

Abstract
The thyroid-stimulating hormone receptor (TSHR) is a heterotrimeric guanine nucleotide–binding protein (G protein)– coupled receptor (GPCR). Autoimmune hyperthyroidism, commonly known as Graves’ disease (GD), is caused by stimulating autoantibodies to the TSHR. We previously described TSHR-specific antibodies (TSHR-Abs) in GD that recognize linear epitopes in the cleavage region of the TSHR ectodomain (C-TSHR-Abs) and induce thyroid cell apoptosis instead of stimulating the TSHR. We found that C-TSHR-Abs entered the cell through clathrin-mediated endocytosis but did not trigger endosomal maturation and failed to undergo normal vesicular sorting and trafficking. We found that stimulating TSHR-Abs (S-TSHR-Abs) activated Gαs and, to a lesser extent, Gαq but that C-TSHR-Abs failed to activate any of the G proteins normally activated in response to TSH. Furthermore, specific inhibition of G proteins in the presence of S-TSHR-mAbs or TSH resulted in a similar failure of endosomal maturation as that caused by C-TSHR-mAbs. Hence, whereas S-TSHR-mAbs and TSH contributed to normal vesicular trafficking of TSHR through the activation of major G proteins, the C-TSHR-Abs resulted in GRK2- and β-arrestin-1–dependent biased signaling, which is interpreted as a danger signal by the cell. Our observations suggest that the binding of antibodies to different TSHR epitopes may decrease cell survival. Antibody-induced cell injury and the response to cell death amplify the loss of self-tolerance, which most likely helps to perpetuate GPCR-mediated autoimmunity.
INTRODUCTION
Heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptors (GPCRs) bind to various signaling molecules, yet they share a common architecture that has been conserved over the course of evolution. The thyroid-stimulating hormone receptor (TSHR) is a GPCR and a major autoantigen in Graves’ disease (GD), an autoimmune disorder that leads to thyroid overactivity through the action of conformationally dependent autoantibodies specific for the TSHR. Ligand binding to the TSHR initiates its coupling to G proteins, which become activated by the dissociation of Gα and Gβγ subunits, including Gi/o, Gαq/11, and Gα12/13 subunits (1–5). Hence, agonist binding to the TSHR activates a number of different G proteins and results in receptor phosphorylation (6, 7) through GPCR kinases (GRKs). This, in turn, leads to an enhanced association between the TSHR and β-arrestin. β-Arrestin acts as an adaptor protein, which modulates the signal most commonly resulting in receptor down-regulation (8–10). Despite the identification of clathrin-coated pits (CCPs) at the cell surface more than 30 years ago, their function in endocytosis has been elusive (11–13). However, CCPs are a major entry portal for GPCRs whereby clathrin assembles to form a lattice around the invaginating buds that have captured endocytic cargo (14, 15). Signaling by transmembrane receptors occurs at the cell surface and throughout the endocytic pathway, which is responsible for recycling the receptor. Signaling from the cell surface may differ in magnitude from the downstream output through positive or negative intracellular modulation of the signal (15, 16). After the internalized receptors are delivered to endosomes, GPCRs may be rapidly recycled, resulting in resensitization, or they may be targeted for lysosomal degradation (down-regulation) (17, 18). Furthermore, GPCRs may continue to signal in a G protein–independent manner after their delivery to endosomes (19).
Antibodies that are specific for the TSHR (TSHR-Abs) in patients with autoimmune thyroid disease may be classified as “stimulating,” “blocking,” or “neutral” in terms of their influence on the TSHR, especially in patients with GD (20–32). Stimulating TSHR-Abs (S-TSHR-Abs) induces thyroid epithelial cell proliferation through Gαs- and Gαq/11-coupled signaling pathways, whereas blocking antibodies inhibit the action of TSH but may also act as weak agonists (20–23). In contrast, the linear (neutral) type of TSHR-Abs recognizes the hinge region of the TSHR, including the “cleavage” region (C-TSHR-Abs) of the receptor ectodomain (amino acid residues 316 to 366). Although they are called neutral because they are unable to stimulate the generation of the second messenger cyclic adenosine monophosphate (cAMP) through the activation of Gαs, these antibodies are capable of initiating a cascade of signaling events that lead to programmed cell death (22, 23, 33–35).
The frequency of C-TSHR-Abs in GD ranges from 30 to 90% based on linear epitope binding to known amino acid residues (23, 24, 33, 35–40). Although the clinical importance of these antibodies has not been well studied, we characterized their pathophysiological role in an in vitro thyrocyte culture system (22, 33). These data showed that C-TSHR-Abs induced apoptosis through multiple stress signaling pathways (22, 23, 33, 35) and that the failure to sustain key signaling pathways (33) led to thyrocyte death. Here, we asked why and how these antibodies fail to maintain thyrocyte homeostasis and have extended our study to begin to dissect the molecular mechanisms involved. Through experiments with a rat thyrocyte model system, we found that C-TSHR-Abs activated the TSHR through clathrin-dependent endocytosis but that the engaged receptors failed to undergo the normal vesicular trafficking and lysosomal degradation processes. C-TSHR-Abs failed to activate any of the major G proteins known to couple to the TSHR, but instead, these antibodies stimulated β-arrestin-1– and GRK-dependent biased signaling. Whereas S-TSHR-Abs contributed to thyrocyte homeostasis by stimulating endocytic maturation and initiating multiple signaling cascades, C-TSHR-Abs induced cell stress and apoptosis as a result of the perturbation of key signaling molecules. These findings suggest that cellular apoptosis induced by C-TSHR-Abs may orchestrate overt intrathyroidal, intraorbital, or epidermal inflammatory autoimmune reactions in GD.
RESULTS
C-TSHR-mAb stimulates β-arrestin–biased signaling in thyrocytes
A feature common to most GPCRs is the cyclical process of signaling, desensitization, internalization, resensitization, and recycling to the plasma membrane. Although C-TSHR–mAbs (monoclonal antibodies) bind to specific sites on the TSHR cleavage region, they do not stimulate cAMP signaling (24), whereas both TSH and S-TSHR-mAbs stimulate cAMP generation. Therefore, to evaluate the influence of C-TSHR-mAbs on TSHR activity, we examined the signaling complex by assessing the amount of GRK2 in rat thyrocytes. The GRKs constitute a group of protein kinases (seven members in mammals) that specifically recognize and phosphorylate agonist-activated GPCRs (41, 42). Upon phosphorylation by GRKs and binding to β-arrestin-1, most GPCRs undergo internalization into clathrin-coated vesicles (CCVs), which leads to the ubiquitination and dephosphorylation of both GRK2 and β-arrestin-1 (43). We found that both C-TSHR-mAb and S-TSHR-mAb led to substantial increases in the amount of phosphorylated GRK2 (pGRK2) with only minor increases in β-arrestin-1 abundance (Fig. 1, A and B); however, consistent with the ability of β-arrestin-1 to activate extracellular signal–regulated kinases 1 and 2 (ERK1/2) (43), we found that ERK1/2 and protein kinase C (PKC) were phosphorylated in a dose-dependent manner, although these effects were not sustained over a 24-hour period (Fig. 1, C and D). In addition to ERK1/2, Akt, and PKC activities, previous studies showed that GRK2- and β-arrestin-1–biased signaling cascades are activated in response to GPCR ligands (44, 45). In our protein array studies, we found that both C-TSHR-mAbs and S-TSHR-mAbs also substantially activated protein phosphatase 2A, p38 mitogen-activated protein kinase (MAPK), the p65 subunit of nuclear factor κB (NF-κB), and the δ isoform of PKC (PKC-δ) in the same thyrocytes over 1 hour (Fig. 1E), whereas the activities of p38, Rho-associated kinase 2 (ROCK2), glycogen synthase kinase 3α, NF-κB p65, and PKC-δ in TSH-stimulated cells were reduced compared to those in cells treated with isotype control antibody (Fig. 1E). C-TSHR-mAbs alone activated ROCK2 robustly. TSH and both types of antibody failed to stimulate any substantial phosphorylation of the known biased signaling molecules cofilin and c-Jun N-terminal kinase (Fig. 1E). These findings suggest that TSHR-mAbs stimulate GRK2- and β-arrestin-1–biased signaling at the TSHR to activate an array of signaling pathways. Because S-TSHR-mAbs, but not C-TSHR-mAbs, stimulated the generation of cAMP, the biased agonism through GRK2- and β-arrestin-1– dependent signaling likely does not involve G proteins. These data suggest that C-TSHR-mAbs represent bona fide biased ligands.
Fig. 1. C-TSHR-mAb stimulates biased signaling in thyrocytes through the activation of GRK2 and β-arrestin-1.
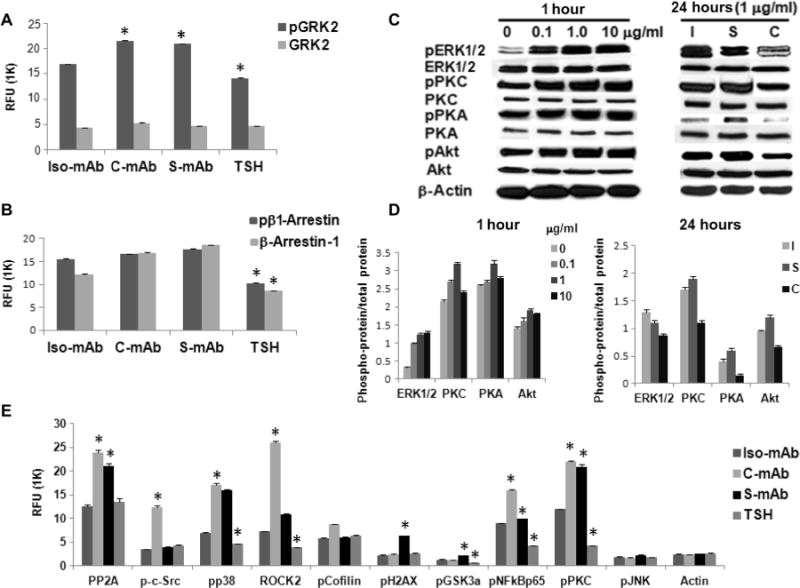
(A) Thyrocytes were treated with thyroid-stimulating hormone (TSH) (1 mU/ml) or with isotype control monoclonal antibody (Iso-mAb), C-TSHR-mAb (C-mAb), or S-TSHR-mAb (S-mAb) (all at 1 μg/ml) for 1 hour. The relative abundances of total heterotrimeric guanine nucleotide–binding protein (G protein)–coupled receptor kinase 2 (GRK2) and phosphorylated GRK2 (pGRK2) were then measured by proteomic array. The relative fluorescence unit (RFU) from bound antibody was then measured. Data are means ± SEM of three independent experiments. *P < 0.02 compared to isotype control mAb–treated cells. (B) Cells treated as described in (A) were analyzed to determine the relative amounts of total and phosphorylated β-arrestin-1 (pβ1-Arrestin). Data are means ± SEM of three independent experiments. *P < 0.006 compared to isotype control mAb–treated cells. (C) Thyrocytes were treated for 1 hour with the indicated concentrations of C-TSHR-mAb in a dose-dependent manner (left) or for 24 hours (right) compared with isotype control mAb (I) and S-TSHR-mAb (S) (all at 1 μg/ml) before being analyzed by Western blotting with antibodies against the indicated proteins. Western blots are representative of three experiments. (D) Densitometric quantification of the Western blots shown in (C). Data are presented as the ratio between the abundances of the indicated total proteins and phosphorylated proteins. β-Actin was used as a loading control. *P < 0.05 compared to untreated or isotype control mAb. (E) Thyrocytes were treated with TSH (1 mU/ml) or with isotype control mAb, C-TSHR-mAb, or S-TSHR-mAb (all at 1 μg/ml) for 1 hour. PP2A, protein phosphatase 2A; pp38, phosphorylated p38; pCofilin; phosphorylated cofilin; pH2AX, phosphorylated histone 2AX; pGSK3a, phosphorylated glycogen synthase kinase 3α; pNFkBp65, phosphorylated NF-κB p65; pJNK, phosphorylated c-Jun N-terminal kinase. The relative abundances of the indicated proteins were then determined by protein array. Data are means ± SEM of three independent experiments. *P < 0.05 compared to isotype control mAb–treated cells.
C-TSHR-mAb stimulates clathrin-mediated endocytosis of the TSHR but without normal vesicular trafficking and sorting
By measuring fluorescently labeled antibody-induced endocytosis and antibody clearance in FRTL-5 thyrocytes, we evaluated the clathrin- mediated endocytosis (CME) and vesicular trafficking of C-TSHR-mAb. Selective inhibitors of CME [Pitstop and phenylarsine oxide (PAO)] and dynamin (dynasore) confirmed that C-TSHR-mAb entered thyrocytes through CME; entry was not influenced by inhibitors of caveolin (genistein) or macropinocytosis (amiloride) (Fig. 2, A to F). In the presence of inhibitors of clathrin and dynamin, there was no endocytosis of labeled C-TSHR-mAb, S-TSHR-mAb, or TSH. There was almost no uptake of labeled TSHR-mAb or TSH by the treated cells. Fluorescence intensity (FI; in arbitrary units) analyses revealed substantially reduced uptake of both labeled TSHR-mAbs and labeled TSH when cells were treated with Pitstop, rottlerin, dynasore, or PAO (Fig. 2G). Furthermore, rottlerin, a nonselective inhibitor of PKC-δ, also inhibited CME (Fig. 2, B and G).
Fig. 2. C-TSHR-mAb induces clathrin-dependent endocytosis but fails to undergo normal vesicular trafficking and sorting.
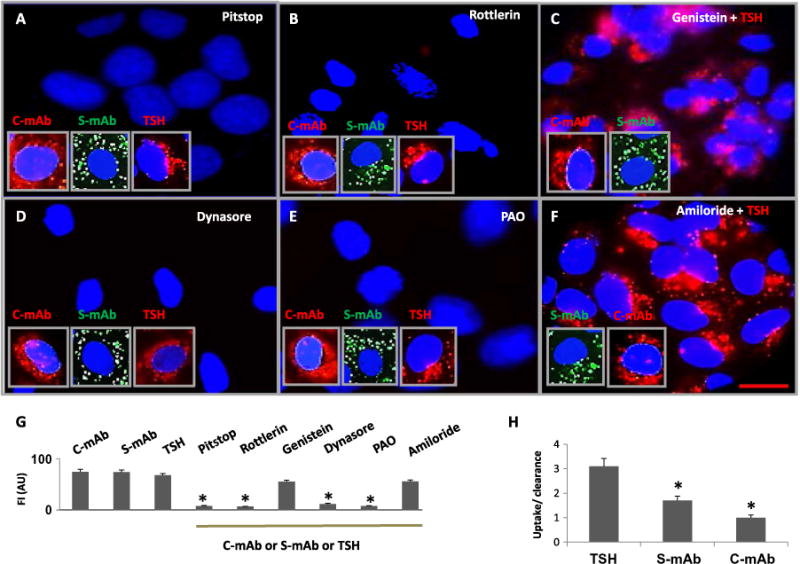
(A to F) Representative live-cell images of thyrocytes are shown in the presence of the indicated labeled TSHR-mAbs or TSH. In addition, the cells were treated with Pitstop (to inhibit clathrin; A), rottlerin [non-selective δ isoform of PKC (PKC-δ) inhibitor; B], genistein (caveolin inhibitor; C), dynasore (dynamin inhibitor; D), PAO (endocytosis inhibitor; E), or amiloride (micropinocytosis inhibitor; F). Endocytosis of labeled C-TSHR-mAb (red, inset), S-TSHR-mAb (green, inset), and TSH (red, inset) was observed. Scale bar, 100 μm. (G) Quantitative evaluation of internalized or nonendocytosed antibodies or TSH was performed in live cells. Data are means ± SEM of three independent experiments with three or more microscopic images analyzed per experiment. *P < 0.006 compared to TSHR-mAb–, TSH-, genistein-, and amiloride-treated cells. FI, fluorescence intensity; AU, arbitrary units. (H) The ratio of the relative abundance of labeled antibody or TSH taken up by the cells (endocytosed) to that cleared by the cell (sorted or secreted into the culture medium) was determined. Data are means ± SEM of three independent experiments. *P < 0.01 compared to TSH.
Because thyrocytes internalize ligand or antibodies by CME and subsequent vesicular sorting, we then investigated how thyrocytes cleared the endocytosed C-TSHR-mAb. After induction of endocytosis with labeled TSHR antibodies or TSH, we quantified the amounts of labeled TSH and antibodies in the cell culture medium over time, which indicated the amount of exocytosis occurring. FI analyses indicated that the cells treated with TSH were more rapidly cleared of labeled ligand than those treated with either S-TSHR-mAb or C-TSHR-mAb, especially when the endocytosis/exocytosis ratio was determined. After 3 hours, more of the TSHR-mAbs than of TSH remained inside the treated cells (Fig. 2H). Furthermore, thyrocytes treated with C-TSHR-mAb cleared less of the antibody than did cells treated with S-TSHR-mAb (Fig. 2H), suggesting that the defect in the sorting mechanisms was specific to C-TSHR-mAbs.
C-TSHR-mAb fails to induce endosomal maturation in thyrocytes
To examine the intracellular sorting mechanisms involved, we first analyzed the endosomal response to TSHR activation. Endosomes are a major intracellular compartment involved in generating sustainable GPCR signaling cascades (12, 13, 46). Once the agonist binds to the TSHR ectodomain, the receptor enters CCVs by CME, as described earlier, which then stimulates endosomal maturation (46, 47). To evaluate endosome formation, we treated thyrocytes with labeled TSHR-mAbs or TSH and examined them for the presence of early endosomes (EEs) and late endosomes (LEs). We found that the binding of S-TSHR-mAb to the TSHR induced endosomal formation (Fig. 3A). We then analyzed multiple live-cell images for the formation of EE or LE spots and also analyzed their colocalization in merged images. Whereas the S-TSHR-mAb stimulated the formation of both EEs and LEs, the C-TSHR-mAb failed to initiate normal endosome formation (Fig. 3, B and C). Whereas clusters of S-TSHR-mAb colocalized with EEs, fewer and deformed endosomes were induced by C-TSHR-mAb (Fig. 3, A to C). In some cases, the endosomes formed in response to C-TSHR-mAb appeared to be dilated and distorted or collapsed, lacking their normal structure. These data suggest that the C-TSHR-mAb was unable to induce normal EE formation and thus failed to undergo normal vesicular trafficking and sorting, consistent with the earlier observations from antibody uptake and clearance assays.
Fig. 3. C-TSHR-mAb fails to induce endosomal maturation in thyrocytes.
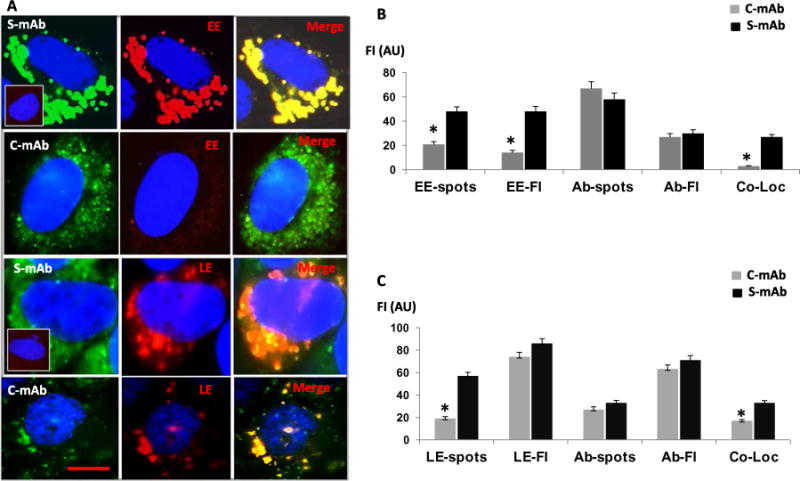
(A) Representative live-cell images of early endosomes (EE; red) induced by S-TSHR-mAb (green) or C-TSHR-mAb (green). The top panel inset shows that no induction of EE formation occurred in the absence of antibody. Representative live-cell images of late endosomes (LE; red) are shown in the bottom two panels. Inset (third row, left column) shows the lack of induction of LE formation in the absence of antibody. Scale bar, 100 μm. (B) EE-spots, EE-FI, Ab-spots, Ab-Fl, and Co-Loc (colocalization) by TSHR-mAbs were quantified by image analysis software and expressed in arbitrary units. *P < 0.005 compared to S-mAb. (C) LE-spots, LE-FI, Ab-spots, Ab-Fl, and Co-Loc in TSHR-mAb–treated thyrocytes were also quantified. Data are means ± SEM of three independent experiments with at least three images per experiment analyzed. *P < 0.005 compared to S-mAb.
C-TSHR-mAb fails to undergo lysosomal degradation
Lysosomes are organelles in the vesicular trafficking systems that degrade endosomal cargoes (48–50). Because the C-TSHR-mAb showed dysfunctional induction of EEs, it was likely that it would also fail to enter the lysosomes. We investigated lysosomal trafficking of TSHR antibodies and TSH with a lysosomal tracker (Lamp1) (Fig. 4A). As predicted, lysosomes failed to mature in C-TSHR-mAb–treated cells. Those still present were reticular-like and devoid of their typical three-dimensional structure. In contrast, S-TSHR-mAb and TSH induced distinctive lysosomes from day 1 (Fig. 4A). Accumulation of labeled C-TSHR-mAb became prominent on day 3 and formed globular-like structures by day 5 when the nucleus of the cells became fragmented, as part of the cell death process (apoptosis) induced by C-TSHR-mAb. As shown earlier, such thyrocytes were unable to induce rapid exocytosis for clearing the internalized cargoes, whereas S-TSHR-mAb– or TSH-treated cells demonstrated peripheral localization within the lysosomes by day 3 and were almost all cleared by day 5. Evaluations on consecutive days revealed a decline in lysosomal protein abundance under all three conditions, but this decline lagged in the C-TSHR-mAb–treated cells (Fig. 4B). Labeled C-TSHR-mAb accumulated in the cytoplasm by day 3 (Fig. 4A). Co-localization of C-TSHR-mAb with lysosomes was almost absent, in contrast with that of S-TSHR-mAb or TSH (Fig. 4C).
Fig. 4. C-TSHR-mAb fails to undergo lysosomal degradation.
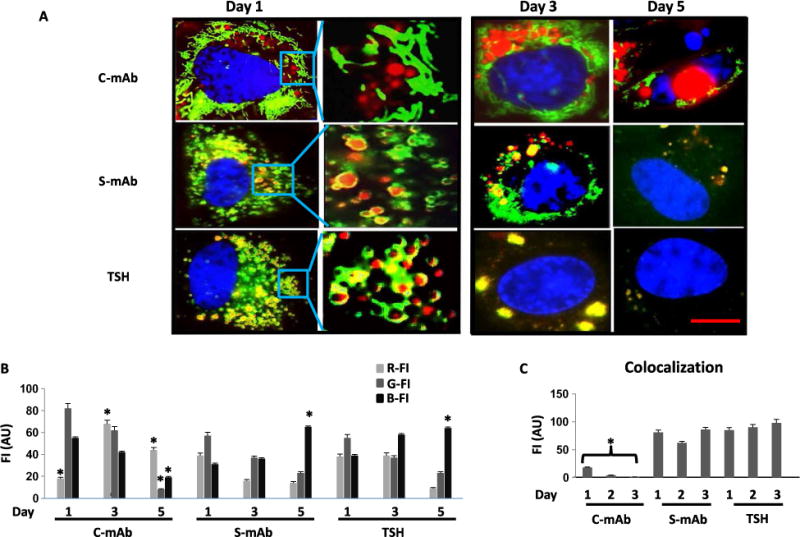
(A) Microscopic analysis of lysosomal three-dimensional structure was visualized in round red organelles encircled in yellow (colocalization of the indicated TSHR-mAbs or TSH; red with green). Accumulated C-TSHR-mAb (red) in the perinuclear region at day 3 is shown in the top panel. DNA fragmentation at day 5 (apoptosis, blue) is shown in the top right panel. Clearance of endocytosed labeled S-TSHR-mAb or TSH (red) on day 5 is accompanied by the lack of DNA fragmentation. Representative live-cell images of lysosomes (green) and labeled TSHR-mAbs (red) or TSH (red) are shown in the insets at higher magnifications. Images are representative of three independent experiments with at least three images analyzed per experiment. Scale bar, 100 μm. (B) Quantitative analysis of the representative images shown in (A) was measured as red FI (R-FI), green FI (G-FI), and blue FI (B-FI) in arbitrary units. Data are means ± SEM of three independent experiments. *P < 0.002 compared to S-mAb or TSH. (C) The colocalization of lysosomes (green) with the indicated TSHR-mAbs (red), which is represented as yellow fluorescence, was also quantified. Data are means ± SEM of three independent experiments. *P < 0.004 compared to S-mAb or TSH.
C-TSHR-mAb induces fewer vesicular proteins than does S-TSHR-Ab or TSH
To help evaluate the lack of vesicular trafficking by C-TSHR-mAb, we also examined protein localization by immunohistochemistry (IHC). The amounts of clathrin light chain (CLTA), EE (EEA1), lysosome (Lamp1), and cathepsin E (CTSE), which are all found in vesicles, were all reduced in the C-TSHR-mAb–treated cells compared to those in S-TSHR-mAb– or TSH-treated cells. CLTA and EEA1 localization in S-TSHR-mAb– or TSH-treated cells was mostly perinuclear (Fig. 5A, left and middle columns), whereas distinctive spots scattered throughout the cytoplasm were observed in C-TSHR-mAb–treated cells (Fig. 5A, right column). These histologic observations were quantified (Fig. 5B) and confirmed by LI-COR immunoblot assay (Fig. 5, C and D). To determine whether these effects, induced by C-TSHR-mAb, were also capable of reducing the abundance of other vesicular proteins, we tested for a Golgi protein (GOPC) and an exocytosis-associated protein (Rab11b) and again observed lower protein abundance when compared with those in control-TSHR-mAb–treated cells (fig. S1). Furthermore, syntaxin binding protein 6 (STXbp6), which binds to components of the SNARE (soluble N-ethylmaleimide–sensitive factor attachment protein receptor) complex for fusion of synaptic vesicles, showed the opposite effect (fig. S1). Together, these findings suggest that vesicular dysregulation in thyrocytes is induced by the C-TSHR-mAb.
Fig. 5. C-TSHR-mAb induces fewer vesicular proteins.
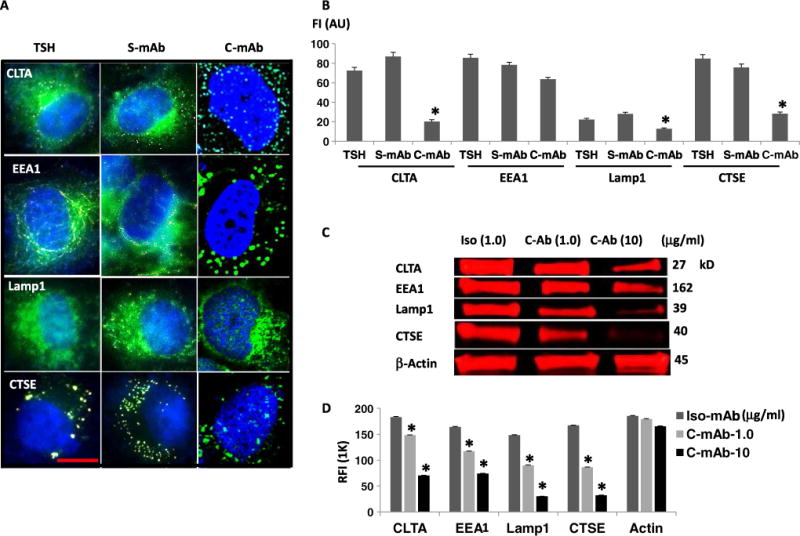
(A) Immunohistochemical analysis of clathrin light chain (CLTA), EE (EEA1), LE (Lamp1), and cathepsin E (CTSE) after the exposure of thyrocytes to TSH or the indicated antibodies for 24 hours. Scale bar, 100 μm. Images are representative of three independent experiments. (B) The relative abundances of CLTA, EEA1, Lamp1, and CTSE in cells treated with the indicated reagents were then quantified by image analysis and expressed as FI in arbitrary units. Data are means ± SEM of three independent experiments. *P < 0.03 compared to S-mAb. (C) LI-COR Western blot analysis of cells after exposure to isotype control mAb [hamster immunoglobulin G2 (IgG2), κ chain] or the indicated concentrations of C-TSHR-mAb. Western blots are representative of three independent experiments. (D) Quantification of the relative FI (RFI) of the bands in the Western blots represented in (C) with Image Studio software. Data are means ± SEM of three independent experiments. *P < 0.008 compared to the isotype control mAb.
C-TSHR-mAb fails to activate major G proteins or sustain signaling
The defects in multiple cellular organelles and signaling events induced by C-TSHR-mAb suggested that the failure of upstream effectors, G proteins, may be involved, as indicated by the known lack of cAMP generation. In luciferase reporter assays [Chinese hamster ovary (CHO)–TSHR overexpression system] for multiple G protein activities (51), C-TSHR-mAb failed to activate any of the G proteins examined (Fig. 6, A to E). Both S-TSHR-mAbs and TSH activated Gαs, Gαq, or both more robustly than they did Gβγ or Gα12 (Fig. 6A). As expected, TSH induced the characteristic activation of all four G proteins in a dose-dependent manner and was substantially different from C-TSHR- mAb (Fig. 6, B to E). The amount of Gβγ activity was substantially lower in response to higher concentrations of C-TSHR-mAb compared to those of TSH or S-TSHR-mAb (Fig. 6C). The S-TSHR-mAb also induced statistically significantly greater Gαq activity than did C-TSHR-mAb (at 100 μg/ml) but much less than did TSH (Fig. 6D). To ascertain the status of selected downstream effectors in another cell type, we also demonstrated the dose-dependent activation of PKC and Akt by C-TSHR-mAb in parallel studies with CHO-TSHR cells, which, although not sustained, was again indicative of G protein–independent signaling (fig. S2, A and B). As expected, the C-TSHR-mAb also failed to generate cAMP in the CHO-TSHR cells (fig. S2C).
Fig. 6. C-TSHR-mAb fails to activate major G proteins.
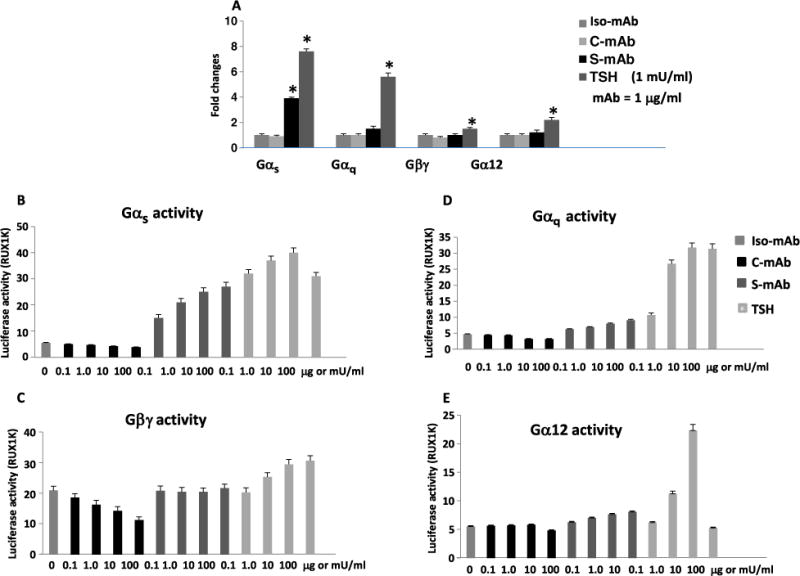
(A) G protein activities were detected by dose-dependent luciferase activity assays in Chinese hamster ovary (CHO)–TSHR cells. The activities of the indicated G proteins (Gαs, Gαq, Gβγ, and Gα12) were measured after treatment of the cells with the indicated TSHR-mAbs (1 μg/ml) or TSH (1 mU/ml). Data are presented as the fold change in luciferase activity and are means ± SEM of three independent experiments. *P < 0.0001 compared to S-TSHR-mAb or TSH. (B to E) Dose-dependent activation or inhibition of the indicated G proteins by the indicated antibodies or TSH was determined by measurement of luciferase activity in the treated CHO-TSHR cells. Data are means ± SEM of three independent experiments.
G protein activation is critical for the formation of endosomes
To determine the mechanism for the failure of endosome formation in C-TSHR-mAb–treated cells, we performed a series of investigations of live thyrocytes using Rab5a protein as an EE indicator. After a 1-hour incubation with labeled S-TSHR-mAb, EE formation was induced and they colocalized with the antibody (Fig. 7A). To determine whether this was secondary to upstream effector activation, we performed experiments with suramin, a known Gα-selective inhibitor (52), which we found prevented S-TSHR-mAb–induced EE formation but not antibody internalization (Fig. 7A). Similarly, Baf-A1, a vacuolar proton– adenosine triphosphatase inhibitor (53), and guanosine diphosphate (GDP), a guanosine 5′-triphosphate (GTP) inhibitor that prevents G protein activation, all prevented S-TSHR-mAb–induced EE formation (Fig. 7A). These data suggest that EE formation in response to S-TSHR-mAb depended on G protein activation. In contrast, galleon (54), a specific Gβγ inhibitor, did not substantially inhibit EE generation (Fig. 7A). These results suggest that endosome formation was G protein–dependent and critical for subsequent vesicular trafficking and sorting. To clarify which G protein was responsible for the induction of endosome formation, we used a TSHR-selective antagonist (anatag3) that inhibits Gαs activation and cAMP generation (55) and a TSHR-nonselective Gαq/11 inhibitor (YM-254890) (56). Surprisingly, of these two, only the TSHR-selective inhibitor of Gαs-cAMP signaling blocked the formation of endosomes (Fig. 7A). The TSHR- nonselective G protein inhibitor, SCH 202676 hydrobromide (57), which inhibits agonist- and antagonist-induced G protein activation, also blocked endosome formation (Fig. 7A). Quantitative FI analyses confirmed these effects on S-TSHR-mAb–induced EE formation in the presence of the pharmacological G protein inhibitors (Fig. 7B). Similar data on G protein activities were obtained from experiments performed with reporter CHO-TSHR cells (fig. S3). These observations suggest that G proteins induce endosomal signaling for thyrocyte homeostasis by alternative mechanisms from the cAMP-PKA–CREB (cAMP response element–binding protein) cascade.
Fig. 7. Lack of G protein activation fails to induce endosomes.
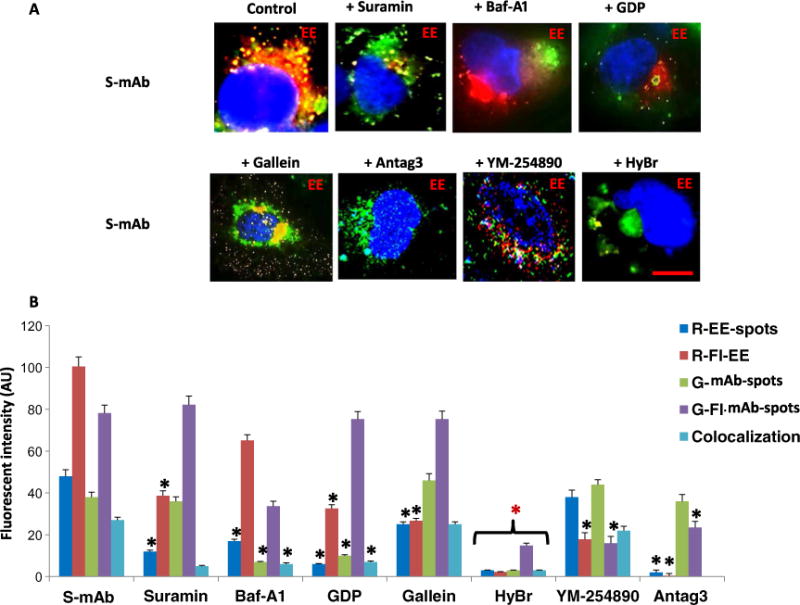
(A) Perturbation of the S-TSHR-mAb–induced formation of early endosomes (red) by the G protein disruptors suramin, hydrobromide (HyBr), and Antag3. Note that Baf-A1 and guanosine diphosphate (GDP) induced malformed EE formation. Antibodies are labeled in green. Scale bar, 100 μm. Images are representative of three independent experiments. (B) Quantification of the images represented in (A) was performed by measuring the FI of red EE-spots (R-EE-spots), R-FI-EE, green mAb-spots (G-mAb-spots), G-Fl mAb-spots, and colocalization. Data are means ± SEM of three experiments with at least three images analyzed per experiment. *P < 0.003 compared to S-mAb.
The Ras-PI3K-Rac pathway downstream of G proteins is a key pathway for vesicle formation
The known major signaling cascades activated by the TSHR include the cAMP-PKA-CREB pathway through Gαs, the phosphatidylinositol 3-kinase (PI3K)–PKC-MAPK-Ca+ signaling pathway through Gαq or Gβγ, and Rho signaling through Gα12 (22–24, 33). To determine which signaling molecules downstream of G proteins were responsible for initiating normal endosome formation, we performed experiments in which thyrocytes were pretreated with pathway-selective pharmacological inhibitors of PI3K (wortmannin), c-Raf (Kobe2602), Ras (FTaseII), or Rac (NSC 23766). Both S-TSHR-mAb and TSH failed to induce the EE and LE formation (fig. S4A). Although some malformed endosomes were produced, labeled S-TSHR-mAb did not colocalize with these endosomes. These inhibitors prevented the uptake of labeled S-TSHR-mAb or TSH, whereas inhibitors of cAMP (SQ22536), PKA (H89), ERK1/2 (PD98059), PKC (Go 6983), and Rho (Y-27632) had no such effect on EE and LE formation (fig. S4, A to C). Because some of these inhibitors blocked endosome formation in response to S-TSHR-mAb, these data suggest a role for Ras, PI3K, and Rac in this process. To further confirm that Ras, PI3K, and Rac were involved in the induction of endosome formation, we performed proteomic array analyses. We found that C-TSHR-mAb activated PI3K to a lesser degree than did either S-TSHR-mAb or TSH and that it failed to activate the Ras proteins Raf1, pRaf1, B-Raf, Rac, or pRac/CDC42 (fig. S4D).
Because Ras family proteins play important roles in cellular homeostasis, including the regulation of vesicular trafficking and sorting and cellular proliferation and differentiation, we still considered that Ras was the likely major effector orchestrating endosomal and lysosomal trafficking. To help evaluate this, we measured the active form of Ras (Ras-GTP) by immunoprecipitation with Ras-GTP-mAb in the lysates of treated cells and found that C-TSHR-mAb inhibited Ras-GTP formation more than did S-TSHR-mAb and TSH (fig. S4, E and F), which is consistent with Ras being one of the main effectors for endosome formation. We then applied pharmacological inhibitors to our CHO-TSHR reporter cells. We found that inhibitors of H-Ras, Ras, PI3K, and PAO, but not Rac, had suppressive effects on all G protein activities in a dose-dependent manner (figs. S5 and S6). Rac and Rho inhibitors substantially suppressed the activities of Gαq, Gβγ, and Gα12, whereas inhibitors of cAMP generation, PKA, and ERK1/2 did not influence any of the G proteins examined. Besides the Rac inhibitor, these results revealed similar effects on endosome formation in thyrocytes. Our proposed model (fig. S7) suggests that C-TSHR-mAb blocks multiple G protein activities, endosome formation, vesicular sorting and trafficking, and signaling, which contributes to apoptosis through stress or reactive oxygen species (ROS) generation. Furthermore, we reproduced these effects of C-TSHR-mAb in experiments with the WRT cell line (from Wistar rat thyroids), the ML-1 line (from a human follicular thyroid cancer), and 3T3L1 cells (derived from mouse embryonic fibroblasts) and found similar apoptotic effects as a final readout (fig. S8).
DISCUSSION
Our current observations demonstrate functional differences between conformational epitope–dependent S-TSHR-Abs and linear- dependent C-TSHR-Abs and uncovered some of the mechanisms explaining why C-TSHR-Abs induce the apoptotic death, rather than the survival, of thyrocytes. Although the C-TSHR-mAb used in our study stimulated the TSHR to activate GRK2- and β-arrestin-1– dependent signaling, it failed to stimulate key regulatory signaling pathways. The activation of GRK2 by TSHR-mAbs and the suppression of GRK2 by TSH indicate that TSHR-mAbs may be only partial agonists. Consistent with this, the contact residues on the TSHR may also discriminate agonist binding from TSHR-Ab binding. Enhanced GRK2 activity has been demonstrated in GD (58). To achieve thyrocyte homeostasis, signaling cascades initiated by multiple G proteins appear to be crucial for cell survival and the synthesis and secretion of thyroid hormones. TSH, the primary ligand for the TSHR, and S-TSHR-Abs exerted these actions in thyrocytes through activation of the cAMP-PKA pathway, whereas C-TSHR-Abs failed to activate this signaling pathway (59). This likely indicates perturbation in the upstream effectors. Further characterization of such biased agonism by TSHR-Abs may, therefore, help determine their precise mechanisms of action in the thyroid glands of GD patients.
Because the C-TSHR-mAb activated the TSHR but was unable to induce sustained signaling, we hypothesized that C-TSHR-mAb fails to initiate an early event in the signaling cascade, such as clathrin- dependent receptor endocytosis. However, CME in thyrocytes treated with C-TSHR-mAb was unchanged when compared to that in thyrocytes exposed to S-TSHR-mAb. We analyzed all known pathways that contribute to receptor- and nonreceptor-mediated endocytosis, including caveolin-dependent receptor endocytosis and macropinocytosis, with multiple selective inhibitors but found no defect. We then investigated the internalization and clearance of the C-TSHR-Ab. Both TSH and S-TSHR-Ab underwent more rapid endocytosis and exo-cytosis when compared to the C-TSHR-Ab, suggesting that this antibody was less able to stimulate key regulatory signals needed for sorting and trafficking. Defects in vesicular trafficking have been observed previously in other diseases, such as Alzheimer’s disease (60), Huntington’s disease (61), amyotrophic lateral sclerosis (62), and other neurodegenerative diseases (60), in which accumulated proteins within the cytoplasm are unable to undergo proper vesicular sorting (60). Our observations suggest that the failure of C-TSHR-mAb to stimulate trafficking signals was responsible for the perturbations in the sorting and clearance mechanisms.
After initiation of the endocytic process through clathrin activation, endosome formation is a key step in vesicular sorting and trafficking. Whereas S-TSHR-mAb and TSH stimulated endosomal maturation, C-TSHR-mAb did not. This is consistent with earlier observations that the ratio between uptake and clearance of labeled C-TSHR-mAb was markedly reduced compared to that of TSH. Mechanisms underlying endocytic dysregulation remain unclear, but mutations in genes encoding certain signaling proteins (61) and cellular redox stress (62) may be involved. A repertoire of proteins is involved in the manufacture of endosomes and the maintenance of their functions (63). Lysosomes eliminate protein complexes, such as antibody-receptor aggregates, through enzymatic degradation (64). However, we found that C-TSHR- mAb complexes did not undergo lysosomal degradation but remained inside the cytoplasm for a prolonged period. Time-chase experiments demonstrated that the increasing accumulation of C-TSHR-mAbs induced cell death by day 5. These findings further confirmed our earlier observations that C-TSHR-mAb was unable to activate essential signaling molecules required for adequate vesicle formation and function, reminiscent of lysosomal storage diseases (65).
These observations of the loss of sustained cAMP-PKA-CREB-Akt signaling in thyrocytes treated with C-TSHR-mAb provided a clue that upstream effectors, most likely G proteins, may be the key regulatory elements for multiple signaling events orchestrating endocytosis and vesicular sorting and trafficking. As expected, C-TSHR-mAb failed to activate any G proteins, whereas TSH and S-TSHR-mAb stimulated G protein activation. Hence, antibodies to the TSHR showed biased agonist activities and played roles that were dissimilar to those of TSH. This biased signaling may induce previously uncharacterized phenotypic signatures and, therefore, may be involved in the differing clinical presentations of GD.
When a ligand binds to its GPCR, it causes a conformational change that enables the receptor to act as a guanine nucleotide exchange factor (GEF). The GPCR thus activates an associated G protein by enhancing its exchange of GDP bound to the α subunit for GTP. The activated G protein α subunit then dissociates from the βγ dimer to further affect intracellular signaling proteins or target functional proteins directly, depending on the α subunit type (66). Our results suggest that C-TSHR-mAb may not be capable of stimulating the GEF activity of the TSHR because of its failure to induce the required conformational changes in the receptor. Understanding how heterotrimeric G proteins are linked to GPCRs is key to clarifying the precise mechanisms played by the different TSHR-mAbs.
The importance of G proteins in the context of endosomal maturation in thyrocytes is unclear. To determine the functions of G proteins in the generation of vesicles, we examined G protein effects on endosome formation. Our results suggest that certain G protein disruptors perturbed endosomal formation. However, with S-TSHR-mAb or TSH, endosomes were formed in the presence of gallein and YM-254890, suggesting that the Gβγ and Gαq/11 proteins were not involved in endosomal maturation. Furthermore, suramin, which selectively disrupts multiple Gα proteins, blocked endosomal maturation, suggesting that Gα may be crucial for this process. Because S-TSHR-mAb stimulated endosomal maturation and also stimulated the activation of Gα (but not Gβγ), this suggests that Gα plays a central role in endosomal formation. In support of this conclusion, a previous study showed that Gα plays a role in both GPCR signaling and vesicular trafficking pathways (67). G protein activities are regulated by factors that control their ability to bind to GTP and catalyze its hydrolysis to GDP, and as we showed, excess GDP inhibited certain G protein actions.
Therefore, our results suggest that G protein activation is the driving force in the signaling necessary for thyrocyte homeostasis. The results of our Ras activation assay, which demonstrated that C-TSHR-mAb failed to generate active Ras (Ras-GTP), are also key to showing the importance of downstream Rab activation for endosomal maturation. Together, these results suggest that Gα protein activation through conformational changes required for the activation of upstream effectors is deficient after C-TSHR-mAb binding, whereas such activities are accelerated by TSH and, to a lesser extent, by S-TSHR-mAb. Clathrin-dependent endosomal formation in response to TSH and S-TSHR-mAb required the activation of Ras, PI3K, and Rac, all of which failed to adequately activate C-TSHR-mAb. Because Ras is required for the activation of Rab proteins and PI3K to induce vesicle formation (13), it is reasonable to assume that they are linked to downstream positive signals that maintain cellular homeostasis.
In conclusion, we showed that G proteins in thyrocytes were required not only for GPCR signaling but also for vesicular trafficking and sorting, processes that were independent of cAMP, PKA, and CREB. Furthermore, we showed that autoantibodies against the TSHR may have a major role in the initiation and continuation of thyroid autoimmune disease and that the mechanisms involved have implications for a wide array of autoimmune dysfunctions. Failure of C-TSHR-Abs to activate G protein–dependent signaling had multiple consequences, including the activation of GRK2- and β-arrestin-1–dependent biased sig-naling, the accumulation of intracellular inclusions, the generation of ROS, and cell death.
MATERIALS AND METHODS
Cell culture and treatments
Synchronized FRTL-5 rat thyroid cells were used (24, 68) and cultured as described previously (24). Before any stimulation experiments, synchronized cells were made quiescent by starvation in bovine calf serum– free basal medium (modified Ham’s F12) containing 0.3% bovine serum albumin for 2 days (24). Before stimulation, the medium was discarded from the 60-mm culture dishes, and the cells were washed three times with fresh medium containing Hanks’ balanced salt solution (HBSS; Life Technologies Inc.). Cells were then treated with different reagents in the basal medium as described previously (24).
Pharmacological inhibitors
Inhibitors of c-Raf (Kobe2602), Ras (FTaseII), Rac (NSC 23766), and Gβγ (gallein) were purchased from Cayman Chemical Co. Inhibitors of cAMP (SQ22536) and PKA (H89), bafilomycin A1, caveolin (genistein), the selective Gα inhibitor suramin, the PI3K inhibitor wortmannin, and GDP were purchased from Sigma-Aldrich. Anatag3 (ML-224) was obtained from DC Chemicals. Inhibitors of ERK1/2 (PD98059), PKC (Go6983), and Rho (Y-27632) and the nonselective PKC-δ inhibitor rottlerin were purchased from Calbiochem/EMD Millipore. The dynamin inhibitor dynasore, the Gβγ inhibitor hydrobromide, and the endocy-tosis inhibitor PAO were from Tocris Bioscience. The clathrin inhibitor Pitstop was from R&D Systems Inc. The Gαq/11 inhibitor YM-254890 was obtained from Wako Chemicals USA Inc.
Detection of intracellular cAMP
FRTL-5 (80% confluent) and CHO cells overexpressing TSHR (JPO9; 3 × 104 per well) were used for the measurement of intracellular cAMP concentrations by enzyme immunoassay (EIA) (Assay Designs Inc. or Amersham cAMP Biotrak EIA System from GE Healthcare Bio-Sciences Corp.) using polyclonal antibody against cAMP to bind, in a competitive manner, the cAMP in the standards or samples or a conjugate molecule to which cAMP was covalently attached. The intensity of the bound color was inversely proportional to the concentration of cAMP in either standards or samples. In each treatment, 2 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich) in the basal medium was used when measuring intracellular cAMP concentrations (24). To ensure reproducibility, all stimulation and inhibition experiments were repeated three times. The basal amounts of the analytes in all assays used, when analyzed either densitometrically or colorimetrically, were comparable (coefficient of variation, 4.6 to 11%).
Signaling molecules analyzed by proteomic array
Using a fixed concentration of TSH (1 mU/ml) or TSHR-mAb (1 μg/ml) (23) in fresh modified Ham’s F12 basal medium, cells were stimulated for 1 hour at 37°C in the incubator; washed twice with ice-cold phosphate-buffered saline (PBS) (pH 7.2) without calcium or magnesium (Mediatech Inc.); scraped into cold lysis buffer (provided by Kinexus Co.) containing different cocktails of phosphatase and protease inhibitors (cOmplete, Mini; Roche Applied Science), phenylmethylsulfonyl fluoride (Sigma-Aldrich), and 1% Triton X-100; and sonicated and analyzed as described previously (23). Known GPCR signaling pathways were investigated by a proteomic array performed at Kinexus Co. Arrays were performed with TSH, isotype control mAb, S-TSHR-Ab, and C-TSHR-mAbs (Tab-16) as described previously (23).
Immunohistochemistry
IHC was performed according to protocols described by Cell Signaling Technology. Briefly, after washing with PBS, adherent cells on slides were fixed in 4% paraformaldehyde, permeabilized in 90% methanol, and blocked for 2 hours in blocking buffer. The cells were then incubated overnight at 4°C with specific antibodies diluted in blocking buffer. After washing, the slides were incubated with fluorescently conjugated secondary antibodies for 1 hour with appropriate dilution, washed three times in PBS, mounted with mounting medium containing nuclear dye [DAPI (4′,6-diamidino-2-phenylindole)] and visualized immediately under digital or confocal microscopy or both (33).
Western blotting
Western blotting was performed as described previously (23, 24). Phospho- and nonphospho-specific antibodies against the following targets were used: ERK (ERK1/2; Thr202/Tyr204) and PKC-ζ/λ (Thr410/Thr403), Akt (Ser473), and PKA-C (Thr197) (Cell Signaling Technology). Mouse mAb against β-actin and unrelated control mAbs [immunoglobulin G2 (IgG2), κ-chain] were obtained from Sigma-Aldrich and BD Biosciences, respectively. To analyze vesicular proteins, including CLTA and EE (EEA1), and to detect lysosomes (Lamp1), Golgi (GOPC), STXbp6, and exocytosis (Rab11b), rabbit polyclonal antibodies from AVIVA Bio-sciences were used and quantitative LI-COR Western blot assays were performed.
Live-cell imaging of internalized labeled antibodies or TSH
Labeling of TSHR-mAbs or TSH was performed according to the manufacturer’s protocol (Molecular Probes, Thermo Fisher Scientific Inc.). Live-cell imaging was performed after each treatment in delta T dishes with labeled antibodies either with fluorescein isothiocyanate/Alexa Fluor 488 (green) or Alexa Fluor 647 (red). Before imaging, cells were washed twice with HBSS, and images were taken in HBSS medium containing Hoechst 33342 nuclear dye. For long-term imaging up to day 6, cells remained incubated with labeled antibodies in the basal medium for subsequent days, and images were taken and analyzed with different software, including CellProfiler and Image-Pro 3D analyzer as described previously (69).
Assay for antibody internalization and clearance
FRTL-5 cells (25,000 cells per well) were grown in 96-well black plates in triplicate for 2 days and starved as described earlier. The cells were then incubated with Alexa Fluor 488–labeled TSHR-mAbs or TSH in basal medium for 30 min and washed with HBSS. After adding fresh basal medium, the treated cells were then chased for 3 hours. Every 30 min, fluorescence readings were taken for both internalized (uptake) and released (supernatant) antibodies or TSH with a fluorescent plate reader (PHERAstar FS, BMG LABTECH). The ratio between the uptake and release of labeled antibodies or TSH was then calculated at different time points.
Live-cell imaging of endocytosis and vesicular trafficking
Live-cell imaging was also performed after expressing protein markers of EEs (RFP-Rab5a, red), LEs (RFP-Rab7a, red), and lysosomes (GFP-Lamp1, green) in a baculovirus expression system according to the manufacturer’s protocol (Molecular Probes). Cells pretreated with different small-molecule inhibitors were incubated with labeled antibodies or TSH and then analyzed under digital (Eclipse TE2000-S, Nikon) or confocal microscope (Zeiss LSM 700). At least five images were captured per experiment, and three images each of treated or untreated cells were analyzed quantitatively by different software.
Quantification of live fluorescence imaging and immunocytochemistry images
The recorded images were loaded into Adobe Photoshop (Adobe Systems) for analysis, in which different colors were visually quantified by two independent observers. The number of cells positive for both the target color and nuclear staining (Hoechst) was considered positive for fluorescence and digitally recorded to prevent multiple counts. FI was calculated using CCTF (corrected total cell fluorescence) in Image-Pro software: CCTF = Integrated density − (Area of selected cell × Mean fluorescence of background readings). The number of cells positive for distinct colocalized colors was also digitally recorded. CellProfiler and Image-Pro software were used to discriminate between different colors, including colocalization, fluorescence dot counts, and average FI measurements.
HTS luciferase reporter assays
For high-throughput screening (HTS), we used cells generated by transfecting a highly selected stable line of CHO cells expressing the human TSHR with a hemagglutinin (HA) tag at the N terminus (CHO-HA-TSHR cells) (50) with the pGL4.29 [luc2P/CRE/Hygro] construct and then selected a stable line with hygromycin. We developed stable CHO-HA-TSHR cell lines expressing various reporter plasmids (CRE-, NFATRE, SRE-, and SRF-RE-) for studying the activation of various G proteins. These stable lines were characterized and optimized for responses with positive controls (TSH, ionomycin, or phorbol 12-myristate 13-acetate) and negative controls as described. Experiments were performed both in 96- and 384-well plate formats with three or four different concentrations of antibodies and TSH with or without pretreatment with the appropriate small- molecule inhibitors.
Ras activation assay
Ras activation assays were performed according to the protocol provided with the kit from NewEast Biosciences. This assay was based on the configuration-specific anti–Ras-GTP mAb to measure the abundance of the active form of Ras (Ras-GTP). Briefly, antibody against Ras-GTP was incubated with lysates of treated cells. The bound Ras-GTP was then pulled down by protein A/G agarose. The precipitated active Ras-GTP was then detected by LI-COR immunoblot analysis with anti-Ras rabbit polyclonal antibody.
Analysis of apoptosis by caspase activity assay
To observe cell death or apoptosis, synchronized FRTL-5, Wrt (Wistar rat thyrocytes), ML-1 (human follicular thyroid cancer cells), and 3T3L1 (mouse embryonic fibroblasts) cells were incubated with increasing concentrations of C-TSHR-mAbs and analyzed by flow cytometry or microscopy to detect annexin V staining as described previously (23). To confirm the effector mechanisms of the cell death process, we used the FAM caspase assay kit, which detects multiple active caspases (33). The methodology was based on a FLICA (fluorometric inhibitor of caspases) assay and performed according to the instructions provided with the kit (Imgenex Corp.).
Statistical analysis
Two-tailed paired t tests were used to evaluate the statistical significance of differences in means for continuous variables. P values for fold changes or percentage increases were calculated after log2 transformation. P ≤ 0.05 was used to determine statistical significance. Data are means ± SEM.
Supplementary Material
Fig. S1. The C-TSHR-mAb disrupts vesicular trafficking.
Fig. S2. The C-TSHR-mAb does not stimulate signaling in CHO-TSHR cells.
Fig. S3. Effects of G protein perturbations on Gαs, Gαq, Gβγ, and Gα12 activities on CHO-TSHR cells.
Fig. S4. The Ras-PI3K-Rac pathway downstream of G proteins is a key signaling pathway for vesicle formation.
Fig. S5. Perturbation of major G proteins by different pharmacological inhibitors in CHO-TSHR cells.
Fig. S6. Dose-dependent suppression of the TSH-induced activities of Gαs, Gαq, Gβγ, and Gα12 by pharmacological inhibitors in CHO-TSHR cells.
Fig. S7. Model depicting the mechanism of action of C-TSHR-mAb in FRTL-5 thyrocytes.
Fig. S8. The C-TSHR-mAb induces apoptosis in thyrocytes and fibroblasts.
Acknowledgments
Funding: This work was supported in part by DK069713 from the NIH, the David Owen Segal Endowment, and the VA Merit Review Program (to T.F.D.).
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencesignaling.org/cgi/content/full/11/514/eaah4120/DC1
Author contributions: S.A.M. performed the experimental design, experimental work, data analysis, and wrote and reviewed the manuscript. R.M. contributed to the microscopic data interpretation and analyses. R.L. helped with writing and reviewing of the manuscript. T.F.D. contributed to the funding acquisition and writing and reviewing of the manuscript. All authors read and discussed the manuscript.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Li J, Ning Y, Hedley W, Saunders B, Chen Y, Tindill N, Hannay T, Subramaniam S. The Molecule Pages database. Nature. 2002;420:716–717. doi: 10.1038/nature01307. [DOI] [PubMed] [Google Scholar]
- 2.Kobilka BK. Agonist-induced conformational changes in the β2 adrenergic receptor. J Pept Res. 2002;60:317–321. doi: 10.1034/j.1399-3011.2002.21062.x. [DOI] [PubMed] [Google Scholar]
- 3.Farid NR, Szkudlinski MW. Minireview: Structural and functional evolution of the thyrotropin receptor. Endocrinology. 2004;145:4048–4057. doi: 10.1210/en.2004-0437. [DOI] [PubMed] [Google Scholar]
- 4.Calebiro D, Nikolaev VO, Lohse MJ. Imaging of persistent cAMP signaling by internalized G protein-coupled receptors. J Mol Endocrinol. 2010;45:1–8. doi: 10.1677/JME-10-0014. [DOI] [PubMed] [Google Scholar]
- 5.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human β2-adrenergic G protein–coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 7.Lohse MJ. Molecular mechanisms of membrane receptor desensitization. Biochim Biophys Acta. 1993;1179:171–188. doi: 10.1016/0167-4889(93)90139-g. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 9.Milligan G, White JH. Protein-protein interactions at G-protein-coupled receptors. Trends Pharmacol Sci. 2001;22:513–518. doi: 10.1016/s0165-6147(00)01801-0. [DOI] [PubMed] [Google Scholar]
- 10.Kristiansen K. Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: Molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol Ther. 2004;103:21–80. doi: 10.1016/j.pharmthera.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Singh RD, Puri V, Valiyaveettil JT, Marks DL, Bittman R, Pagano RE. Selective caveolin-1-dependent endocytosis of glycosphingolipids. Mol Biol Cell. 2003;14:3254–3265. doi: 10.1091/mbc.E02-12-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott DB, Michailidis I, Mu Y, Logothetis D, Ehlers MD. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J Neurosci. 2004;24:7096–7109. doi: 10.1523/JNEUROSCI.0780-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murphy JE, Padilla BE, Hasdemir B, Cottrell GS, Bunnett NW. Endosomes: A legitimate platform for the signaling train. Proc Natl Acad Sci USA. 2009;106:17615–17622. doi: 10.1073/pnas.0906541106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 15.McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 16.Le Roy C, Wrana JL. Signaling and endocytosis: A team effort for cell migration. Dev Cell. 2005;9:167–168. doi: 10.1016/j.devcel.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 17.de Duve C. The lysosome turns fifty. Nat Cell Biol. 2005;7:847–849. doi: 10.1038/ncb0905-847. [DOI] [PubMed] [Google Scholar]
- 18.Luzio JP, Pryor PR, Bright NA. Lysosomes: Fusion and function. Nat Rev Mol Cell Biol. 2007;8:622–632. doi: 10.1038/nrm2217. [DOI] [PubMed] [Google Scholar]
- 19.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. β-arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 20.Kimura T, Van KA, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: A critical evaluation of in vitro models. Endocr Rev. 2001;22:631–656. doi: 10.1210/edrv.22.5.0444. [DOI] [PubMed] [Google Scholar]
- 21.Medina DL, Santisteban P. Thyrotropin-dependent proliferation of in vitro rat thyroid cell systems. Eur J Endocrinol. 2000;143:161–178. doi: 10.1530/eje.0.1430161. [DOI] [PubMed] [Google Scholar]
- 22.Morshed SA, Davies TF. Graves’ disease mechanisms: The role of stimulating, blocking, and cleavage region TSH receptor antibodies. Horm Metab Res. 2015;47:727–734. doi: 10.1055/s-0035-1559633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morshed SA, Ando T, Latif R, Davies TF. Neutral antibodies to the TSH receptor are present in Graves’ disease and regulate selective signaling cascades. Endocrinology. 2010;151:5537–5549. doi: 10.1210/en.2010-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morshed SA, Latif R, Davies TF. Characterization of thyrotropin receptor antibody-induced signaling cascades. Endocrinology. 2009;150:519–529. doi: 10.1210/en.2008-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ando T, Davies TF. Monoclonal antibodies to the thyrotropin receptor. Clin Dev Immunol. 2005;12:137–143. doi: 10.1080/17402520500078238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ando T, Latif R, Davies TF. Concentration-dependent regulation of thyrotropin receptor function by thyroid-stimulating antibody. J Clin Invest. 2004;113:1589–1595. doi: 10.1172/JCI21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ando T, Latif R, Davies TF. Thyrotropin receptor antibodies: New insights into their actions and clinical relevance. Best Pract Res Clin Endocrinol Metab. 2005;19:33–52. doi: 10.1016/j.beem.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 28.Ando T, Latif R, Davies TF. Antibody-induced modulation of TSH receptor post-translational processing. J Endocrinol. 2007;195:179–186. doi: 10.1677/JOE-07-0058. [DOI] [PubMed] [Google Scholar]
- 29.Ando T, Latif R, Pritsker A, Moran T, Nagayama Y, Davies TF. A monoclonal thyroid-stimulating antibody. J Clin Invest. 2002;110:1667–1674. doi: 10.1172/JCI16991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanders J, Allen F, Jeffreys J, Bolton J, Richards T, Depraetere H, Nakatake N, Evans M, Kiddie A, Premawardhana LDKE, Chirgadze DY, Miguel RN, Blundell TL, Furmaniak J, Smith BR. Characteristics of a monoclonal antibody to the thyrotropin receptor that acts as a powerful thyroid-stimulating autoantibody antagonist. Thyroid. 2005;15:672–682. doi: 10.1089/thy.2005.15.672. [DOI] [PubMed] [Google Scholar]
- 31.Sanders J, Jeffreys J, Depraetere H, Evans M, Richards T, Kiddie A, Brereton K, Premawardhana LD, Chirgadze DY, Núñez MR, Blundell TL, Furmaniak J, Smith BR. Characteristics of a human monoclonal autoantibody to the thyrotropin receptor: Sequence structure and function. Thyroid. 2004;14:560–570. doi: 10.1089/1050725041692918. [DOI] [PubMed] [Google Scholar]
- 32.Sanders J, Jeffreys J, Depraetere H, Richards T, Evans M, Kiddie A, Brereton K, Groenen M, Oda Y, Furmaniak J, Smith BR. Thyroid-stimulating monoclonal antibodies. Thyroid. 2002;12:1043–1050. doi: 10.1089/105072502321085135. [DOI] [PubMed] [Google Scholar]
- 33.Morshed SA, Ma R, Latif R, Davies TF. How one TSH receptor antibody induces thyrocyte proliferation while another induces apoptosis. J Autoimmun. 2013;47:17–24. doi: 10.1016/j.jaut.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morshed SA, Latif R, Davies TF. Delineating the autoimmune mechanisms in Graves’ disease. Immunol Res. 2012;54:191–203. doi: 10.1007/s12026-012-8312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vlase H, Graves PN, Magnusson RP, Davies TF. Human autoantibodies to the thyrotropin receptor: Recognition of linear, folded, and glycosylated recombinant extracellular domain. J Clin Endocrinol Metab. 1995;80:46–53. doi: 10.1210/jcem.80.1.7829638. [DOI] [PubMed] [Google Scholar]
- 36.Ueda Y, Sugawa H, Akamizu T, Okuda J, Ueda M, Kosugi S, Ohta C, Kihou Y, Mori T. Thyroid-stimulating antibodies in sera from patients with Graves’ disease are heterogeneous in epitope recognition. Eur J Endocrinol. 1995;132:62–68. doi: 10.1530/eje.0.1320062. [DOI] [PubMed] [Google Scholar]
- 37.Endo T, Kogai T, Nakazato M, Saito T, Kaneshige M, Onaya T. Autoantibody against Na+/I− symporter in the sera of patients with autoimmune thyroid disease. Biochem Biophys Res Commun. 1996;224:92–95. doi: 10.1006/bbrc.1996.0989. [DOI] [PubMed] [Google Scholar]
- 38.Nagy EV, Burch HB, Mahoney K, Lukes YG, Morris JC, III, Burman KD. Graves’ IgG recognizes linear epitopes in the human thyrotropin receptor. Biochem Biophys Res Commun. 1992;188:28–33. doi: 10.1016/0006-291x(92)92345-x. [DOI] [PubMed] [Google Scholar]
- 39.Fan JL, Desai RK, Seetharamaiah GS, Dallas JS, Wagle NM, Prabhakar BS. Heterogeneity in cellular and antibody responses against thyrotropin receptor in patients with Graves’ disease detected using synthetic peptides. J Autoimmun. 1993;6:799–808. doi: 10.1006/jaut.1993.1065. [DOI] [PubMed] [Google Scholar]
- 40.Takai O, Desai RK, Seetharamaiah GS, Jones CA, Allaway GP, Akamizu T, Kohn LD, Prabhakar BS. Prokaryotic expression of the thyrotropin receptor and identification of an immunogenic region of the protein using synthetic peptides. Biochem Biophys Res Commun. 1991;179:319–326. doi: 10.1016/0006-291x(91)91372-j. [DOI] [PubMed] [Google Scholar]
- 41.Penela P, Ribas C, Mayor F., Jr Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell Signal. 2003;15:973–981. doi: 10.1016/s0898-6568(03)00099-8. [DOI] [PubMed] [Google Scholar]
- 42.Premont RT, Gainetdinov RR. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- 43.Buchanan FG, DuBois RN. Emerging roles of β-arrestins. Cell Cycle. 2006;5:2060–2063. doi: 10.4161/cc.5.18.3212. [DOI] [PubMed] [Google Scholar]
- 44.Evron T, Daigle TL, Caron MG. GRK2: Multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol Sci. 2012;33:154–164. doi: 10.1016/j.tips.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsvetanova NG, Irannejad R, von Zastrow M. G protein-coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes. J Biol Chem. 2015;290:6689–6696. doi: 10.1074/jbc.R114.617951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baratti-Elbaz C, Ghinea N, Lahuna O, Loosfelt H, Pichon C, Milgrom E. Internalization and recycling pathways of the thyrotropin receptor. Mol Endocrinol. 1999;13:1751–1765. doi: 10.1210/mend.13.10.0360. [DOI] [PubMed] [Google Scholar]
- 47.van den Hove MF, Croizet-Berger K, Tyteca D, Selvais C, de Diesbach P, Courtoy PJ. Thyrotropin activates guanosine 5′-diphosphate/guanosine 5′-triphosphate exchange on the rate-limiting endocytic catalyst, Rab5a, in human thyrocytes in vivo and in vitro. J Clin Endocrinol Metab. 2007;92:2803–2810. doi: 10.1210/jc.2006-2351. [DOI] [PubMed] [Google Scholar]
- 48.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Appelqvist H, Wäster P, Kågedal K, Öllinger K. The lysosome: From waste bag to potential therapeutic target. J Mol Cell Biol. 2013;5:214–226. doi: 10.1093/jmcb/mjt022. [DOI] [PubMed] [Google Scholar]
- 50.Platt FM, Boland B, van der Spoel AC. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–734. doi: 10.1083/jcb.201208152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Latif R, Ali MR, Ma R, David M, Morshed SA, Ohlmeyer M, Felsenfeld DP, Lau Z, Mezei M, Davies TF. New small molecule agonists to the thyrotropin receptor. Thyroid. 2015;25:51–62. doi: 10.1089/thy.2014.0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Freissmuth M, Boehm S, Beindl W, Nickel P, Ijzerman AP, Hohenegger M, Nanoff C. Suramin analogues as subtype-selective G protein inhibitors. Mol Pharmacol. 1996;49:602–611. [PubMed] [Google Scholar]
- 53.Johnson LS, Dunn KW, Pytowski B, McGraw TE. Endosome acidification and receptor trafficking: Bafilomycin A1 slows receptor externalization by a mechanism involving the receptor’s internalization motif. Mol Biol Cell. 1993;4:1251–1266. doi: 10.1091/mbc.4.12.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lehmann DM, Seneviratne AMPB, Smrcka AV. Small molecule disruption of G protein βγ subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol Pharmacol. 2008;73:410–418. doi: 10.1124/mol.107.041780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neumann S, Nir EA, Eliseeva E, Huang W, Marugan J, Xiao J, Dulcey AE, Gershengorn MC. A selective TSH receptor antagonist inhibits stimulation of thyroid function in female mice. Endocrinology. 2014;155:310–314. doi: 10.1210/en.2013-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, Kobori M. A novel Gαq/11-selective inhibitor. J Biol Chem. 2004;279:47438–47445. doi: 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- 57.Lewandowicz AM, Vepsäläinen J, Laitinen JT. The ‘allosteric modulator’ SCH-202676 disrupts G protein-coupled receptor function via sulphydryl-sensitive mechanisms. Br J Pharmacol. 2006;147:422–429. doi: 10.1038/sj.bjp.0706624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Voigt C, Holzapfel HP, Meyer S, Paschke R. Increased expression of G-protein-coupled receptor kinases 3 and 4 in hyperfunctioning thyroid nodules. J Endocrinol. 2004;182:173–182. doi: 10.1677/joe.0.1820173. [DOI] [PubMed] [Google Scholar]
- 59.Strachan RT, Sun J-p, Rominger DH, Violin JD, Ahn S, Rojas Bie Thomsen A, Zhu X, Kleist A, Costa T, Lefkowitz RJ. Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR) J Biol Chem. 2014;289:14211–14224. doi: 10.1074/jbc.M114.548131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nixon RA. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiol Aging. 2005;26:373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 61.Li X, Standley C, Sapp E, Valencia A, Qin ZH, Kegel KB, Yoder J, Comer-Tierney LA, Esteves M, Chase K, Alexander J, Masso N, Sobin L, Bellve K, Tuft R, Lifshitz L, Fogarty K, Aronin N, DiFiglia M. Mutant huntingtin impairs vesicle formation from recycling endosomes by interfering with Rab11 activity. Mol Cell Biol. 2009;29:6106–6116. doi: 10.1128/MCB.00420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Otomo A, Pan L, Hadano S. Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol Res Int. 2012;2012:498428. doi: 10.1155/2012/498428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-β-arrestin complexes after receptor endocytosis*. J Biol Chem. 2001;276:19452–19460. doi: 10.1074/jbc.M101450200. [DOI] [PubMed] [Google Scholar]
- 64.Irannejad R, von Zastrow M. GPCR signaling along the endocytic pathway. Curr Opin Cell Biol. 2014;27:109–116. doi: 10.1016/j.ceb.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winchester B, Vellodi A, Young E. The molecular basis of lysosomal storage diseases and their treatment. Biochem Soc Trans. 2000;28:150–154. doi: 10.1042/bst0280150. [DOI] [PubMed] [Google Scholar]
- 66.Neves SR, Ram PT, Iyengar R. G protein pathways. Science. 2002;296:1636–1639. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- 67.Rosciglione S, Thériault C, Boily MO, Paquette M, Lavoie C. Gαs regulates the post-endocytic sorting of G protein-coupled receptors. Nat Commun. 2014;5:4556. doi: 10.1038/ncomms5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iacovelli L, Capobianco L, Salvatore L, Sallese M, D’Ancona GM, De BA. Thyrotropin activates mitogen-activated protein kinase pathway in FRTL-5 by a cAMP-dependent protein kinase A-independent mechanism. Mol Pharmacol. 2001;60:924–933. doi: 10.1124/mol.60.5.924. [DOI] [PubMed] [Google Scholar]
- 69.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The C-TSHR-mAb disrupts vesicular trafficking.
Fig. S2. The C-TSHR-mAb does not stimulate signaling in CHO-TSHR cells.
Fig. S3. Effects of G protein perturbations on Gαs, Gαq, Gβγ, and Gα12 activities on CHO-TSHR cells.
Fig. S4. The Ras-PI3K-Rac pathway downstream of G proteins is a key signaling pathway for vesicle formation.
Fig. S5. Perturbation of major G proteins by different pharmacological inhibitors in CHO-TSHR cells.
Fig. S6. Dose-dependent suppression of the TSH-induced activities of Gαs, Gαq, Gβγ, and Gα12 by pharmacological inhibitors in CHO-TSHR cells.
Fig. S7. Model depicting the mechanism of action of C-TSHR-mAb in FRTL-5 thyrocytes.
Fig. S8. The C-TSHR-mAb induces apoptosis in thyrocytes and fibroblasts.