

Abstract
Excessive and/or persistent activation of calcium-calmodulin protein kinase II (CaMKII) is detrimental in acute and chronic cardiac injury. However, intrinsic regulators of CaMKII activity are poorly understood. We find that cellular retinoic acid binding protein 1 (CRABP1) directly interacts with CaMKII, and uncover a functional role for CRABP1 in regulating CaMKII activation. We generated Crabp1 null mice (CKO) in C57BL/6J background for pathophysiological studies. CKO mice develop hypertrophy as adults, exhibiting significant left ventricular dilation with reduced ejection fraction at the baseline cardiac function. Interestingly, CKO mice have elevated basal CaMKII phosphorylation at T287, and phosphorylation on its substrate phospholamban (PLN) at T17. Acute isoproterenol (ISO) challenge (80 mg/kg two doses in 1 day) causes more severe apoptosis and necrosis in CKO hearts, and treatment with a CaMKII inhibitor KN-93 protects CKO mice from this injury. Chronic (30 mg/kg/day) ISO challenge also significantly increases hypertrophy and fibrosis in CKO mice as compared to WT. In wild type mice CRABP1 expression is increased in early stages of ISO challenge and eventually reduces to the basal level. Mechanistically, CRABP1 directly inhibits CaMKII by competing with calmodulin (CaM) for CaMKII interaction. This study demonstrates increased susceptibility of CKO mice to ISO-induced acute and chronic cardiac injury due to, at least in part, elevated CaMKII activity. Deleting Crabp1 results in reduced baseline cardiac function and aggravated damage challenged with acute and persistent β-adrenergic stimulation. This is the first report of a physiological role of CRABP1 as an endogenous regulator of CaMKII, which protects the heart from ISO-induced damage.
Keywords: CaMKII, Crabp1, heart failure, cardiac remodeling
Introduction
Neuroendocrine stimulation of β-adrenergic receptors (β-AR) in the heart augments muscle contractility and heart rate, thereby increasing cardiac output during fight-or-flight response (Mann and Bristow 2005). Excess and/or persistent stimulation by catecholamines, e.g. isoproterenol (ISO)―a synthetic agonist of β-ARs, causes decompensation and left ventricular remodeling marked by cell death, inflammation, fibrosis, hypertrophy, and cardiac dilation, leading to arrhythmias and heart failure (Garg and Khanna 2014, Nichtova et al. 2012). β-adrenergic stimulation activates cAMP-protein kinase A (PKA) signaling and CaMKII (Grimm and Brown 2010) through multiple mechanisms.
Initial activation of CaMKII by Ca2+/calmodulin (CaM) induces autophosphorylation at T287 that provides molecular memory to sustain its activity. CaMKII substrates include ryanodine receptor 2a (RyR2a) at S2814 and phospholamban (PLN) at T17 on the sarcoplasmic reticulum (SR) membrane to augment excitation-contraction coupling (Mattiazzi et al. 2015). Ca2+ surge caused by excessive and/or persistent CaMKII activity leads to diastolic SR Ca2+ leak and arrhythmias (Chelu et al. 2009, Ferrero et al. 2007). Indeed, CaMKII inhibition by specific inhibitors and genetic ‘loss-of-function’ in animal models have demonstrated that excessive CaMKII activation underlines β-AR stimulated cardiac damage (Kreusser et al. 2016, Zhang et al. 2005), ischemia/reperfusion injury (Ling et al. 2013, Said et al. 2011), workload and pressure overload injury (Grimm et al. 2015), hypertrophy (Sucharov et al. 2006), and cell apoptosis and necrosis (Vila-Petroff et al. 2007). Consistently, CaMKII expression and its autonomous activity are significantly elevated in hypertrophy and heart failure (Mollova et al. 2015, Zhang et al. 2003). Therefore, a consensus has emerged that CaMKII activation underlines various heart diseases and can be a therapeutic target for heart failure. To this end, several small molecule and peptide inhibitors have been tested experimentally, but not for clinical use due to concerns over the inhibition of other kinases and channels (Pellicena and Schulman 2014). Understanding physiological regulation of CaMKII is important for managing the cardiovascular system. This study attempts to address a newly discovered negative physiological control of CaMKII activity in the heart.
Cellular retinoic acid (RA) binding protein 1 (CRABP1) is a highly-conserved cytosolic protein that can bind RA with a high affinity (Fiorella et al. 1993). Its biological activity is classically measured by specific RA binding in vitro and implicated in modulating the biological availability of RA (Donovan et al. 1995, Venepally et al. 1996). However, its physiological function has remained elusive due to the lack of apparent phenotype of Crabp1-null mice (de Bruijn et al. 1994, Gorry et al. 1994). Nevertheless, transgenic mice ectopically expressing CRABP1 exhibited abnormal cell growth and differentiation in the eye and the liver (Perez-Castro et al. 1993, Wei and Chen 1991), indicating certain physiological function of CRABP1 that remains to be uncovered.
In a proteomic screening, we found CRABP1 directly interacts with CaMKII and confirmed its interaction with endogenous CaMKII in cardiomyocytes. We thus generated Crabp1-null mice (CKO) in C57BL/6J background for physiological assessment. Interestingly, the basal cardiac function of CKO mice is significantly reduced and their hearts display evidence of adverse remodeling. We conclude that CKO mice are more susceptible to both acute and chronic ISO challenges due to significantly elevated CaMKII activity in cardiomyocyte, and that a physiological function of CRABP1 is to modulate CaMKII activity through direct interaction with CaMKII and competing with CaM for CaMKII activation. This study reports a physiological role for apo-CRABP1 in modulating heart health.
Materials and Methods
Plasmid constructs and cell culture
Mouse CaMKII alpha Venus was a gift from Steven Vogel (Addgene plasmid # 29427) (Thaler et al. 2009). Mouse cDNA for Camk2D (MMM1013-202805730, GE Dharmacon) was amplified with PCR and subcloned into a modified pCMX-PL1 vector containing Flag tag. Mouse Crabp1 cDNA was PCR amplified with primers for cDNA and cloned into a modified pCMX-PL1 vector containing Flag tag and into pET-15b vector containing His tag for Flag-CRABP1 and His-CRABP1, respectively. Calmodulin was cloned through PCR of mouse cDNA into pGEX-2T containing GST tag. HEK293T cells were maintained in DMEM supplanted with 10% fetal bovine serum and 1% penicillin-streptomycin. Expression vectors for GFP-CaMKIIα and Flag-CRABP1 were transfected by calcium phosphate method.
Experimental mice, ISO injection and tissue preparation
Experimental procedures were conducted according to NIH guidelines and approved by the University of Minnesota Institutional Animal Care and Use Committee. We used a Crabp1-targeted DE3 (ES) clone 22 to generate CKO mice. CKO Crabp1 allele contains 5 bp insertion generating a Not1 restriction site (Gorry et al. 1994). Male WT and CKO mice (16-20 weeks of age) were divided into two groups each: control and ISO groups. We applied two ISO treatment strategies: acute and chronic injection. For acute treatment, ISO groups of WT and CKO mice were intraperitoneally injected with 80 mg/kg of ISO (I6504, Sigma-Aldrich) twice with 8 h interval and were sacrificed at 24 h after first injection. For chronic treatment, ISO groups of WT and CKO mice were treated with daily intraperitoneal injections of ISO (30 mg/kg/day) for 15 days or 5 weeks. Control groups were treated with daily injections of 0.9% saline. KN-92 and KN-93 (5 μmol/kg, 2 doses, MedChem Express, Monmouth Junction, NJ) (Zhang et al. 2005) were injected 30 min before ISO injection (80 mg/kg, 2 doses) into CKO mice. At the end of ISO injection period, hearts were quickly excised, cross-cut in the middle of ventricles, and ventricles were weighed after blood, aorta, atria were removed. Base part was quickly frozen in liquid nitrogen for protein and genetic analyses, and apex part embedded in Tissue-Plus O.C.T. Compound (Fisher Healthcare) without fixation was snap frozen for histological staining.
Echocardiography
Cardiac dimensions are acquired in 2-D-guided M mode images using Vevo 2100 (VisualSonics Inc. Toronto, Canada) on a parasternal short axis view of the heart. All measurements were performed under anesthesia with 1.5–2% isoflurane mixed with 0.5 L/min 100% O2. Heart rate was maintained above at least 300 bpm and body temperatures were maintained between narrow ranges (37.0±1.0 °C) to avoid confounding effects of hypothermia. Measurements were averaged over 3 consecutive beats (Zhang et al. 2005).
Protein extraction, western blot and immunoprecipitation
Cytosolic lysates including membranous fractions were obtained from frozen hearts in Triton X-100 lysis buffer (10 mM Tis-HCl, pH 7.9, 50 mM NaCl, 0.5 M sucrose, 0.1 mM EDTA, and 0.5% Triton X-100) supplemented immediately before homogenization with protease inhibitor cocktail, 1 mM PMSF, 1mM NaF, 1 mM dithiothreitol, 17.5 mM β-glycerophosphate, and 1 mM Na3VO4. Samples for CaMKII (50 μg) were separated in 8 or 10% SDS-polyacrylamide gel and for PLN (20 μg) in 13.8%. The primary antibodies used for western blots are as follows: P-CaMKII (sc-32289), CaMKIIδ (sc-5392), P-PLN at T17 (sc-17024-R), GAPDH (sc-32233), His-probe (sc-8036), and GFP (sc-8334) were from Santa Cruz Biotechnology. Other antibodies are P-PLN at S16 (Ab15000, Abcam), GST (2624S, Cell Signaling), CRABP1 (C1608, Sigma-Aldrich), and Flag (F1804, Sigma-Aldrich). For co-immunoprecipitation, ventricular extracts (300 μg) were precipitated with anti-CaMKIIδ antibody (Santa Cruz) and protein G beads in the binding buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.2% NP40, 10% glycerol, and 1 mM EDTA) at cold room overnight. Or proteins from HEK293T ectopically expressing GFP-CaMKIIα with or without Flag-CRABP1 were extracted in RIPA buffer (25 mM Tis-HCl, pH 7.4, 150 mM NaCl, 0.1% SDS, 1% NP-40, and 0.5% sodium deoxycholate). GFP-CaMKIIα was precipitated with anti-GFP antibody and protein G beads. After washing with the binding buffer, the beads were resuspended in 2X SDS loading buffer followed by western blot analysis with anti-CRABP1 or anti-Flag antibodies.
Serum lactate dehydrogenase Assay
Blood samples were collected and centrifuged for 10 min at 1,000g to obtain serum. Lactate dehydrogenase (LDH) reaction was performed with 3 μl serum at 37 °C and spectrophotometrically assayed at 405 nm using a kit according to the manufacturer’s instruction (Sigma-Aldrich).
Histology and cell size analysis
For IgG staining, freshly frozen tissue sections (7 μm) were rehydrated in 1× PBS, blocked with 5% BSA, washed with PBS and stained with 1:200 goat anti-mouse IgG-Cy3 (Abcam) and 1:50 wheat germ agglutinin (WGA, Alexa Fluor 488 conjugate, ThermoFisher Scientific) for 1 h. The slides were mounted with MM83 mounting media (CBG Biotech, Oakwood Village, OH). The automatic montaged images of whole heart sections were acquired at 10× magnification on a Leica DM5500B microscope (Leica, Buffalo Grove, IL) using LAS Suite AF software (3.1.0 build 8587 version, Leica). The IgG-positive area was calculated using ImageJ (% = the number of pixels in the thresholded IgG channel/the number of pixels of myocardial tissue × 100) (Strakova et al. 2014, Townsend et al. 2008).
For cell size analysis, tissue slides were stained with WGA Alexa 488 conjugate as above. Four images of short-axis cross-sectional areas from each heart were taken at a 40× magnification on a Leica DMIRE2 microscope or a 20× magnification on a Leica DM5500B microscope. The sizes of about 100 cardiomyocytes were quantitated using ImageJ to determine the relative sizes of cardiomyocytes.
For sirius red-fast green (SRFG) staining, freshly frozen tissue slides were fixed in cold acetone for 3 h. The slides were washed with 70% ethanol and water, and stained with SRFG mixture (0.1% direct red 80 and 0.1% fast green FCF in 1.2% picric acid solution) (365548 and F7252, respectively, Sigma-Aldrich) for 15 min with gentle swirling. The washed slides were dehydrated with 70% ethanol, 100% ethanol, and formula 83 (CBG Biotech). Images were acquired with an Olympus BX60 microscope equipped with a UPlanFl 10×/0.3 NA objective and a Blackfly 2.3 MP color camera. Images were captured using Panotiq software version 4.0. (ViewsIQ). The fibrotic areas were quantitated using ImageJ (% = the number of pixels in the thresholded sirius red channel/the number of pixels of myocardial tissue × 100).
TUNEL staining
Frozen heart sections were used. We performed terminal deoxynucleotide transferase-mediated dUTP nick-end labeling (TUNEL) staining using CF™ Dye TUNEL Assay Apoptosis Detection Kit (Biotium, Hayward, CA) according to the manufacturer’s protocol. DAPI was used for nuclear staining. Images were obtained using Leica DM5500B microscope under 20× magnification. TUNEL signals overlapped with DAPI from ten random fields were counted and expressed as the percentage fraction of the approximate number of cells per field.
ADP-Glo kinase assay
ADP-Glo™ kinase assay (Promega) was performed according to the manufacturer’s instruction. His-CRABP1 (50 μM) was prepared in 1× kinase buffer (40 mM Tris, pH 7.5, 20 mM MgCl2, 0.1 mg/ml BSA and 50 μM DTT). Enzyme mixture was prepared with recombinant 0.3 ng/μl human CaMKIID (ThermoFisher Scientific), 0.03 μg/μl CaM (Sigma-Aldrich, C4874), and 0.1 mM CaCl2 in 1x kinase buffer. Fifty microgram of kinase substrate autocamtide-2 (Santa Cruz Biotechnology) and 25 μM ATP were diluted in 1x kinase buffer. Kinase reaction was initiated with addition of 1 μl of CRABP1, 2 μl of enzyme mixture and 2 μl of autocamtide-2 mixture in a 384 well plate at room temperature for 1 h followed by adding 5 μl of ADP-Glo™ reagent for 40 min and then 10 μl of kinase detection reagent for another 1 h. Luminescence was detected in Tecan microplate reader (Infinite M1000 Pro, Tecan).
In vitro interaction and competition and semi-in vitro kinase assay
His pulldown assay: purified His-CRABP1 was captured by Ni-nitrilotriacetic acid (NTA) agarose beads (Qiagen) for 2 h and used as bait. Flag-CaMKIIδ was synthesized with TnT T7-coupled reticulocyte lysate system (Promega). His-CRABP1/Ni-NTA agarose beads were incubated with Flag-CaMKIIδ in TnT-synthesized protein products in the presence or absence of 1 mM CaCl2 in the binding buffer overnight. M2 pulldown assay: Flag-CaMKIIδ was precipitated with M2 agarose beads (Sigma-Aldrich) for 2 h. Flag-CaMKIIδ/M2 agarose beads were incubated with purified 2 μM His-CRABP1. Competition assay: similarly, Flag-CaMKIIδ/M2 agarose beads were incubated with 1.5 μM His-CRABP1 in the presence of increased amounts of GST-CaM (0.1-2.0 μM) or with 0.5 μM GST-CaM in the presence of increased amounts of His-CRABP1 (0.5-4.0 μM) for 2 h followed by western blot.
For Semi-in vitro kinase assay, Flag-CaMKIIδ were used as enzyme and substrate source for autophosphorylation. Flag-CaMKIIδ/M2 agarose beads were prepared as above. After washing, the beads were incubated in the total volume of 35 μl reaction buffer (50 mM HEPES, pH 7.4, 10 mM MgCl2, 10 mM MnCl2, 0.1 mM ATP, 10 mM β-glycerophosphate, and 1 mM CaCl2) containing 0.5 μM GST-CaM and 1.5 μM His-CRABP1 (or increasing amounts indicated) at 30 °C for 15 minutes. At the end of the reaction, SDS loading buffer was added and boiled. CaMKII phosphorylation at T286 was detected by western blot.
Quantitative real time PCR
Total mRNA was extracted from frozen heart tissue with Trizol (Invitrogen) and 2μg of mRNA was reverse transcribed to cDNA using the Omniscript RT kit (Qiagen) followed by real time PCR reaction in triplicates of each sample. The relative values were normalized to Gapdh and Actb, and the data normalized to Actb are presented as ΔΔCq methods. All the primers are described in Table 1.
Table 1.
Primers used for quantitative real time PCR.
| Gene name | Primer orientation | Primer Sequences | NCBI Reference |
|---|---|---|---|
| Nppa | Forward | CACAGATCTGATGGATTTCAAGA | NM_008725.3 |
| Reverse | CCTCATCTTCTACCGGCATC | ||
| Nppb | Forward | GTCTGGCCGGACACTCAG | NM_008726.5 |
| Reverse | TGCACTGGTGTCTTCAACAAC | ||
| Col1a1 | Forward | GAGCGGAGAGTACTGGATCG | NM_007742.4 |
| Reverse | TACTCGAACGGGAATCCATC | ||
| Col3a1 | Forward | ACGTAAGCACTGGTGGACAG | NM_009930.2 |
| Reverse | AGCTGCACATCAACGACATC | ||
| Gapdh | Forward | GGGTTCCTATAAATACGGACTGC | NM_008084.3 |
| Reverse | CCATTTTGTCTACGGGACGA | ||
| Actb | Forward | TGGCCTTAGGGTTCAGGGGG | NM_007393.5 |
| Reverse | GTGGGCCGCTCTAGGCACCA |
Statistical analysis
Comparisons among groups were evaluated with Student’s t-test or ANOVA with two-way (Bonferroni) when appropriate. Differences were considered significant at P<0.05. All values are presented as mean±SEM.
Results
CRABP1 directly interacts with CaMKII
Our unpublished preliminary studies indicated interaction of CRABP1 with CaMKII. Using a reconstituted HEK293T system co-expressing GFP-CaMKIIα and Flag-CRABP1, the association of CRABP1 with CaMKII is detected in co-immunoprecipitated complex (Fig. 1A). Importantly, in normal hearts without any cardiac stimulation or pathological insult, endogenous CRABP1 constitutively associates with CaMKIIδ (Fig. 1B). Since CaMKIIδ is the predominant CaMKII isoform in the heart and its over-activation is a major determinant of various cardiac diseases (Mattiazzi et al. 2015), we assessed interaction of CRABP1 with CaMKIIδ in a reciprocal in vitro pulldown assay using TnT-synthesized Flag-CaMKIIδ and purified His-CRABP1. The data show direct interaction independent of calcium (Fig. 1C and D).
Figure 1.
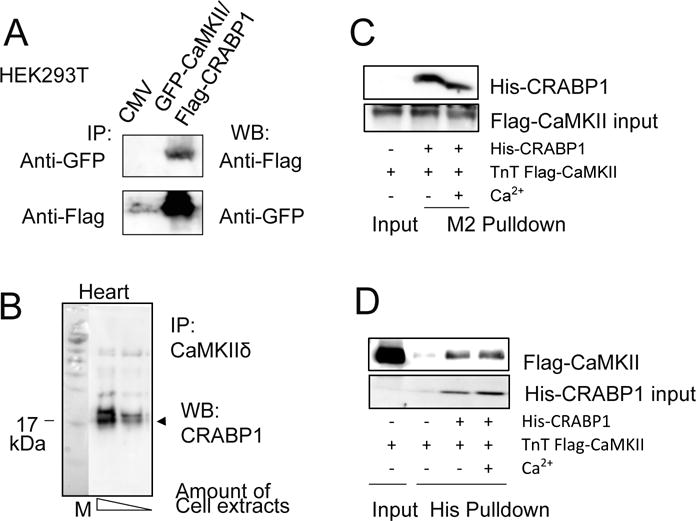
Direct interaction of CRABP1 with CaMKII. A, Co-immunoprecipitation (IP) of GFP-CaMKII and Flag-CRABP1 in HEK293T cells. B, Co-immunoprecipitation of endogenous CaMKIIδ and CRABP1 in ventricular lysates of healthy normal WT mice. C-D, In vitro interaction assay. Flag-CaMKII captured by M2 beads from TnT synthesized products interacted with His-CRABP1 independent of Ca2+ (C). Reciprocally, His-CRABP1 captured by Ni-NTA beads interacted with Flag-CaMKII independent of Ca2+ (D). TnT refers to synthesis by in vitro transcription and translation.
Deleting Crabp1 reduces cardiac function and increases CaMKII activation
Direct interaction of CRABP1 with CaMKII suggests that CRABP1 may modulate CaMKII activity and heart function. To assess the physiological role of CRABP1, we generated Crabp1-null mice (CKO) in the C57BL/6J background using a Crabp1-targeted DE3 (ES) clone containing an interrupted Crabp1 allele with a 5 bp insertion generating a Not1 site (Fig. 2A). Genotype was confirmed by PCR amplification of 783 bp (WT allele) or 788 bp (CKO allele); further, Not1 digestion of the CKO allele generated 638 bp and 150 bp fragments. As predicted, CRABP1 expression is absent in CKO mice (Fig. 2A, bottom).
Figure 2.
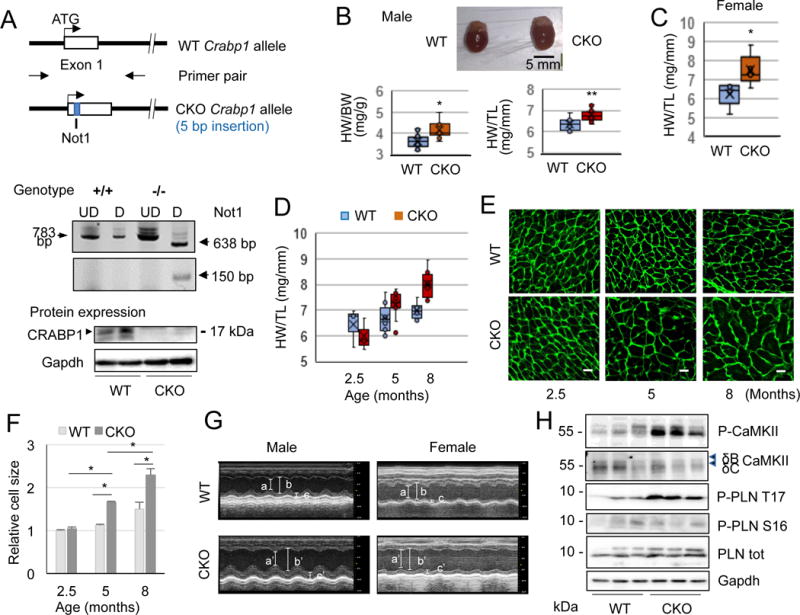
Reduced basal cardiac function and elevated CaMKII in CKO mice. A, Crabp1 allele was interrupted by a 5 bp insertion containing a Not1 site and CKO genotype is confirmed by Not1 digestion. CRABP1 is absent in CKO mice generated in C57BL/6J background. UD: undigested, D: digested. B, Representative heart images of male normal WT and CKO mice (top). Ratios of heart-to-body weight (HW/BW) and -tibia length (HW/TL) of male WT and CKO mice at age 16-20 weeks (n=10 each). *P<0.05, **P<0.01 vs. WT (t-test). C, HW/TL ratio of female WT and CKO mice (n=5 each). *P<0.05 vs. WT (t-test). D, HW/TL ratio of male WT and CKO mice at the indicated age. *P<0.05, **P<0.01 (t-test; n=3 each for 2.5 months, n=10-11 for 5 months, and n=6 each for 8 months). E, Representative microphotographs of WGA staining (n=3 each). Scale bar: 20 μm. F, Summary of relative cell size in D. *P<0.05 (t-test). G, Representative M-mode echocardiographs (a, a’: end-systolic LVID, b, b’: end-diastolic LVID, c, c’: end-diastolic LVPW). H, Representative western blots of ventricular lysates from normal WT and CKO mice probed with antibodies against phospho-CaMKII at T287 and phospho-PLN at T 17 and S16.
Interestingly, male CKO mice have larger hearts (Fig. 2B, upper) and significant increases in heart-to-body weight and heart-to-tibia length ratios (Fig. 2B, bottom). Female CKO mice show a similar phenotype (Fig. 2C). There is no significant difference in body weight between age-matched WT and CKO mice (data not shown). The hypertrophic phenotype results in significantly increased heart mass that is evident at 5-months of age and remains until the last point of examination at 8-months of age (Fig. 2D). This is further verified by wheat germ agglutinin (WGA) staining of sarcolemma, showing significant increases in the size of cardiac myocyte in CKO mice (Fig. 2E and F). We suspect that this hypertrophic phenotype of CKO mice could have reduced baseline heart function. M-mode echocardiography in mice at 4-5 months of age (Fig. 2G and Table 2) show that both male and female CKO mice have significantly reduced systolic performance based upon percent ejection fraction (EF) (from 69.12±1.6% in WT to 50.7±2.68% in CKO of male, P<0.001, and from 56.99±2.09% in WT to 45.67±1.6% in CKO of female, P<0.001). Declines in the cardiac contractile function are associated with left ventricular (LV) dilation. An increase in LV posterior wall thickness is also observed in female CKO mice. This echocardiographic evaluation indicates that loss of CRABP1 is associated with contractile dysfunction and LV dilation in both sexes, suggestive of a dilated cardiomyopathy (DCM). During these characterizations we found no evidence of a significant sex difference in the overall cardiac phenotype. To limit the confounding factor of sex in the molecular mechanism of signaling or signal integration with the hormonal system, we have focused on male mice in subsequent evaluations of molecular and pathological changes.
Table 2.
Echocardiographic parameters of male and female WT vs. CKO mice at basal level.
| Male | Female | |||||
|---|---|---|---|---|---|---|
| WT (n=13) | CKO (n=15) | P = | WT (n=13) | CKO (n=11) | P = | |
| HR (bpm) | 496.85±12.66 | 476.1±9.69 | 0.2 | 528.31±10.81 | 513.09±12.52 | 0.37 |
| EF (%) | 69.12±1.6 | 50.7±2.68* | 0.001 | 56.99±2.09 | 45.67±1.6* | 0.001 |
| FS (%) | 37.96±0.77 | 25.65±1.79* | 0.001 | 29.89±1.46 | 22.47±0.9* | 0.001 |
| LVIDs (mm) | 2.01±0.09 | 2.95±0.1* | 0.001 | 2.43±0.1 | 3.13±0.15* | 0.001 |
| LVIDd (mm) | 3.23±0.1 | 3.96±0.07* | 0.001 | 3.47±0.11 | 4.02±0.16* | 0.001 |
| LVPWd (mm) | 0.74±0.05 | 0.79±0.06 | 0.51 | 0.6±0.03 | 0.75±0.04* | 0.01 |
| LVVs (ml) | 22.3±2.28 | 34.92±2.85* | 0.01 | 21.69±2.15 | 40.16±4.68* | 0.001 |
| LVVd (ml) | 52.07±4.26 | 68.95±2.83* | 0.01 | 50.85±3.62 | 72.78±6.82* | 0.001 |
| SV (ml) | 29.31±1.91 | 34.03±1.82 | 0.085 | 29.08±1.46 | 32.62±2.52 | 0.29 |
| CO (ml/min) | 14.37±0.77 | 16.4±0.99 | 0.12 | 15.39±1.17 | 16.72±1.32 | 0.46 |
Data were expressed as mean ± SEM. HR: heart rate; EF: ejection fraction; FS: fractional shortening; LVIDs: end-systolic left ventricular internal diameter; LVIDd: end-diastolic left ventricular internal diameter; LVPWd: end-diastolic left ventricular posterior wall thickness; LVVs: end-systolic left ventricular volume; LVVd: end-diastolic left ventricular volume; SV: stroke volume; CO: cardiac output.
statistically significant (student t-test).
It is known that CaMKII expression and phosphorylation at T287 are enhanced in DCM (Fischer et al. 2014). To determine whether loss of CRABP1 affects the baseline activity of CaMKII, we monitored phosphorylation of CaMKII at T287 and phosphorylation of its substrate PLN at T17 in the ventricular cytosolic extracts of age-matched WT and CKO mice. It appears that both CaMKII and PLN phosphorylation are significantly elevated in CKO hearts as compared to WT hearts (Fig. 2H). Importantly, phosphorylation at S16, a PKA target site, is not affected (Fig. 2H), indicating that deleting Crabp1 specifically affects autonomous activation of CaMKII. Presumably, depleting CRABP1 from the cells releases CaMKII from its baseline inhibition.
Deleting Crabp1 exacerbates acute β-AR stimulated apoptosis and necrosis in the heart
Excessive β-AR stimulation has been known to lead to detrimental cardiac remodeling (Nichtova et al. 2012, Xu et al. 2016). We examined cardiac remodeling in WT and CKO mice on acute β-AR stimulation with ISO (80 mg/kg, two doses with an 8 h interval). Acute ISO insult induces apoptosis in both genotypes, but CKO mice exhibit significantly increased cardiomyocyte apoptosis as compared to WT mice (Fig. 3A and B). Lactate dehydrogenase (LDH) assay shows ISO treatment causes greater leakage of LDH in serum in CKO mice (from 183.3±23.4 mU/ml in WT to 311.2±36.4 mU/ml in CKO, P<0.05) (Fig. 3C), indicating necrotic cytotoxicity. Specifically, the extent of necrotic lesions in ISO-challenged hearts is confirmed by an examination of immunoglobulin G (IgG) deposit in the center of individual cardiac myocytes counter-stained with WGA (Strakova et al. 2014, Townsend et al. 2008). The results indicate that ISO challenge provokes severe membrane disintegration in larger areas in CKO hearts (from 0.8±0.12% in WT to 2±0.5% in CKO, P<0.05) (Fig. 3D and E). The control hearts of both WT and CKO exhibit no necrosis (data not shown). Taken together, ISO challenge is more detrimental for the CKO hearts, causing more severe apoptosis and necrosis.
Figure 3.
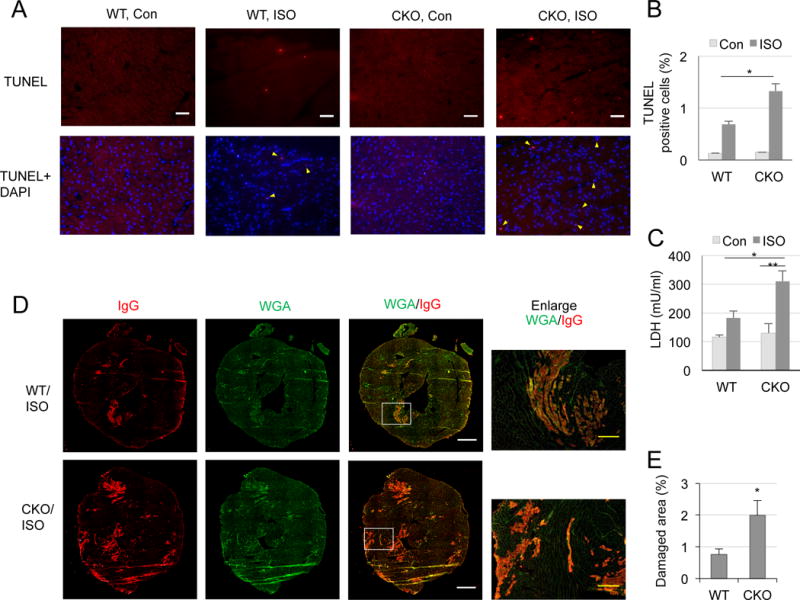
Exacerbated ISO-induced cardiomyocyte apoptosis and necrosis in CKO hearts. A, Representative microphotographs of TUNEL staining of hearts from WT and CKO mice acutely treated with ISO. Scale bar: 50 μm. B, TUNEL positive cells (yellow triangles) from non-overlapping 10 random areas of each slide were counted and summarized. *P<0.05 vs. WT ISO-treated mice (two-way ANOVA, n=3). C, LDH cytotoxicity assay in serum. *P<0.01 and ** P<0.001 (two-way ANOVA, n=7-11). D, Representative microphotographs of IgG (red) staining with WGA (green) background. Scale bar: 1 mm. The areas of white boxes are enlarged. Yellow scale bar: 200 μm. E, Quantitation of IgG staining. *P<0.05 vs. WT ISO-treated mice (t-test, n=7-8).
Deleting Crabp1 increases CaMKII activity in baseline and further in ISO challenge
At the molecular level, excessive and/or persistent activation of CaMKII is responsible for various heart diseases (Said et al. 2011, Vila-Petroff et al. 2007, Zhang et al. 2005). Thus, we monitored CaMKII phosphorylation and PLN phosphorylation in the ventricular cytosolic fraction of WT and CKO mice challenged with acute ISO (80 mg/kg, two doses with an 8 h interval). Indeed, while CaMKII phosphorylation at T287 is markedly increased in control CKO hearts, it is elevated to an even greater degree in acute ISO-challenged CKO hearts (from 2.2±0.3 fold in WT to 5±0.5 fold in CKO, P<0.01) (Fig. 4A). Consistently, phosphorylation of PLN at T17, a CaMKII target site, is significantly increased in CKO hearts with ISO insult (from 4.9±0.5 fold in WT to 7.2±1.0 fold in CKO, P<0.05) (Fig. 4B), indicating greater activation of CaMKII. This is consistent with histological changes associated with apoptosis and necrosis.
Figure 4.
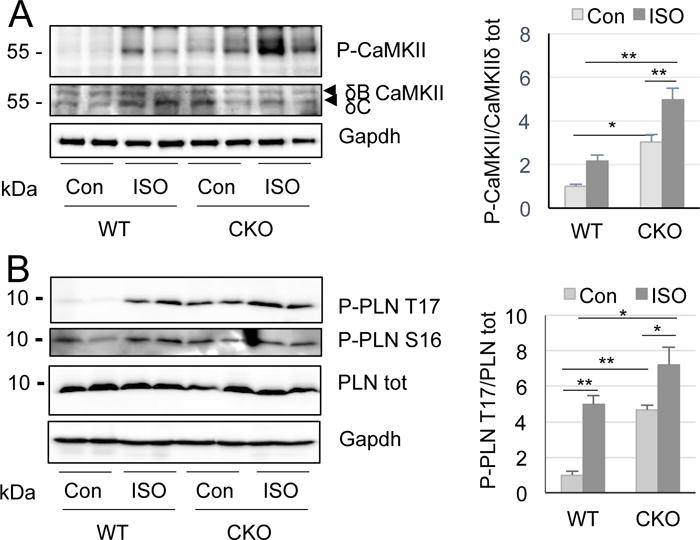
Increased ISO-induced CaMKII activation in CKO hearts. A, Autophosphorylation of CaMKII at T287 in ventricular lysates from WT and CKO mice in basal level and ISO treatment. Quantification at right. *P<0.05 and **P<0.01. B, Phosphorylation of PLN at T17. Quantitation at right. *P<0.05 and **P<0.001 (two-way ANOVA, n=4-10).
Inhibiting CaMKII prevents ISO-induced cardiac damage in CKO mice
We then verified if loss of CRABP1 caused more severe ISO-induced cardiac damage due to elevated CaMKII by treating CKO mice with a CaMKII inhibitor KN-93. As shown in Fig. 5A, KN-93 abolishes acute ISO effect on PLN phosphorylation at T17, but KN-92, an inactive derivative of KN-93, does not (Fig. 5B), while PLN phosphorylation at S16 is not affected by either KN-92 or KN-93 (Fig. 5C). These results indicate that inhibition of CaMKII blocks CRABP1 depletion effect on elevation of PLN phosphorylation with ISO challenge. Further, inhibiting CaMKII decreased IgG staining (Fig. 5D and E) and LDH leakage in serum (Fig. 5F) to the level of WT after ISO insult as shown in Fig. 4E and C, respectively. Thus, KN-93 prevents increase in ISO-induced necrotic damage in CKO mice, supporting the notion that depleting CRABP1 sensitizes myocardium to acute ISO insult due to elevating CaMKII.
Figure 5.
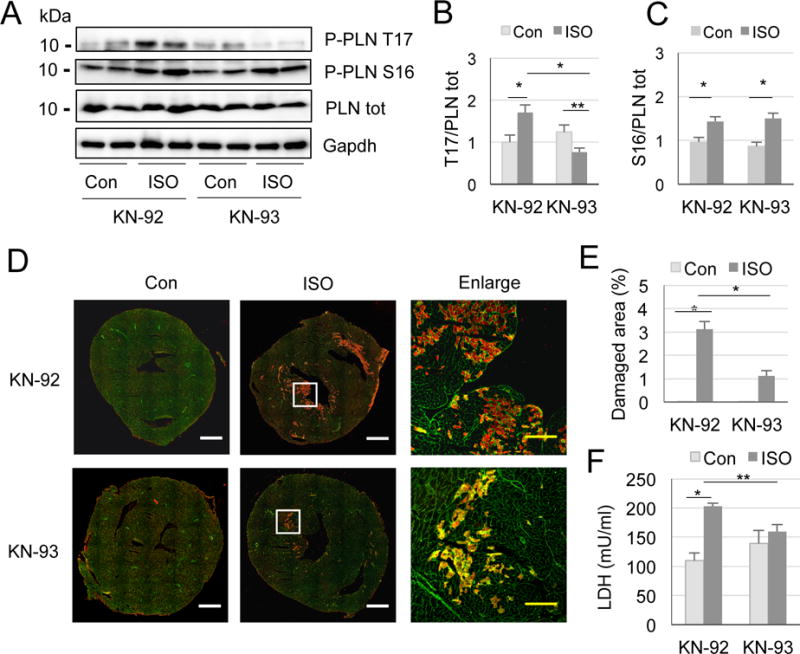
Inhibiting CaMKII prevents ISO induced damage in CKO mice. A, Phosphorylation of PLN at T17 (B), *P<0.01, **P<0.05, and at S16 (C), *P<0.01 (two-way ANOVA, n=4-6). D, Representative microphotographs of IgG/WGA staining of CKO mice injected with 0.9% saline or ISO in the pre-treatment of KN-92 or KN-93. White rectangles are enlarged at right. White scale bar: 1 mm, yellow scale bar: 200 μm. E, Quantitation of IgG staining. *P<0.001 (n=2, 6, 3, 9, respectively). F, LDH cytotoxicity assay in serum. *P<0.001, **P=0.01 (two-way ANOVA, n=3-5).
Deleting Crabp1 increases susceptibility to chronic ISO triggered heart hypertrophy and fibrosis
We then examined chronic (30 mg/kg/day for 15 days) ISO-triggered hypertrophy. Apparently, chronic ISO challenge causes heart enlargement in both WT and CKO mice, but CKO hearts are significantly bigger than WT hearts (Fig. 6A). Consistently, while basal expressions of hypertrophic markers such as atrial natriuretic peptide (Nppa) and brain natriuretic peptide (Nppb) are already elevated in CKO mice as compared to WT mice; ISO treatment further elevates the expressions of these markers in CKO mice (Fig. 6B and C). This hypertrophic event is caused, at least in part, by increased cardiac myocyte dimension as determined by WGA staining (Fig. 6D and E). As shown in Fig. 2E, Crabp1 deletion alone (without ISO challenge) readily increases cardiac myocyte dimension, but ISO challenge further significantly increases cell size in CKO mice. These results suggest that deleting Crabp1 predisposes these heart muscles to exaggerated stress response to maintain cardiac output. This is consistent with changes in myocyte dimension.
Figure 6.
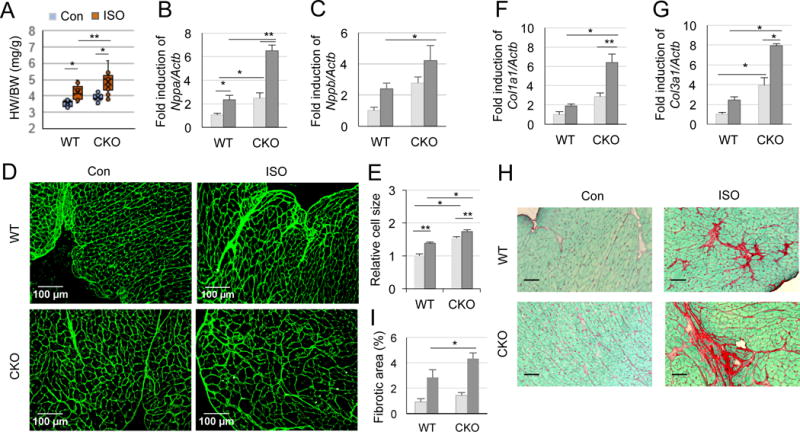
Increased chronic ISO-induced hypertrophy and fibrosis in CKO mice. WT and CKO mice were treated with 0.9% saline or ISO for 15 days (A-G) or for 35 days (H-I). A, Ratio of heart-to-body weight. *P<0.01 and **P<0.05 (two-way ANOVA, n=12-13). B-C, Relative expression of hypertrophic markers normalized to reference gene Actb: Nppa (B) and Nppb (C). *P<0.05 and **P<0.001 (two-way ANOVA, n=4-8). D, Representative microphotographs illustrate cross-sectional cardiomyocytes stained with WGA in WT and CKO mice. E, Summary of cell size from average 100 cells in 4 areas of a heart in D. *P<0.05 and **P<0.001 (two-way ANOVA, n=3-6). F-G, Relative expressions of Col1a1 (F) and Col3a1 (G) normalized to Actb. *P<0.001 and **P<0.01 (two-way ANOVA, n=3-8). H, Representative microphotographs of sirius red-fast green (SRFG) staining of hearts after ISO treatment for 5 weeks. Scale bar: 50 μm. I, Quantitation of fibrotic areas (red) from non-overlapping average 27 areas of each slide in H. *P<0.05 (two-way ANOVA, n=4-6).
Chronic β-AR stimulation also leads to cardiac fibrosis, a major determinant of cardiac stiffness that is mostly caused by the deposition of collagen 1 and 3 in the extracellular matrix (Fan et al. 2012, Garg and Khanna 2014). Upon examining the expression of fibrosis gene markers such as Col1a1 and Col3a1, we found that ISO stress for 15 days significantly elevates the expression levels of Col1a1 and Col3a1 in CKO mice, indicative of more severe fibrosis (Fig. 6F and G). Interestingly, Col3a1 (less abundant than Col1a1 in heart) is already elevated in CKO mice without β-AR stimulation (Fig. 6G). In a 5-week ISO challenge, both WT and CKO mice develop fibrosis (SRFG staining), but CKO hearts reveal more severe fibrosis than WT hearts (Fig. 6H and I). These findings show that deleting Crabp1 significantly increases the myocardial susceptibility to hypertrophy and fibrosis during β-AR stimulation and at the basal level. The effects of ablating CRABP1 on CaMKII activation, as detected in the context of acute β-AR insult and chronic challenge, would suggest that CRABP1 likely acts to modulate CaMKII activity.
CRABP1 inhibits CaMKII activation by competing with CaM for CaMKII binding
CaMKIIδ expression and activity increase in response to pressure overload, hypertrophy, and heart failure (Anderson et al. 2011, Hoch et al. 1999, Zhang et al. 2003). Interestingly, CRABP1 expression is also elevated at early stage (until day 15), but then suppressed at later stages (day 35) during ISO challenge (Fig. 7A). This suggests a compensatory response in animals by first elevating CRABP1 levels as a protecting mechanism in order to dampen CaMKII activation; ultimately a reduction in CRABP1 results in the loss of this CaMKII-modulating protective mechanism in late stages. We then rigorously evaluated if CRABP1 directly inhibits CaMKII activity using in vitro ADP-Glo kinase assay (Fig. 7B). The result shows that CRABP1 has a IC50 ~3 μM, confirming its direct inhibitory effect on CaMKII activation. This is consistent with the significant reduction in CaMKII autophosphorylation by adding purified CRABP1 to the kinase reaction mixture (Fig. 7C). In vitro competition assay (CaMKIIδ incubated with increasing amounts of CaM in the presence of 1.5 μM CRABP1, Fig. 7D top) shows that CRABP1 constitutively interacts with CaMKII, independent of Ca2+, and is competed out by increasing amounts of CaM. In the reciprocal reaction, CaM (0.5 μM) binding to CaMKII in the presence of Ca2+ is gradually reduced by increasing amounts of CRABP1 (Fig. 7E, top panel). Of note, this activity of CRABP1 is detected in the absence of RA, suggesting a new functional role for apo-CRABP1, which is to compete with CaM for binding to CaMKII thereby dampening Ca2+-CaM induced CaMKII activation.
Figure 7.
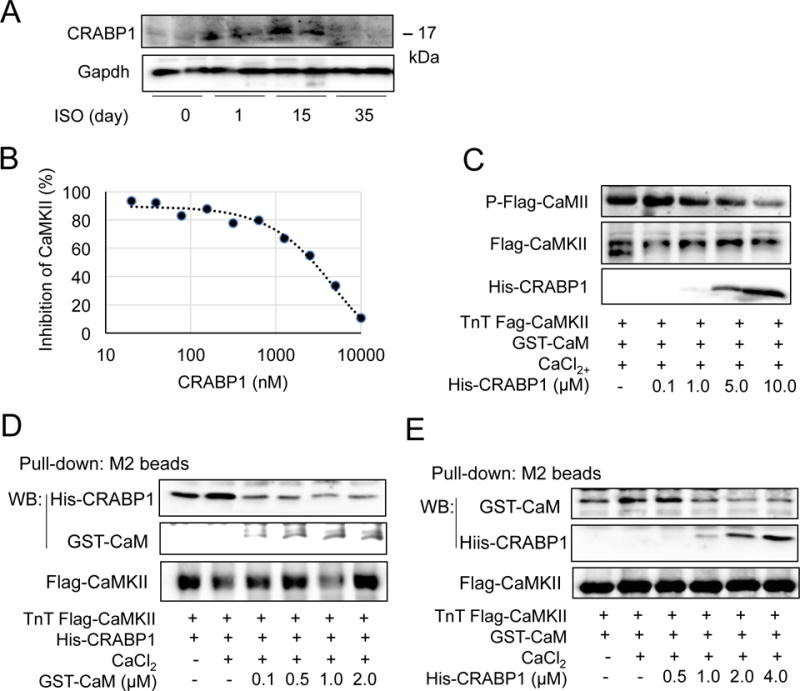
CRABP1 directly inhibits CaMKII by competing with CaM for CaMKII binding. A, Expression of CRABP1 in the ventricles of WT mice injected with ISO for the indicated days. B, ADP-Glo kinase assay. Increasing amounts of CRABP1 were added to the kinase reaction mixture containing 0.3 ng/μl CaMKII, 0.03 μg/μl CaM and ATP. Percent activity was calculated from illumined luminescence. C, CaMKII phosphorylation was decreased by increasing His-CRABP1 (0.1-10.0 μM) in CaMKII kinase reaction mixture. D, In vitro competition. Increasing amounts of GST-CaM (0.1-2.0 μM) decreased His-CRABP1 (1.5 μM) binding to Flag-CaMKII/M2 bead complex. E, Increasing amounts of His-CRABP1 (0.5-4.0 μM) decreased GST-CaM (0.5 μM) binding to the CaMKII complex.
Discussion
This is the first report revealing the physiological activity of apo-CRABP1 in protecting pathological cardiac remodeling by dampening CaMKII activation. CKO mice exhibit declines in systolic function and ventricular dilation. The underlying mechanism of action of CRABP1 is mediated by its direct interaction with, and dampening of, CaMKII through competing with its principal upstream signaling molecule, CaM. Importantly, this interaction occurs without the addition of RA. To this end, CRABP1 has also been implicated as pro-tumorigenic and pro-metastatic in mesenchymal and pancreatic neuroendocrine tumors, which seems to be also independent of RA binding (Kainov et al. 2014), supporting certain physiological roles of apo-CRABP1.
CaMKII inhibition and gene knockout protect animals from various heart diseases and failure (Gray and Heller Brown 2014, Grimm et al. 2015, Mattiazzi et al. 2015, Said et al. 2011). CaMKII transgenic mice develop hypertrophy and heart failure (Zhang et al. 2002, Zhang et al. 2003). It is also recognized that excessive and/or persistent β-AR stimulation causes CaMKIIδ over-activation and maladaptive cardiac remodeling. Since CRABP1 negatively modulates CaMKII, CKO mice might have reduced baseline cardiac function. This is confirmed, evidenced by the development of a hypo-contractile dilated phenotype of the left ventricle characterized by significantly decreased ejection fraction and LV dilation, similar to the DCM phenotype of CaMKII transgenic mice (Japp et al. 2016, Zhang et al. 2002, Zhang et al. 2003). Female CKO mice also exhibit compromised systolic performance with DCM phenotype. Nonetheless, at the ages we examined both male and female CKO mice show preserved stroke volume and cardiac output. CKO mice develop hypertrophic phenotype at approximately 4-5 months old. This phenotype is similar to that of a CaMKII transgenic mouse model (Zhang et al. 2002, Zhang et al. 2003), suggesting that CRABP1 may prevent cardiac hypertrophy and failure, in part, by dampening the activity of CaMKII. This study reveals that CRABP1 is a new physiological regulator of CaMKII and is critical to heart function. It is tempting to speculate that CRABP1 plays a protective role by increasing the threshold for CaMKII activation, and that CKO mice may be more vulnerable to β-AR stimulation (ISO treatment) either by acute administration with a high dose or chronic treatment with a low dose. These are experimentally demonstrated in this report.
It is known that excessive CaMKII activation and elevated intracellular Ca2+ result in mitochondrial Ca2+ uptake and release of cytochrome c to activate intrinsic apoptotic pathway (Feng and Anderson 2017). CaMKIIδ overexpression or activation with doxorubicin induces necrosis through its oxidation and autonomous phosphorylation and downstream target RIP3 phosphorylation (Zhang et al. 2016). In the acute ISO stress model, CRABP1 depletion significantly aggravates acute cardiac damage characterized by increased apoptosis, necrosis, and LDH leakage into serum. Specifically, IgG accumulation within the cardiac myocytes reveals local sarcolemma disruption that allows IgG to enter the myocyte (Strakova et al. 2014, Townsend et al. 2008). WGA provides counterstaining of the extracellular matrix surrounding the cardiomyocytes. Thus, positive IgG staining of the hearts together with elevated serum LDH in CKO mice (an indicator of general tissue damage or hemolytic condition and a pathologic marker of necrosis) are consistent with a model where CRABP1 depletion causes more severe necrosis under acute ISO insults. The chronic ISO stress results also confirm that CRABP1 depletion exacerbates cardiac hypertrophic responses and fibrotic remodeling. Since regulation of CaMKII is crucial to the maintenance of normal heart function, over-activation of CaMKII (such as by depleting CRABP1) would promote not only acute injury like apoptosis and necrosis but also chronic remodeling. Our data clearly support this notion. It is possible that the pathological outcome of ISO treatment in CKO mice may be mediated, directly, through CRABP1’s effects on CaMKII activation, or by other indirect mechanisms. Our data showing that KN-93 treatment abolishes the effect of depleting CRABP1 on CaMKII activation and ISO-induced necrosis as assessed by IgG staining and LDH cytotoxicity assay (compare Fig. 5E and F to Fig. 3E and C, respectively) would support the notion that CRABP1 may directly regulate CaMKII activation. However, there may be other potential mechanisms of CRABP1 action in protecting the heart, which remains to be examined.
IC50 for apo-CRABP1, determined from in vitro ADP-Glo kinase assay, is approximately 3 μM with regards to CaMKII inhibition. Although CRABP1 expression level in the heart of healthy individuals is relatively low (https://www.proteomicsdb.org/proteomicsdb/#human/proteinDetails/P29762/expression), it is induced during ISO, but eventually is reduced to the basal level when the heart develops serious remodeling (Fig. 7A). The low level of Crabp1 expression in early stages of disease development reduces the potential of targeting Crabp1 to suppress CaMKII activation, but the induced Crabp1 expression during the course of disease progression may still provide a window for intervention. Importantly, the eventual loss of CRABP1 in later stages of heart diseases may contribute to the unrestricted activation of CaMKII. This may be related to eventual pathological outcome in a chronically stimulated condition. An interesting possibility is that the status of the endogenous RA, a CRABP1 ligand, may modulate CRABP1 function in the heart. To this end, RA has been shown to have a protective effect in heart failure (Choudhary et al. 2008, Zhu et al. 2015). Thus, it will be an important topic in further studies to examine whether and how RA, or other retinoid-like compounds, may affect the action of CRABP1.
Mechanistically, CRABP1 interacts with, and negatively modulates, CaMKII. While a lack of PLN phosphorylation at S16 rules out that CRABP1 affects PKA-activation, elevation of PLN phosphorylation at T17 supports CRABP1’s specificity in dampening CaM-induced CaMKII activation. CaMKIIδ activation can be achieved via O-glycosylation at S280, oxidation at M281/282, nitrosylation at C273 and C290, and phosphorylation at T287 (Feng and Anderson 2017). Our data clearly show that CRABP1 dampens CaMKII autophosphorylation at T287. It remains to be determined whether CRABP1 can also modulate other modifications.
Secretoneurin has received increasing attention for drug development because pathologically induced internalized ecretoneurin interacts with both CaM and CaMKII to attenuate CaMKII activity, which may be a prognostic biomarker for, and protect hearts from, failure (Ottesen et al. 2015). CRABP1 expressed in heart represents the first physiological, cell autonomous, negative modulator of CaMKII in cardiomyocytes, which is also protective.
It is important that this specific activity of CRABP1 is independent of RA. Our data suggest a model where constitutively CaMKII-bound CRABP1 raises the threshold (or decrease the sensitivity) of CaMKII to Ca2+/CaM by directly competing with Ca2+/CaM. This results in the need for higher concentrations of Ca2+/CaM to activate CaMKII (Fig. 7). Intriguingly, calcineurin and CaMKII bind Ca2+/CaM with different affinities: calcineurin has a very high Ca2+/CaM affinity (Kd ~ 28 pM), whereas CaMKII has a relatively low Ca2+/CaM affinity (Kd 10-50 nM) (Saucerman and Bers 2008, 2012). Thus, CaMKII activation requires relatively high local [Ca2+]i and free CaM. However, whether CRABP1 interaction with CaMKII may reduce CaM affinity remains to be determined. CaMKII inhibition is a therapeutic strategy for various cardiac diseases. CRABP1 is likely to provide an endogenous modulator of CaMKII to safe-guard its activation. For direct modulation of CaMKII activity, CaMKIIN and related peptide inhibitors and small molecule inhibitors have been developed, but they are used only as experimental tools due to the low specificity and unwanted effects (Pellicena and Schulman 2014). Therefore, if RA or other more selective ligands affect the activity of CRABP1 with regards to CaMKII modulation, specifically and conditionally targeting CRABP1 may provide a potentially viable therapeutic strategy to specifically dampen CaMKII for various heart diseases, especially heart failure.
Summary.
CRABP1, a retinoic acid binding protein with unknown physiological function, is found to regulate CaMKII activity in cardiomyocyte, thereby protecting animals from drug-induced heart failure.
Acknowledgments
We thank Drs. Joseph M. Metzger and Jop van Berlo for professional discussion. We especially thank Dr. Berlo for proofreading the manuscript. We also thank Kurt W. Prins, Yi-Wei Lin, Bomi Lee, Yu-Lung Lin, and Jennifer Nhieu for technical support.
Sources of funding
This work was supported by NIH grants DK54733, DK60521, DK60521–12S1, and the Dean’s Commitment and the Distinguished McKnight Professorship of University of Minnesota to LNW.
Footnotes
Disclosures
None.
References
- Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. Journal of Molecular Cellular Cardiology. 2011;51:468–473. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. Journal of Clinical Investigation. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary R, Palm-Leis A, Scott RC, 3rd, Guleria RS, Rachut E, Baker KM, Pan J. All-trans retinoic acid prevents development of cardiac remodeling in aortic banded rats by inhibiting the renin-angiotensin system. American Journal of Physiology-Heart and Circulatory Physiology. 2008;294:H633–644. doi: 10.1152/ajpheart.01301.2007. [DOI] [PubMed] [Google Scholar]
- de Bruijn DR, Oerlemans F, Hendriks W, Baats E, Ploemacher R, Wieringa B, Geurts van Kessel A. Normal development, growth and reproduction in cellular retinoic acid binding protein-I (CRABPI) null mutant mice. Differentiation. 1994;58:141–148. doi: 10.1046/j.1432-0436.1995.5820141.x. [DOI] [PubMed] [Google Scholar]
- Donovan M, Olofsson B, Gustafson AL, Dencker L, Eriksson U. The cellular retinoic acid binding proteins. Journal of Steroid Biochemistry Molecular Biology. 1995;53:459–465. doi: 10.1016/0960-0760(95)00092-e. [DOI] [PubMed] [Google Scholar]
- Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis and Tissue Repair. 2012;5:15. doi: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng N, Anderson ME. CaMKII is a nodal signal for multiple programmed cell death pathways in heart. Journal of Molecular Cellular Cardiology. 2017;103:102–109. doi: 10.1016/j.yjmcc.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero P, Said M, Sanchez G, Vittone L, Valverde C, Donoso P, Mattiazzi A, Mundina-Weilenmann C. Ca2+/calmodulin kinase II increases ryanodine binding and Ca2+-induced sarcoplasmic reticulum Ca2+ release kinetics during beta-adrenergic stimulation. Journal of Molecular Cellular Cardiology. 2007;43:281–291. doi: 10.1016/j.yjmcc.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorella PD, Giguere V, Napoli JL. Expression of cellular retinoic acid-binding protein (type II) in Escherichia coli. Characterization and comparison to cellular retinoic acid-binding protein (type I) Journal of Biological Chemistry. 1993;268:21545–21552. [PubMed] [Google Scholar]
- Fischer TH, Eiringhaus J, Dybkova N, Forster A, Herting J, Kleinwachter A, Ljubojevic S, Schmitto JD, Streckfuss-Bomeke K, Renner A, et al. Ca(2+)/calmodulin-dependent protein kinase II equally induces sarcoplasmic reticulum Ca(2+) leak in human ischaemic and dilated cardiomyopathy. European Journal of Heart Failure. 2014;16:1292–1300. doi: 10.1002/ejhf.163. [DOI] [PubMed] [Google Scholar]
- Garg M, Khanna D. Exploration of pharmacological interventions to prevent isoproterenol-induced myocardial infarction in experimental models. Therapeutic Advances in Cardiovascular Disease. 2014;8:155–169. doi: 10.1177/1753944714531638. [DOI] [PubMed] [Google Scholar]
- Gorry P, Lufkin T, Dierich A, Rochette-Egly C, Decimo D, Dolle P, Mark M, Durand B, Chambon P. The cellular retinoic acid binding protein I is dispensable. Proc Natl Acad Sci U S A. 1994;91:9032–9036. doi: 10.1073/pnas.91.19.9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray CB, Heller Brown J. CaMKIIdelta subtypes: localization and function. Frontiers in Pharmacology. 2014;5:15. doi: 10.3389/fphar.2014.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. Journal of Molecular Cellular Cardiology. 2010;48:322–330. doi: 10.1016/j.yjmcc.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm M, Ling H, Willeford A, Pereira L, Gray CB, Erickson JR, Sarma S, Respress JL, Wehrens XH, Bers DM, et al. CaMKIIdelta mediates beta-adrenergic effects on RyR2 phosphorylation and SR Ca(2+) leak and the pathophysiological response to chronic beta-adrenergic stimulation. Journal of Molecular Cellular Cardiology. 2015;85:282–291. doi: 10.1016/j.yjmcc.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circulation Research. 1999;84:713–721. doi: 10.1161/01.res.84.6.713. [DOI] [PubMed] [Google Scholar]
- Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK. The Diagnosis and Evaluation of Dilated Cardiomyopathy. Journal of the American College of Cardiology. 2016;67:2996–3010. doi: 10.1016/j.jacc.2016.03.590. [DOI] [PubMed] [Google Scholar]
- Kainov Y, Favorskaya I, Delektorskaya V, Chemeris G, Komelkov A, Zhuravskaya A, Trukhanova L, Zueva E, Tavitian B, Dyakova N, et al. CRABP1 provides high malignancy of transformed mesenchymal cells and contributes to the pathogenesis of mesenchymal and neuroendocrine tumors. Cell Cycle. 2014;13:1530–1539. doi: 10.4161/cc.28475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreusser MM, Lehmann LH, Wolf N, Keranov S, Jungmann A, Grone HJ, Muller OJ, Katus HA, Backs J. Inducible cardiomyocyte-specific deletion of CaM kinase II protects from pressure overload-induced heart failure. Basic Research in Cardiology. 2016;111:65. doi: 10.1007/s00395-016-0581-2. [DOI] [PubMed] [Google Scholar]
- Ling H, Gray CB, Zambon AC, Grimm M, Gu Y, Dalton N, Purcell NH, Peterson K, Brown JH. Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circulation Research. 2013;112:935–944. doi: 10.1161/CIRCRESAHA.112.276915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DL, Bristow MR. Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation. 2005;111:2837–2849. doi: 10.1161/CIRCULATIONAHA.104.500546. [DOI] [PubMed] [Google Scholar]
- Mattiazzi A, Bassani RA, Escobar AL, Palomeque J, Valverde CA, Vila Petroff M, Bers DM. Chasing cardiac physiology and pathology down the CaMKII cascade. American Journal of Physiology-Heart and Circulatory Physiology. 2015;308:H1177–1191. doi: 10.1152/ajpheart.00007.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollova MY, Katus HA, Backs J. Regulation of CaMKII signaling in cardiovascular disease. Frontiers in Pharmacology. 2015;6:178. doi: 10.3389/fphar.2015.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichtova Z, Novotova M, Kralova E, Stankovicova T. Morphological and functional characteristics of models of experimental myocardial injury induced by isoproterenol. General Physiology and Biophysics. 2012;31:141–151. doi: 10.4149/gpb_2012_015. [DOI] [PubMed] [Google Scholar]
- Ottesen AH, Louch WE, Carlson CR, Landsverk OJ, Kurola J, Johansen RF, Moe MK, Aronsen JM, Hoiseth AD, Jarstadmarken H, et al. Secretoneurin is a novel prognostic cardiovascular biomarker associated with cardiomyocyte calcium handling. Journal of the American College of Cardiology. 2015;65:339–351. doi: 10.1016/j.jacc.2014.10.065. [DOI] [PubMed] [Google Scholar]
- Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Frontiers in Pharmacology. 2014;5:21. doi: 10.3389/fphar.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Castro AV, Tran VT, Nguyen-Huu MC. Defective lens fiber differentiation and pancreatic tumorigenesis caused by ectopic expression of the cellular retinoic acid-binding protein I. Development. 1993;119:363–375. doi: 10.1242/dev.119.2.363. [DOI] [PubMed] [Google Scholar]
- Said M, Becerra R, Valverde CA, Kaetzel MA, Dedman JR, Mundina-Weilenmann C, Wehrens XH, Vittone L, Mattiazzi A. Calcium-calmodulin dependent protein kinase II (CaMKII): a main signal responsible for early reperfusion arrhythmias. Journal Molecular Cellular Cardiology. 2011;51:936–944. doi: 10.1016/j.yjmcc.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucerman JJ, Bers DM. Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophysical Journal. 2008;95:4597–4612. doi: 10.1529/biophysj.108.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucerman JJ, Bers DM. Calmodulin binding proteins provide domains of local Ca2+ signaling in cardiac myocytes. Journal of Molecular Cellular Cardiology. 2012;52:312–316. doi: 10.1016/j.yjmcc.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strakova J, Dean JD, Sharpe KM, Meyers TA, Odom GL, Townsend D. Dystrobrevin increases dystrophin’s binding to the dystrophin-glycoprotein complex and provides protection during cardiac stress. Journal of Molecular Cellular Cardiology. 2014;76:106–115. doi: 10.1016/j.yjmcc.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucharov CC, Mariner PD, Nunley KR, Long C, Leinwand L, Bristow MR. A beta1-adrenergic receptor CaM kinase II-dependent pathway mediates cardiac myocyte fetal gene induction. American Journal of Physiology-Heart and Circulatory Physiology. 2006;291:H1299–1308. doi: 10.1152/ajpheart.00017.2006. [DOI] [PubMed] [Google Scholar]
- Thaler C, Koushik SV, Puhl HL, 3rd, Blank PS, Vogel SS. Structural rearrangement of CaMKIIalpha catalytic domains encodes activation. Proceedings of the National Academy of Sciences of the United Sates of America. 2009;106:6369–6374. doi: 10.1073/pnas.0901913106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend D, Yasuda S, Li S, Chamberlain JS, Metzger JM. Emergent dilated cardiomyopathy caused by targeted repair of dystrophic skeletal muscle. Molecular Therapy. 2008;16:832–835. doi: 10.1038/mt.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venepally P, Reddy LG, Sani BP. Characterization of cellular retinoic acid-binding protein I from chick embryo and its ligand binding properties. Archives of Biochemistry and Biophysics. 1996;336:231–239. doi: 10.1006/abbi.1996.0553. [DOI] [PubMed] [Google Scholar]
- Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, Hajjar RJ, Kranias EG, Mundina-Weilenmann C, Mattiazzi A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovascular Research. 2007;73:689–698. doi: 10.1016/j.cardiores.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Wei LN, Chen GJ. Production and analyses of transgenic mice with ectopic expression of cellular retinoic acid-binding protein. Biochemical and Biophysical Research Communications. 1991;179:210–216. doi: 10.1016/0006-291x(91)91356-h. [DOI] [PubMed] [Google Scholar]
- Wang Xu S, Zhang P, Gong H, Gutierrez Cortes G, Zhu N, Yoon W, Tian YR, Wang W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nature Communications. 2016;7:13189. doi: 10.1038/ncomms13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nature Medicine. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, Belke DD, Dillmann WH, Rogers TB, Schulman H, et al. The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. Journal of Biological Chemistry. 2002;277:1261–1267. doi: 10.1074/jbc.M108525200. [DOI] [PubMed] [Google Scholar]
- Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circulation Research. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, Liu Y, Zheng W, Shang H, Zhang J, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nature Medicine. 2016;22:175–182. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zhu J, Zhao X, Yang K, Lu L, Zhang F, Shen W, Zhang R. All-Trans Retinoic Acid Ameliorates Myocardial Ischemia/Reperfusion Injury by Reducing Cardiomyocyte Apoptosis. PLoS ONE. 2015;10:e0133414. doi: 10.1371/journal.pone.0133414. [DOI] [PMC free article] [PubMed] [Google Scholar]