the x-ray coordinate file, whereas those displayed in green belong to Subunit 1. Residue labels ending with an asterisk correspond to Subunit 1. GDP-3-deoxyperosamine is highlighted in gold bonds. Potential hydrogen bonds are indicated by the dashed lines. Ser 32* adopts two conformations, one of which is in the proper orientation for hydrogen bonding to the guanine base of the ligand.
One immediate question is why was it possible to observe the binding of GDP-3-deoxyperosamine to the wild-type enzyme in the crystalline lattice, but not GDP-perosamine, given the strikingly similar manners in which these two ligands are accommodated within the active site clefts of their respective proteins (Figures 2b and 3b). An answer to this comes from the above-described kinetic assays, which probed the forward reactions for the enzyme, namely the formation of GDP-perosamine from GDP-4-keto-6-deoxymannose (or GDP-3-deoxyperosamine from GDP-4-keto-3,6-dideoxymannose). Clearly, from the results presented in Table 3, the loss of the 3′-OH group from the substrate perturbs the catalytic efficiency of the enzyme by approximately two orders of magnitude. Given the caveat that the x-ray crystallographic analyses exploit the back reaction (Scheme 1), it still is possible to use the kinetic results to provide a possible answer to this question. In the case of GDP-perosamine binding which resulted in the formation of the external aldimine, the x-ray results clearly indicate the presence of a hydrogen bond between the pyranosyl 3′-OH group and a PLP phosphoryl oxygen (Figure 2b). Presumably this interaction also occurs when the substrate, GDP-4-keto-6-deoxymannose binds. Such an interaction cannot occur when GDP-4-keto-3,6,-dideoxymannose is the substrate. This perturbation of the hydrogen-bonding network defining the active site results in the unnatural substrate binding to the catalytic pocket in a manner that simply is not conducive for efficient catalysis, and this would explain why we observed GDP-3-deoxyperosamine but not the natural product GDP-perosamine in the wild-type crystal soaking experiments. Most likely, GDP-perosamine reacted quickly with the enzyme in the crystalline lattice to form the substrate, which then dissociated from the enzyme.
Comparison of GDP-Perosamine Synthase with ColD
ColD catalyzes a dehydration reaction at C-3′ whereas GDP-perosamine synthase catalyzes an amination reaction at C-4′. (Scheme 1). These are two very different chemical outcomes, and yet the overall three-dimensional architectures of ColD and GDP-perosamine synthase are strikingly similar such that their subunits superimpose with a root-mean-square deviation of 1.5 Å for 327 structurally equivalent α-carbons. What then are the structural features that allow for these different reactivities with the same substrate? A superposition of the ligand binding sites for GDP-perosamine synthase and ColD is presented in Figure 4. Note that the PLP and pyranosyl moieties of the ligands are accommodated in the respective active sites of GDP-perosamine synthase and ColD in nearly identical manners. The amino acids surrounding these groups are either identical or chemically similar with the one exception of Asn 188 in GDP-perosamine synthase which forms a hydrogen bond with the ring oxygen. This does not occur in ColD due to its replacement with Ser 187. The major differences in ligand binding between these two proteins begin at the pyrophosphoryl groups and are propagated through to the ribosyl moieties and the guanine bases (Figure 4).
Figure 4.

Superposition of the active site pockets for GDP-perosamine synthase and ColD. The nucleotide-linked sugar ligand and surrounding residues in GDP-perosamine synthase are highlighted in gold bonds, whereas those in ColD are presented in red. The top and bottom amino acid labels refer to GDP-perosamine synthase and ColD, respectively. Residues labeled with an asterisk refer to the second subunit of the dimer. Note that Met 27 in ColD adopts multiple conformations. X-ray coordinates for ColD were reported in (10) and deposited in the protein data bank (accession no. 3B8X).
There are three major insertions in ColD relative to GDP-perosamine synthase, but only one of these is near the active site cleft. Specifically, there is a large 17-residue insertion after Asn 220 in ColD, which results in an additional α-helical turn and the insertion of Arg 219 into the active site (Figure 4). The guanidinium group of Arg 219 participates in electrostatic interactions with the phosphoryl groups of the nucleotide-linked sugar with the net effect of shifting the pyrophosphoryl moiety of the ligand more towards the interior of the pocket relative to that observed in GDP-perosamine synthase. A strictly homologous residue for Arg 219 does not exist in GDP-perosamine synthase due to the lack of this 17-residue insertion. However, in GDP-perosamine synthase, there is an arginine located at position 220 that forms electrostatic interactions with the pyrophosphoryl groups of the nucleotide-linked sugar. Notably in ColD the homologous residue for this arginine is Ser 239 (Figure 4).
It was originally suggested in the initial study of ColD that perhaps the observed boat conformation of the sugar favored a dehydration rather than an amination reaction (10). The structures described in this report of the nucleotide-linked sugars bound to GDP-perosamine synthase, however, also show the boat conformations. As such, it can be concluded that the pyranosyl conformation has nothing to do with whether an enzyme functions as a dehydratase or an aminotransferase. Are the different reactions catalyzed by these two enzymes then simply the result of ColD having an active site histidine and GDP-perosamine synthase containing an active site lysine? Recent investigations from our laboratory demonstrate that this is also not the case. First, a site-directed mutant protein of ColD was constructed whereby His 188 was replaced with a lysine (as in GDP-perosamine synthase) (9). Functional assays demonstrated that this mutant form of ColD could not catalyze the dehydration reaction nor could it function to aminate the nucleotide-linked sugar. In a parallel study, a site-directed mutant protein of GDP-perosamine synthase was constructed whereby Lys 186 was replaced with a histidine. Again, the catalytic activity of the K186H mutant protein was below the detection limit of the assay.
From the investigations presented here, it is clear that a combination of subtle factors determine whether a particular enzyme functions as a simple aminotransferase or a more complicated dehydratase. These studies highlight that the enzymatic differences between ColD and GDP-perosamine synthase are not simply confined to the manner in which the pyranose is accommodated in the active site pocket. Likewise our experiments confirm that it is not simply the replacement of an active site lysine for a histidine that results in ColD being a dehydratase. The explanations for the differences in reactions catalyzed by these two enzymes most likely extend from the immediate active site to additional residues involved in accommodating the pyrophosphoryl and nucleotide moieties of the substrates (Figure 4). Site-directed mutagenesis experiments designed to test these hypotheses are presently underway.
Acknowledgments
Results in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by the University of Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. The insightful comments of Drs. Martin St. Maurice and W. W. Cleland are gratefully acknowledged. We thank Dr. James B. Thoden for assistance with the x-ray data collection.
Abbreviations
- GDP
guanosine 5′-diphosphate
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- MES
2-(N-morpholino)ethanesulfonic acid
- NADP+
nicotinamide adenine dinucleotide phosphate
- NMP
nucleotide monophosphate
- PLP
pyridoxal-5′-phosphate
- PMP
pyridoxamine 5′-phosphate
- Tris
tris-(hydroxymethyl)aminomethane
Footnotes
This research was supported in part by an NIH grant (DK47814 to H. M. H.)
X-ray coordinates have been deposited in the Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, N. J. (accession nos. 3DR4 and 3DR7).
References
- 1.Lerouge I, Vanderleyden J. O-antigen structural variation: mechanisms and possible roles in animal/plant-microbe interactions. FEMS Microbiol Rev. 2001;26:17–47. doi: 10.1111/j.1574-6976.2002.tb00597.x. [DOI] [PubMed] [Google Scholar]
- 2.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu HW, Thorson JS. Pathways and mechanisms in the biogenesis of novel deoxysugars by bacteria. Annu Rev Microbiol. 1994;48:223–256. doi: 10.1146/annurev.mi.48.100194.001255. [DOI] [PubMed] [Google Scholar]
- 4.Thibodeaux CJ, Melancon CE, Liu HW. Unusual sugar biosynthesis and natural product glycodiversification. Nature. 2007;446:1008–1016. doi: 10.1038/nature05814. [DOI] [PubMed] [Google Scholar]
- 5.He X, Liu HW. Mechanisms of enzymatic C-O bond cleavages in deoxyhexose biosynthesis. Curr Opin Chem Biol. 2002;6:590–597. doi: 10.1016/s1367-5931(02)00367-8. [DOI] [PubMed] [Google Scholar]
- 6.He XM, Liu HW. Formation of unusual sugars: mechanistic studies and biosynthetic applications. Annu Rev Biochem. 2002;71:701–754. doi: 10.1146/annurev.biochem.71.110601.135339. [DOI] [PubMed] [Google Scholar]
- 7.Stroeher UH, Karageorgos LE, Brown MH, Morona R, Manning PA. A putative pathway for perosamine biosynthesis is the first function encoded within the rfb region of Vibrio cholerae O1. Gene. 1995;166:33–42. doi: 10.1016/0378-1119(95)00589-0. [DOI] [PubMed] [Google Scholar]
- 8.Cook PD, Thoden JB, Holden HM. The structure of GDP-4-keto-6-deoxy-D-mannose-3-dehydratase: a unique coenzyme B6-dependent enzyme. Protein Sci. 2006;15:2093–2106. doi: 10.1110/ps.062328306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cook PD, Holden HM. A Structural Study of GDP-4-Keto-6-Deoxy-D-Mannose-3-Dehydratase: Caught in the Act of Geminal Diamine Formation. Biochemistry. 2007;46:14215–14224. doi: 10.1021/bi701686s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook PD, Holden HM. GDP-4-keto-6-deoxy-D-mannose 3-dehydratase, accommodating a sugar substrate in the active site. J Biol Chem. 2008;283:4295–4303. doi: 10.1074/jbc.M708893200. [DOI] [PubMed] [Google Scholar]
- 11.Cook PD, Holden HM. GDP-Perosamine Synthase: Structural Analysis and Production of a Novel Trideoxysugar. Biochemistry. 2008;47:2833–2840. doi: 10.1021/bi702430d. [DOI] [PubMed] [Google Scholar]
- 12.Jansonius JN. Structure, evolution and action of vitamin B6-dependent enzymes. Curr Opin Struct Biol. 1998;8:759–769. doi: 10.1016/s0959-440x(98)80096-1. [DOI] [PubMed] [Google Scholar]
- 13.Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 14.Paiardini A, Bossa F, Pascarella S. Evolutionarily conserved regions and hydrophobic contacts at the superfamily level: The case of the fold-type I, pyridoxal-5′-phosphate-dependent enzymes. Protein Sci. 2004;13:2992–3005. doi: 10.1110/ps.04938104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 16.Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr Sect D. 1999;55(Pt 2):484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 17.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 18.Tronrud DE, Ten Eyck LF, Matthews BW. An efficient general-purpose least-squares refinement program for macromolecular structures. Acta Crystallogr Sect A. 1987;43:489–501. [Google Scholar]
- 19.Zhao G, Liu J, Liu X, Chen M, Zhang H, Wang PG. Cloning and characterization of GDP-perosamine synthetase (Per) from Escherichia coli O157:H7 and synthesis of GDP-perosamine in vitro. Biochem Biophys Res Commun. 2007;363:525–530. doi: 10.1016/j.bbrc.2007.08.184. [DOI] [PubMed] [Google Scholar]
- 20.Albermann C, Piepersberg W. Expression and identification of the RfbE protein from Vibrio cholerae O1 and its use for the enzymatic synthesis of GDP-D-perosamine. Glycobiology. 2001;11:655–661. doi: 10.1093/glycob/11.8.655. [DOI] [PubMed] [Google Scholar]
- 21.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]