
Figure 1.
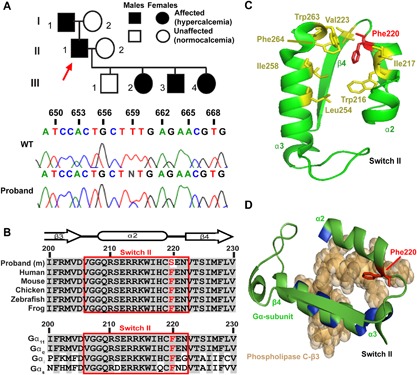
Identification and structural assessment of a Phe220Ser Gα11 mutation in a family with familial hypocalciuric hypercalcemia type 2. (A) The family comprised of 5 affected and 3 unaffected members (top). The proband (individual II.1) is indicated by an arrow. A heterozygous T‐to‐C transition at nucleotide c.659, predicted to result in a missense substitution of the WT Phe to a mutant (m) Ser at codon 220 of the Gα11 protein, was identified in the proband and confirmed to cosegregate with hypercalcemia by Sanger DNA sequence analysis. (B) Multiple protein sequence alignment of residues comprising the β3‐α2 loop, α2‐helix, and α2‐β4 loop that form switch II (red box) of Gα11 orthologs (top) and Gα‐subunit paralogs (bottom). Conserved residues are shown in gray, and WT (Phe, F) and mutant (Ser, S) residues are shown in red. (C) Structural model showing that Phe220 of Gα11 is part of a hydrophobic cluster of amino acids (yellow) within switch II and the α3‐helix, which stabilize the region in a conformation that facilitates effector binding. (D) Close‐up view of Phe220 residue, within the switch II region of the GTPase domain of Gα11, in complex with phospholipase C‐β3 (brown space‐filling model), with directly interacting residues of the α2 and α3 helices colored blue.