

Summary
Background
Plasma kallikrein is a serine protease that plays an integral role in many biological processes including coagulation, inflammation, and fibrinolysis. The main function of kallikrein in coagulation is the amplification of activated factor XIIa (FXIIa) generation which ultimately leads to thrombin generation and fibrin clot formation. Kallikrein is generated by FXIIa-mediated cleavage of the zymogen, prekallikrein which is usually complexed with the nonenzymatic cofactor, high-molecular-weight kininogen (HK). HK also serves as a substrate for kallikrein to generate the pro-inflammatory peptide, bradykinin (BK). Interestingly, prekallikrein-deficient mice are protected from thrombotic events while retaining normal hemostatic capacity. Therefore, therapeutically targeting kallikrein may provide a safer alternative to traditional anticoagulants with anti-inflammatory benefits.
Objectives
We sought to isolate and characterize an RNA aptamer that binds and inhibits plasma kallikrein and to elucidate its mechanism of action.
Methods and Results
Using convergent Systematic Evolution of Ligands by Exponential Enrichment (SELEX), we isolated an RNA aptamer that targets kallikrein. This aptamer, Kall1-T4, specifically binds to both prekallikrein and kallikrein with similar subnanomolar binding affinities and dose-dependently prolongs fibrin clot formation in an aPTT coagulation assay. In a purified in vitro system, Kall1-T4 inhibits the reciprocal activation of prekallikrein and FXII primarily by reducing the rate of FXIIa-mediated prekallikrein activation. Additionally, Kall1-T4 significantly reduces kallikrein-mediated HK cleavage and subsequent BK release.
Conclusions
We have isolated a specific and potent inhibitor of prekallikrein/kallikrein activity that serves as a powerful tool for further elucidating the role of kallikrein in thrombosis and inflammation.
Keywords: anticoagulant, bradykinin, plasma kallikrein, prekallikrein, RNA aptamer
Introduction
Prekallikrein, a single-chain monomeric glycoprotein and the inactive precursor of the serine protease plasma kallikrein, is an important component of many biological systems including the contact activation system (CAS) [1], kallikrein-kinin system (KKS) [2], renin-angiotensin system (RAS) [3], fibrinolytic system [4] and the alternative complement pathway [5]. Disease states associated with aberrant prekallikrein/kallikrein activity include thrombosis, hereditary angioedema (HAE), diabetic retinopathy, intracerebral hemorrhage and septicemia [4, 6, 7].
The CAS is comprised of prekallikrein, factor XII (FXII), factor XI (FXI), and high-molecular-weight kininogen (HK) [8, 9]. At the amino acid level, prekallikrein is ~58% homologous to FXI, and both proteins circulate in non-covalent complexes with HK [10–12]. The CAS is activated when a small amount of FXII is converted to activated FXII (FXIIa) via contact with a negatively charged surface [8]. Prekallikrein and FXI, both anchored to the contact surface by HK, are then cleaved by FXIIa resulting in kallikrein and activated FXI (FXIa) formation, respectively [13, 14]. CAS-generated kallikrein subsequently activates additional FXII to produce FXIIa in a feedback loop known as reciprocal activation [13] while FXIa generation triggers a series of proteolytic cleavage reactions that ultimately results in thrombin generation and thrombus formation [8]. Despite their importance to the contact pathway in vitro, FXII, prekallikrein and HK deficiencies do not manifest with a bleeding phenotype [8]. These observations led to the hypothesis that the CAS is not required for hemostasis but may play a central role in thrombosis in vivo [15]. In vivo CAS-knockout mouse models were protected from occlusion in arterial and venous thrombosis models without significantly increased bleeding risk [16–18]. Targeting CAS components for anticoagulant development may therefore provide a safer alternative to factor X- and thrombin-targeted anticoagulants which induce a significant bleeding risk [19].
Prekallikrein, kallikrein and HK are also important components of the KKS. Single-chain HK serves as an endogenous substrate for kallikrein cleavage, releasing the inflammatory nonapeptide bradykinin (BK) [20]. While KKS activation and BK release can result from CAS activation, prekallikrein can also be activated by other activators such as prolylcarboxypeptidase, heat shock protein 90 and polyphosphates [21–24].
Kallikrein activity in vivo is regulated by C1-esterase inhibitor (C1-inh) and C1-inh replacement therapy is used clinically to treat HAE. However, C1-inh infusion poses the risk of thromboembolic events [25]. Other kallikrein inhibitors have been described, including soybean trypsin inhibitor (SBTI) [26], ecallantide [27] and DX-2930 [28] and are mainly active site inhibitors developed for HAE treatment. Interestingly, very few of these inhibitors have been explored as anticoagulants [29] even though prekallikrein-deficient mice are protected against thrombosis [16, 30].
Due to the central roles of both prekallikrein and kallikrein in the CAS and KKS systems, we sought to develop and characterize a high affinity RNA aptamer inhibitor to both proteins [31]. We hypothesized that targeting prekallikrein/kallikrein with an aptamer could provide an alternate mechanism of inhibition relative to the previously developed kallikrein active site inhibitors since coagulation factor aptamers typically block macromolecular interactions [31, 32]. Additionally, aptamer activity can be rapidly modulated by a complementary antidote sequence or universal antidote resulting in an improved safety profile [33, 34].
We identified a RNA aptamer, Kall1-T4, which exhibits potent anticoagulant effects via inhibition of reciprocal activation of prekallikrein by primarily blocking FXIIa-mediated prekallikrein activation. Additionally, Kall1-T4 reduces HK cleavage and BK release and thus may represent an anti-inflammatory agent. We have developed a highly specific and potent agent that targets prekallikrein/kallikrein and works at the interface between coagulation and inflammation.
Experimental Procedures
Materials
Prekallikrein, kallikrein, HK, α-FXIIa, corn trypsin inhibitor (CTI) and all chromogenic substrates (Pefachrome PK and Pefachrome 6017) were purchased from Enzyme Research Laboratories, FXII was from Haematologic Technologies, SBTI from ATCC, PEG-8000 from Fluka Biochemika and dextran sulfate from Pharmacia Biotech. Human pooled normal and prekallikrein-deficient plasmas were purchased from George King Bio-Medical, HK-depleted plasma from Affinity Biologicals, TriniCLOT aPTT S and PT Excel reagents from Trinity Biotech, APTT-XL reagent from Thermo Scientific and CK Prest 5 (kaolin reagent) from Diagnostica Stago.
Aptamer generation
Convergent SELEX was performed as previously described [34, 35]. Briefly, a 2′-fluoro pyrimidine RNA library was generated by in vitro transcription using a modified T7 polymerase (Y639F) and selection was performed against human plasma in HEPES-based buffers. After five rounds of selection to the plasma proteome, four rounds of SELEX were performed against purified kallikrein. The fourth round was cloned and sequenced by standard methods as previously described [35]. All RNA sequences used in subsequent experiments were transcribed and purified as previously described [35] and were refolded prior to use by heating to 65 °C for 5 minutes followed by 3 minutes at room temperature in the specified reaction buffer.
Nitrocellulose filter binding assay
A double-filter nitrocellulose binding assay was used to determine the apparent binding affinity (Kd) of [γ32P]-labeled aptamer to protein as previously described [34, 35]. The bound and unbound aptamers were separated by passing the mixture over a nitrocellulose filter (Protran BA 85, GE Healthcare) overlaid on a nylon filter (Gene Screen Plus, Perkin Elmer) and the fraction of bound vs unbound aptamer was quantified with a Storm 825 phosphorimager (GE Healthcare). Data were fit with a non-linear regression for one-site binding using GraphPad Prism software and the apparent Kd was calculated.
Plasma clotting assays
Blood samples were drawn from normal, healthy volunteers in accordance with the Duke University Institutional Review Board (IRB). After discarding the initial 3 mL, blood was collected into a syringe preloaded with CTI and slowly dripped into a citrated tube such that the final blood concentrations of citrate and CTI were 11 mM and 50 µg/mL, respectively. The CTI concentration used is sufficient to inhibit FXIIa formed during the plasma preparation but also allows for robust FXII activation upon addition of aPTT activators [36]. Plasma was collected and stored at −80 °C until use.
Activated partial thromboplastin time (aPTT) and prothrombin time (PT) were performed on a STart4 coagulometer. For both assays, RNAs were diluted into a HEPES-based buffer [20 mM HEPES pH 7.4, 150 mM NaCl, 2 mM CaCl2] and refolded. For aPTT and PT assays, RNA (5 µL) was added to plasma (50 µL) and incubated for 5 minutes at 37 °C. For aPTT, 50 µL of TriniCLOT aPTT S reagent (micronized silica), ellagic acid or kaolin reagent was added to plasma/RNA mixture and incubated at 37 °C for the time indicated. Clotting was initiated with 25 mM CaCl2(50 µL) for aPTT and TriniCLOT PT Excel reagent (100 µL) for PT. Aptamer concentrations represent the final reaction concentrations.
Chromogenic assays
All the chromogenic assays described were performed in 96-well flat-bottom microtiter plates (Greiner Bio-One) at 37 °C in reaction buffer [20 mM HEPES pH 7.4, 150 mM NaCl, 2 mM CaCl2, 0.01% BSA and 0.1% PEG-8000] +/− 10 µM ZnCl2 [24] in a 100 µL final reaction volume. Aptamer and protein concentrations represent final reaction concentrations and substrate cleavage was measured at 405 nm at 37 °C using a SpectraMax i3 multi-mode microplate reader (Molecular Devices). All measurements were performed in duplicate and repeated at least three times. Data were analyzed using SoftMax Pro (Molecular Devices) and GraphPad Prism software and fit to linear and/or non-linear regression analysis unless indicated otherwise.
Kallikrein small peptide substrate cleavage
Michaelis-Menton kinetics were determined by incubating Kall1-T4 (500 nM) with human kallikrein (10 nM) for 10 minutes prior to adding varying concentrations (0–500 µM) of kallikrein chromogenic substrate, Pefachrome PK, and monitoring substrate cleavage. The velocity for each reaction was determined by linear regression and plotted against substrate concentration and the Km was calculated by non-linear regression analysis.
For kallikrein active site cleavage experiments, kallikrein (10 nM) was incubated with varying concentrations (0–500 nM) of aptamer, mutant control or SBTI for 10 minutes before addition of Pefachrome PK (0.25 mM).
Reciprocal and FXIIa activation of prekallikrein
Prekallikrein (10 nM) and HK (10 nM) were incubated with varying concentrations (0–500 nM) of aptamer, mutant control or SBTI for 10 mins. To initiate the reactions, FXII (2 nM) and dextran sulfate (2 µg/mL) were added for reciprocal activation while α-FXIIa (0.05 nM) was added for FXIIa-initiated activation. The reaction was incubated for 20 minutes and FXIIa activity was quenched by CTI (1 µM) addition. Kallikrein formation was assessed by Pefachrome PK (0.25 mM) addition. The relative reaction rate was determined by normalizing the kallikrein formation rate for all three conditions (Kall1-T4, Kall1-T4M1 and SBTI) to the rate without inhibitor/RNA. For kinetic reactions, FXIIa activity was quenched at various time-points by CTI addition and kallikrein formation was determined.
Kallikrein activation of FXII
Kallikrein (10 nM) and HK (10 nM) were incubated with varying concentrations (0–500 nM) of aptamer, mutant control or SBTI for 10 mins prior to FXII (100 nM) addition. The reaction was incubated for 20 minutes and kallikrein activity was quenched by SBTI addition (50 µg/mL). FXIIa formation was assessed by Pefachrome 6017 (1 mM) addition. For kinetic reactions, kallikrein activity was quenched at various time-points by SBTI addition and FXIIa formation was evaluated.
Prekallikrein autoactivation
Prekallikrein (100 nM) and HK (100 nM) were incubated with varying concentrations (0–2 µM) of aptamer or mutant control for 10 mins. Long-chain polyphosphate (10 µM) was added to the reactions and incubated for 1 hour. Kallikrein formation was assessed by Pefachrome PK (0.25 mM) addition. The relative reaction rate was determined by normalizing the kallikrein formation rate for both conditions to the rate without RNA.
Bradykinin release
The bradykinin concentration released from kallikrein cleavage of HK was determined using a bradykinin Enzyme-Linked Immunosorbent Assay (ELISA) kit (Enzo Life Sciences). Kallikrein (1 nM) was incubated with varying concentrations of folded RNAs or SBTI in reaction buffer [20 mM HEPES pH 7.4, 150 mM NaCl, 2 mM CaCl2, 0.01% BSA and 0.1% PEG-8000] for 10 minutes at 37 °C. HK (10 nM) was added and the reaction was incubated at 37 °C for 2 hours in a 100 µL total reaction volume. Reactions were precipitated with ethanol at −80 °C overnight followed by centrifugation at 10,000× g for 1 hour at 4 °C. The BK-containing supernatant was evaporated to almost complete dryness (~ 20 µL remained). ELISA assay buffer (400 µL) was added to each sample and the ELISA was performed as described. The data were analyzed using GraphPad Prism, a BK standard curve was generated and the values were log transformed to generate a linear regression for the standard curve. The BK generated for each reaction condition was interpolated from the standard curve.
Statistical analysis
GraphPad Prism was used to determine all linear and non-linear regressions unless indicated otherwise. For IC50 determination, Kall1-T4 concentrations were logarithmic transformed using the equation:×= log(x+1), since an RNA concentration of zero was used in the assay.
Results
Selection and truncation of a prekallikrein/kallikrein binding aptamer
To identify an RNA that recognizes prekallikrein and kallikrein, convergent SELEX was performed using a modified 2′-fluoropyrimidine RNA library initially focused on the plasma proteome followed by further enrichment against purified kallikrein [35]. A few high-affinity species were identified and Kall1, an 81-mer, was chosen as the lead aptamer based on its superior binding affinity (supplemental Table 1). We determined that full-length Kall1 binds to both prekallikrein and kallikrein with subnanomolar affinities and apparent Kd of 0.12 ± 0.01 nM and 0.26 ± 0.02 nM, respectively (Fig. 1A and B). To determine the minimal RNA sequence required for binding and functionality, nucleotide deletions were systematically performed to obtain a final truncate of 54 nucleotides, termed Kall1-T4 (Fig. 1C). Kall1-T4 binds to prekallikrein and kallikrein with only modestly reduced affinity (0.28 ± 0.02 and 0.88 ± 0.05 nM, respectively) compared to the parent molecule (Fig. 1A and B). A mutant control sequence designed by mutating three nucleotides (Kall1-T4M1, Fig. 1C, gray nucleotides), retains a similar overall predicted structure while abolishing binding to both prekallikrein and kallikrien (Fig. 1A and B).
Figure 1.
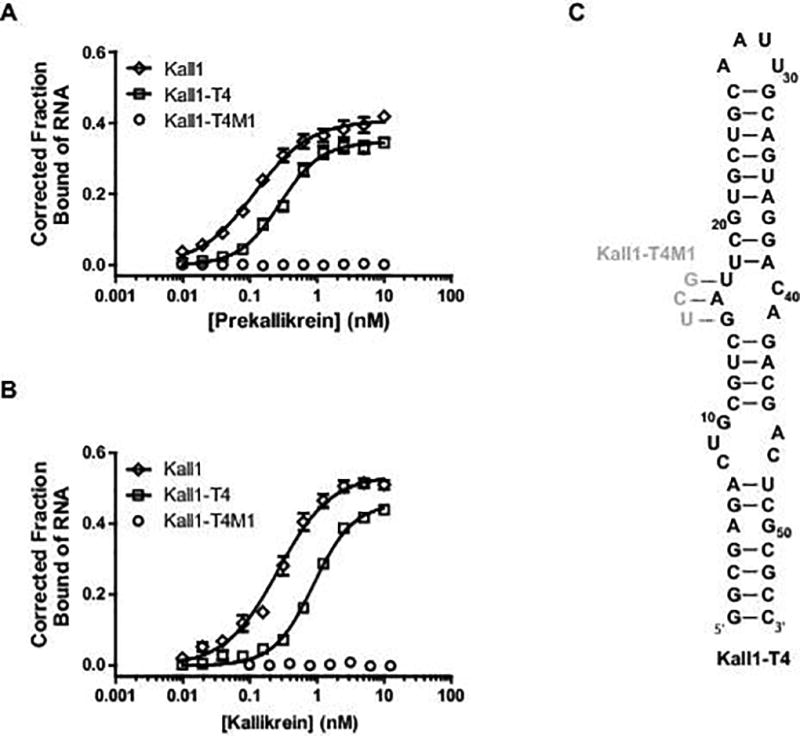
Kall1 and truncate, Kall1-T4, bind with high affinity to prekallikrein and kallikrein. Double-filter nitrocellulose binding of Kall1 (◊), Kall1-T4 (□) and Kall1-T4M1 (○) to prekallikrein (A) and kallikrein (B). The corrected fraction bound represents the mean ± SEM of at least three independent experiments. (C) M-fold predicted secondary structure model of Kall1-T4 highlighting the nucleotides that were mutated (in gray) in the design of the non-binding mutant control, Kall1-T4M1.
In addition to binding with high affinity, Kall1-T4 also demonstrated specificity for prekallikrein and kallikrein, exhibiting little to no binding (apparent Kd > 1 µM) to other coagulation proteins (HK, FXIIa, FXIa, FIXa and FIIa) even at very high protein concentrations (Fig. S1). Each of the coagulation proteins tested were either functionally similar to kallikrein (serine proteases such as FXIa, FIXa and thrombin), or form macromolecular interactions with prekallikrein and/or kallikrein (such as FXIIa and HK). The specificity data indicates that Kall1-T4 binds to a particular region of both prekallikrein and kallikrein with greater than 1000-fold selectivity over related coagulation proteins.
Kall1-T4 has anticoagulant activity
To determine the anticoagulant potential of Kall1-T4 in plasma, we performed a clinical coagulation assay, aPTT, using micronized silica to activate the contact pathway. Kall1-T4 dose-dependently increased aPTT clotting time of CTI-citrated plasma compared to the mutant RNA, Kall1-T4M1 (Fig. 2) with a 7-fold increase over baseline at 4 µM Kall1-T4. As expected, both RNAs (Kall1-T4 and T4M1) displayed no effect in the PT clotting assay which activates via the extrinsic pathway using tissue factor (Fig. 2). These results indicate that Kall1-T4 prolongs clot time by inhibiting thrombin generation via a contact pathway-specific protein.
Figure 2.
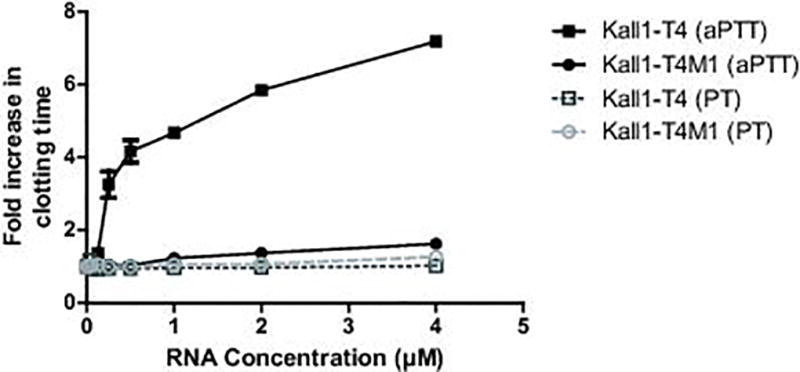
Kall1-T4 dose-dependently prolongs clotting time in CTI-citrated human plasma in an aPTT clotting assay but has no effect in a PT clotting assay. APTT assay with Kall1-T4 (■) and Kall1-T4M1 (●) activated with micronized silica, and PT assay with Kall1-T4 (□) and Kall1-T4M1 (○) activated with TF. The data were normalized to the baseline clotting time in the absence of RNA and are represented as the mean ± SEM of at least three independent experiments.
Kallikrein aptamer does not inhibit active site function
To determine whether Kall1-T4 inhibits small substrate binding to and enzymatic function of kallikrein’s active site, we incubated kallikrein with a small chromogenic substrate and varying concentrations of Kall1-T4, Kall1-T4M1 and SBTI (a known kallikrein active site inhibitor) [26]. In the presence of Kall1-T4 or Kall1-T4M1, cleavage of the small chromogenic substrate occurred, while SBTI dose-dependently inhibited cleavage of the same substrate (Fig. 3). Additionally, the presence of Kall1-T4 or Kall1-T4M1 did not prevent SBTI dose-dependent inhibition of kallikrein active site function in the absence or presence of HK (Fig. S2). These results indicate that Kall1-T4 does not bind to and block kallikrein’s active site or impact active site function in an allosteric manner. Thus, the observed anticoagulant activity of Kall1-T4 is likely due to inhibition of macromolecular substrates and/or cofactor binding to prekallikrein and/or kallikrein and this mechanism of action would be consistent with that of previously described aptamers to coagulation factors [31, 32, 37–39].
Figure 3.
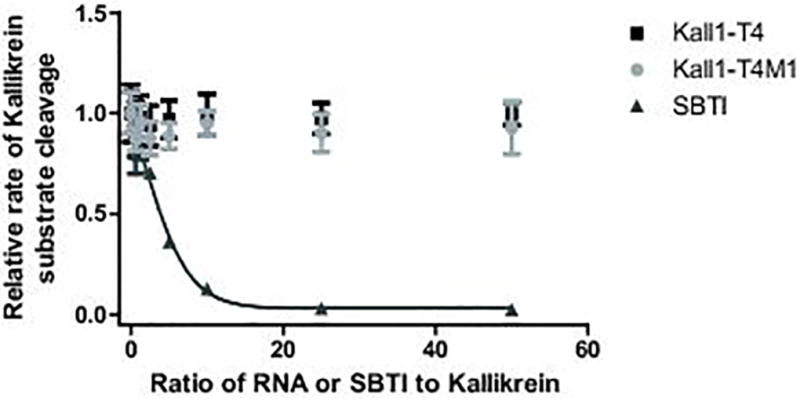
Kall1-T4 does not inhibit kallikrein cleavage of a small chromogenic substrate. Varying concentrations of Kall1-T4 (■), Kall1-T4M1 (●) and SBTI (▲) were incubated with kallikrein and cleavage of the substrate was monitored. The data were normalized to the rate of substrate cleavage in the absence of RNA or SBTI and represent the mean ± SD of at least three independent experiments.
Aptamer-mediated inhibition of reciprocal activation of prekallikrein
In plasma, FXIIa activates prekallikrein to kallikrein which then serves to amplify FXIIa generation by activating FXII in a feedback loop known as reciprocal activation (Fig. 4A) [8]. Prekallikrein can also be auto-activated to form kallikrein independent of FXIIa in the presence of negatively charged surfaces and long-chain polyphosphates [24]. The ability of Kall1-T4 to inhibit this feedback loop was examined by assessing the rate and amount of kallikrein generated in the absence and presence of either Kall1-T4 or the kallikrein active site inhibitor, SBTI. This purified protein assay uses FXII and stoichiometric equivalents of prekallikrein and HK, and is activated by dextran sulfate addition. Dextran sulfate preferentially induces FXII auto-activation [40] generating small amounts of FXIIa that activates prekallikrein to kallikrein. As a consequence, significant kallikrein formation in this assay requires concurrent FXIIa generation via the reciprocal activation feedback loop. Kall1-T4 significantly and dose-dependently decreased reciprocal kallikrein amplification by greater than 90% in comparison to equivalent concentrations of the control RNA, Kall1-T4M1, and at similar levels to the SBTI dose-dependent decrease of kallikrein activity (Fig. 4A). We subsequently determined an IC50 value for Kall1-T4 inhibition for the reciprocal activation reaction of 18.5 nM (Fig. 5A) which is similar to the IC50 value obtained in the absence of HK (13 nM, Fig. S3).
Figure 4.
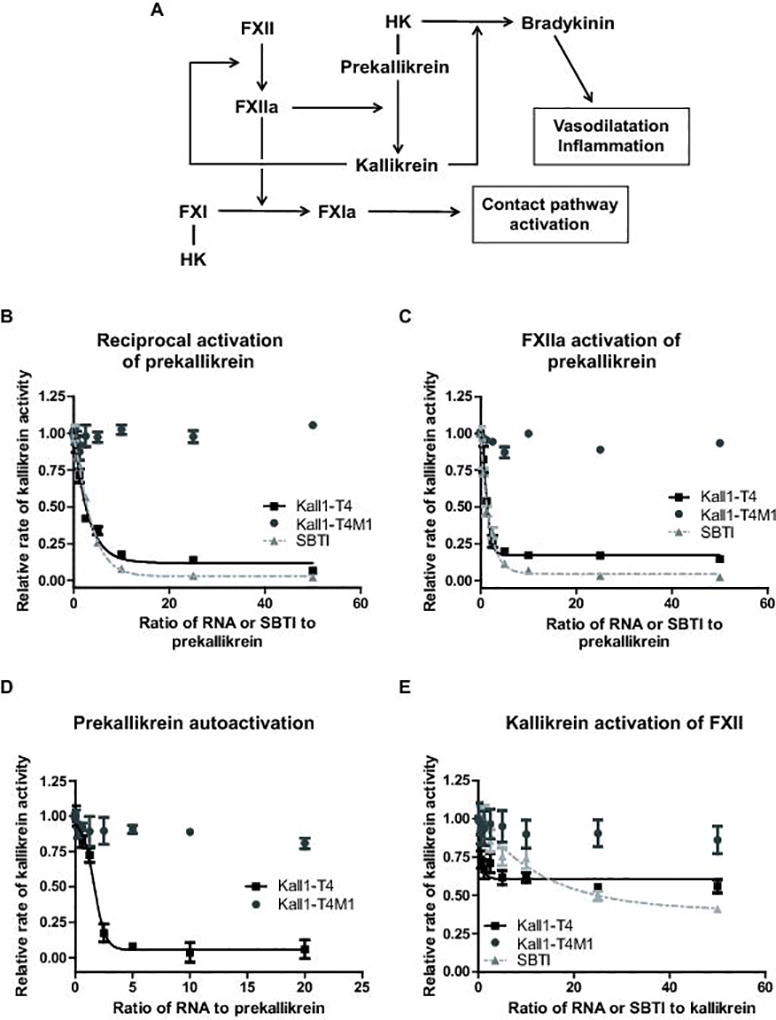
Kall1-T4 dose-dependently inhibits reciprocal activation of prekallikrein by primarily inhibiting FXIIa activation of prekallikrein. (A) Schematic illustrating the contact pathway and interaction with the kallikrein-kinin system. (B-E) Varying concentrations of Kall1-T4 (■), Kall1-T4M1 (●) and SBTI (▲) [for B, C and E] were incubated with (B) prekallikrein and HK prior to addition of FXII and dextran sulfate to initiate FXII activation, (C) prekallikrein and HK prior to addition of purified α-FXIIa, (D) prekallikrein and HK prior to addition of long-chain polyphosphate and (E) kallikrein and HK prior to addition of FXII. For (B), (C) and (D), the reaction rate represents the rate of cleavage of the kallikrein substrate by the kallikrein generated during the reaction whereas for (E), the reaction rate represents the rate of cleavage of the FXIIa substrate by the FXIIa generated during the reaction. The relative rate of kallikrein activity was obtained by normalizing each rate to the rate of substrate cleavage in the absence of RNA or SBTI and represent the mean ± SD of at least three independent experiments.
Figure 5.
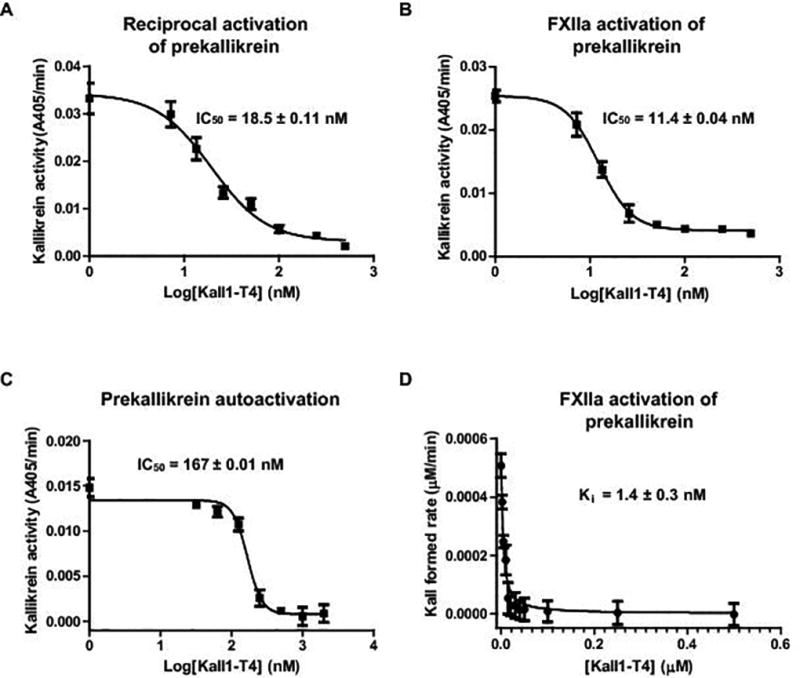
Kinetics of Kall1-T4 inhibition in the context of the reciprocal activation, FXIIa activation of prekallikrein and prekallikrein autoactivation reactions. Kall1-T4 at varying concentrations (0, 6.25, 12.5, 25, 50, 100, 250, 500 nM) were incubated with prekallikrein prior to addition of (A) FXII and dextran sulfate to initiate FXII activation, (B) α-FXIIa or (C) long-chain polyphosphates. IC50 values were obtained by non-linear regression analysis of the rate of kallikrein activity vs log transformation of the Kall1-T4 concentrations. (D) Prekallikrein was activated by FXIIa and the reaction was quenched at varying time-points by CTI and assayed for the amount of kallikrein formed in the presence of varying concentrations (0, 2.5, 5, 10, 15, 20, 30, 40, 50, 100, 250, 500 nM) of Kall1-T4. Ki was determined by non-linear regression analysis (Morrison Ki analysis) in GraphPad Prism. For all reactions, the data are represented as the mean ± SD of three independent experiments.
To further explore the mechanism of Kall1-T4 inhibition of reciprocal activation, we used a purified protein system to test inhibition of FXIIa-mediated prekallikrein activation and determined the rate and amount of kallikrein formed in the absence and presence of varying concentrations of Kall1-T4. Kall1-T4 dose-dependently decreased FXIIa activation of prekallikrein by greater than 85% in comparison to the control mutant aptamer (Kall1-T4M1) and we observed a similar decrease in kallikrein generation in the presence of varying SBTI concentrations (Fig. 4C). We determined an IC50 of 11.4 nM for Kall1-T4 inhibition of FXIIa-mediated prekallikrein activation (Fig. 5B) which is similar to the IC50 value (18.5 nM) obtained for inhibition of the reciprocal activation reaction. We also assessed the effect of zinc ions on Kall1-T4 inhibition of reciprocal activation since it has been suggested that some contact reactions require zinc ions for maximal activity. In the presence of 10 µM ZnCl2, we observed only a modest 2-fold increase in the IC50 value (18.5 vs 35.1 nM, Fig. S4).
To further characterize the kinetics of Kall1-T4-mediated inhibition of FXIIa-dependent prekallikrein activation, we next determined a Michaelis constant (Km) of 188.7 ± 15 µM for kallikrein cleavage of the chromogenic substrate in the presence of Kall1-T4 (data not shown). Using this Km, we obtained an apparent equilibrium inhibition constant (Ki) of 1.4 nM (Fig. 5D) which is approximately 8-fold lower than the IC50 (11.4 nM) determined for Kall1-T4 inhibition of FXIIa-mediated prekallikrein activation (Fig. 5B). However, the Ki obtained is much closer to the apparent Kd of Kall1-T4 binding to prekallikrein (0.28 nM) and is likely a more accurate representation of the inhibition kinetics rather than the IC50 values.
We then examined the effect of Kall1-T4 on prekallikrein autoactivation in the presence of long-chain polyphosphates [24]. We observed a similar dose-dependent decrease in kallikrein formation in the presence of Kall1-T4 in comparison to control RNA (Fig. 4D). We determined an IC50 value of 167 nM for the reaction (Fig. 5C) which is 10-fold higher than that obtained for both the reciprocal and FXIIa-mediated prekallikrein activation. This result was unsurprising since we had to use a 10-fold greater concentration of prekallikrein and HK to observe appreciable autoactivation in this assay.
Finally, we assessed the effect of Kall1-T4 on kallikrein-mediated FXII activation. We observed an initial 40% decrease in FXIIa generated at low concentrations of Kall1-T4 (6.25–25 nM) compared to control RNA, Kall1-T4M1 (Fig. 4E). This decreased rate of FXIIa generation remained constant even in the presence of much higher concentrations of Kall1-T4. Taken together, the data suggest that Kall1-T4 inhibits reciprocal activation in a concentration dependent manner primarily through inhibition of FXIIa-mediated prekallikrein activation. Although, a small contribution may exist from the inhibition of kallikrein-mediated FXII activation, the lack of concentration dependence suggests that this mode of inhibition is secondary.
Aptamer-mediated inhibition of HK cleavage and bradykinin release
In addition to propagation of thrombin generation via reciprocal activation, plasma kallikrein is also involved in activating the innate immune response via BK liberation (Fig. 4A). To determine whether Kall1-T4 has an effect on HK cleavage, we tested various Kall1-T4 concentrations (0–500 nM) in a kallikrein-mediated BK release assay. Kall1-T4 dose-dependently reduced BK release with up to ~ 65% decrease observed at the highest Kall1-T4 concentrations tested (250–500 nM) (Fig. 6A). We then directly compared the amount of BK released using specific concentrations of Kall1-T4, Kall1-T4M1 and SBTI (Fig. 6B). Compared to a control reaction without inhibitor or RNA, Kall1-T4 and SBTI at concentrations of 50 and 500 nM, significantly decreased HK cleavage and BK release. Specifically, 50 nM of Kall1-T4 decreased BK release by ~33% (p < 0.01) while the equivalent SBTI concentration resulted in ~25% decrease (p < 0.05). Greater inhibition of BK release was observed at the higher concentrations of Kall1-T4 and SBTI with ~65% and ~70% inhibition, respectively compared to control (p < 0.001). In contrast, the mutant aptamer, did not result in a significant change in BK release.
Figure 6.
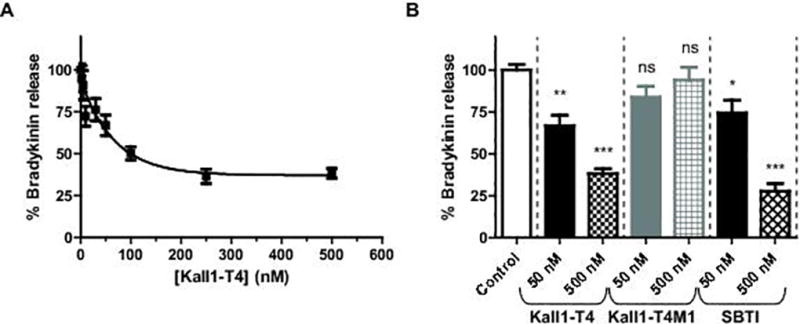
Kall1-T4 dose-dependently decreases HK cleavage and release of BK. (A) Varying concentrations of Kall1-T4 were incubated with kallikrein prior to addition of HK and initiation of cleavage reaction. (B) Percent bradykinin release in the presence of Kall1-T4, control aptamer and SBTI at 50 and 500 nM were compared to a control reaction without inhibitor or RNA. For both panels, the amount of BK generated was quantified by a BK ELISA and percent BK release was calculated by normalizing the amount of BK produced for each reaction condition to the amount of BK produced in the absence of RNA or inhibitor. The data are represented as the mean ± SEM of at least three independent experiments and the data were analyzed using one-way ANOVA with Dunnett’s post hoc test used to adjust for multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001, and NS indicates not significant.
Discussion
The relative importance of the contact pathway in hemostasis and thrombosis has long been debated and is thought to be a possible surface-activated defense system [1]. Interest in FXII inhibitors has increased recently due to many studies highlighting that FXIIa inhibition provides protection from arterial and venous thrombosis without significant increase in bleeding [16, 41]. Other studies indicate that inhibition of other contact pathway mediators such as kallikrein and HK, may also provide protection from thrombosis in vivo [16]. Since most of the current kallikrein inhibitors target and block active site cleavage and serine protease specificity in coagulation is mainly derived from the macromolecular interactions at exosites [15, 42], we sought to develop an exosite-targeting aptamer that could bind to and inhibit both prekallikrein and kallikrein.
Here we describe the development and characterization of a novel prekallikrein/kallikrein-targeting RNA aptamer that has both anticoagulant and anti-inflammatory activities (Fig. 4A). The Kall1-T4 aptamer binds to prekallikrein preventing thrombin generation via inhibition of reciprocal activation of prekallikrein and FXII primarily by inhibiting FXIIa-mediated prekallikrein activation. Additionally, Kall1-T4 binds to kallikrein and reduces HK cleavage and BK release without inhibiting active site function.
Kall1-T4 treatment of CTI-citrated plasma results in potent anticoagulation (Fig. 2). Despite circulating as a complex with HK, these results are likely not HK-dependent since Kall1-T4 also dose-dependently increases the clotting time of HK-depleted plasma (Fig. S5A). Prekallikrein-deficient plasma displays differential aPTT clotting times based on aPTT reagent with silica reagent resulting in prolonged clotting time while ellagic acid normalizes clotting time [30, 43]. We compared all 3 activation reagents in prekallikrein-deficient plasma and only observed prolongation of clotting time with the silica reagent (Fig. S5B). We also observed that Kall1-T4 does not prolong the clotting time in an ellagic-acid or kaolin triggered aPTT assay using CTI-citrated plasma (Fig. S5C). This observation is also consistent with the notion that the relative contribution of prekallikrein to coagulation in the aPTT assay is dependent on the reagent used to initiate FXII activation [43, 44]. Interestingly, we observed no clotting time prolongation in normal citrated plasma upon treatment with Kall1-T4 (Fig. S6) which suggests that enough contact pathway activation occurs during normal plasma preparation and produces sufficient FXIIa to bypass the anticoagulant effects of Kall1-T4. Consistent with this observation, addition of FXIIa also bypasses the anticoagulant effect of Kall1-T4 (data not shown).
Although, Kall1-T4 effectively and dose-dependently blocks FXIIa-mediated prekallikrein activation, Kall1-T4 only has a small effect (~40% decrease) on kallikrein-mediated FXII activation. One explanation for this observation is that FXII easily undergoes auto-activation on artificial surfaces and in the presence of many biological substances [45]. FXII auto-activation could account for the FXIIa activity observed in the presence of both Kall1-T4 and SBTI. Another explanation for incomplete Kall1-T4 inhibition of kallikrein-mediated FXII activation is that our current assay was not sensitive enough to detect complete inhibition of FXII activation since SBTI at 2.5 µM (a concentration that should produce almost complete inhibition [26]) only resulted in ~65% decrease of FXII activation in this assay (data not shown).
The mechanism of Kall1-T4 suggests that the aptamer binds to a specific patch on prekallikrein that is involved in macromolecular contacts with FXIIa and this site may either be in close proximity to or induce allosteric changes in the HK binding site. Since Kall1-T4 binds to both prekallikrein and kallikrein with relatively similar affinities and prekallikrein conversion to kallikrein results in active site conformational changes, the Kall1-T4 binding site is less likely to be located on the light-chain containing the active site. Given that HK does not appear to compete with Kall1-T4 for binding on prekallikrein or kallikrein (data not shown) and that prekallikrein homology modeling suggest that the FXIIa binding site is located on the heavy chain of prekallikrein [46], we hypothesize that Kall1-T4 binds to the heavy chain of both prekallikrein and kallikrein at an exosite that prevents FXIIa binding. Based on previous aptamer-protein crystal structures where aptamer binding sites have mostly been confined to basic patches on the target proteins [31], Kall1-T4 likely binds a basic surface on kallikrein which allosterically reduces HK binding and/or cleavage.
Further testing of Kall1-T4 in vivo would be required in order to determine its potency as an anticoagulant. Targeting prekallikrein/kallikrein could provide therapeutic value based on studies showing that prekallikrein-deficient mice were protected from occlusion after FeCl3- or Rose Bengal-induced vessel injury [17]. However, FXIIa active site inhibition more effectively prevented occlusion compared to kallikrein active site inhibition in vivo [17]. In light of these studies, Kall1-T4, may not be as potent as a FXIIa inhibitor to prevent thrombosis in vivo especially in instances where robust anticoagulation is required. One solution would be to pair the kallikrein-targeting aptamer with the previously developed FXII/FXIIa-targeting aptamer, R4cXII-1t, either separately or by linking them to create a bivalent aptamer [39, 47]. Since R4cXII-1t inhibits FXII auto-activation and FXIIa-mediated FXI activation, combining both aptamers would effectively eliminate the initiation of the contact pathway in vivo and provide a viable option for safely preventing thrombosis.
Supplementary Material
Essentials.
Kallikrein amplifies contact activation and is a potential target for preventing thrombosis.
Developed and characterized a kallikrein aptamer using convergent evolution and kinetic assays.
Kall1-T4 prolongs intrinsic clotting time by inhibiting FXIIa-mediated prekallikrein activation.
Kall1-T4 decreases high-molecular-weight kininogen (HK) cleavage and bradykinin (BK) release.
Acknowledgments
This work was supported in part by NIH grant R01 HL065222 (BAS).
Disclosure
B. A. Sullenger reports funding from Regado Biosciences/Tobira, outside the submitted work.
All authors have a patent submission pending.
Footnotes
Addendum
K-A. Steen-Burrell designed and performed research, analyzed data, and wrote the manuscript. J. Layzer performed research, analyzed data, and wrote the manuscript. B. A. Sullenger designed and coordinated research, analyzed data, and wrote the manuscript.
References
- 1.Colman RW, Schmaier AH. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood. 1997;90:3819–43. [PubMed] [Google Scholar]
- 2.Schmaier AH. Assembly, activation, and physiologic influence of the plasma kallikrein/kinin system. Int Immunopharmacol. 2008;8:161–5. doi: 10.1016/j.intimp.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shariat-Madar Z, Schmaier AH. The plasma kallikrein/kinin and renin angiotensin systems in blood pressure regulation in sepsis. J Endotoxin Res. 2004;10:3–13. doi: 10.1179/096805104225003807. [DOI] [PubMed] [Google Scholar]
- 4.Bjorkqvist J, Jamsa A, Renne T. Plasma kallikrein: the bradykinin-producing enzyme. Thromb Haemost. 2013;110:399–407. doi: 10.1160/TH13-03-0258. [DOI] [PubMed] [Google Scholar]
- 5.DiScipio RG. The activation of the alternative pathway C3 convertase by human plasma kallikrein. Immunology. 1982;45:587–95. [PMC free article] [PubMed] [Google Scholar]
- 6.Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14:28–39. doi: 10.1111/jth.13194. [DOI] [PubMed] [Google Scholar]
- 7.Bjorkqvist J, Sala-Cunill A, Renne T. Hereditary angioedema: a bradykinin-mediated swelling disorder. Thromb Haemost. 2013;109:368–74. doi: 10.1160/TH12-08-0549. [DOI] [PubMed] [Google Scholar]
- 8.Maas C, Oschatz C, Renne T. The plasma contact system 2.0. Semin Thromb Hemost. 2011;37:375–81. doi: 10.1055/s-0031-1276586. [DOI] [PubMed] [Google Scholar]
- 9.Thompson RE, Mandle R, Jr, Kaplan AP. Association of factor XI and high molecular weight kininogen in human plasma. J Clin Invest. 1977;60:1376–80. doi: 10.1172/JCI108898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujikawa K, Chung DW, Hendrickson LE, Davie EW. Amino acid sequence of human factor XI, a blood coagulation factor with four tandem repeats that are highly homologous with plasma prekallikrein. Biochemistry. 1986;25:2417–24. doi: 10.1021/bi00357a018. [DOI] [PubMed] [Google Scholar]
- 11.Wuepper KD, Cochrane CG. Plasma prekallikrein: isolation, characterization, and mechanism of activation. J Exp Med. 1972;135:1–20. doi: 10.1084/jem.135.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mandle RJ, Colman RW, Kaplan AP. Identification of prekallikrein and high-molecular-weight kininogen as a complex in human plasma. Proc Natl Acad Sci U S A. 1976;73:4179–83. doi: 10.1073/pnas.73.11.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cochrane CG, Revak SD, Wuepper KD. Activation of Hageman factor in solid and fluid phases. A critical role of kallikrein. J Exp Med. 1973;138:1564–83. doi: 10.1084/jem.138.6.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ivanov I, Shakhawat R, Sun MF, Dickeson SK, Puy C, McCarty OJ, Gruber A, Matafonov A, Gailani D. Nucleic acids as cofactors for factor XI and prekallikrein activation: Different roles for high-molecular-weight kininogen. Thromb Haemost. 2017;117:671–81. doi: 10.1160/TH16-09-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gailani D. Future prospects for contact factors as therapeutic targets. Hematology Am Soc Hematol Educ Program. 2014;2014:52–9. doi: 10.1182/asheducation-2014.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Revenko AS, Gao D, Crosby JR, Bhattacharjee G, Zhao C, May C, Gailani D, Monia BP, MacLeod AR. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood. 2011;118:5302–11. doi: 10.1182/blood-2011-05-355248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kokoye Y, Ivanov I, Cheng Q, Matafonov A, Dickeson SK, Mason S, Sexton DJ, Renne T, McCrae K, Feener EP, Gailani D. A comparison of the effects of factor XII deficiency and prekallikrein deficiency on thrombus formation. Thromb Res. 2016;140:118–24. doi: 10.1016/j.thromres.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Renne T, Schmaier AH, Nickel KF, Blomback M, Maas C. In vivo roles of factor XII. Blood. 2012;120:4296–303. doi: 10.1182/blood-2012-07-292094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Gao BB, Clermont AC, Blair P, Chilcote TJ, Sinha S, Flaumenhaft R, Feener EP. Hyperglycemia-induced cerebral hematoma expansion is mediated by plasma kallikrein. Nat Med. 2011;17:206–10. doi: 10.1038/nm.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan AP, Joseph K, Silverberg M. Pathways for bradykinin formation and inflammatory disease. J Allergy Clin Immunol. 2002;109:195–209. doi: 10.1067/mai.2002.121316. [DOI] [PubMed] [Google Scholar]
- 21.Shariat-Madar Z, Mahdi F, Schmaier AH. Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J Biol Chem. 2002;277:17962–9. doi: 10.1074/jbc.M106101200. [DOI] [PubMed] [Google Scholar]
- 22.Joseph K, Tholanikunnel BG, Kaplan AP. Heat shock protein 90 catalyzes activation of the prekallikrein-kininogen complex in the absence of factor XII. Proc Natl Acad Sci U S A. 2002;99:896–900. doi: 10.1073/pnas.022626899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maas C, Govers-Riemslag JW, Bouma B, Schiks B, Hazenberg BP, Lokhorst HM, Hammarstrom P, ten Cate H, de Groot PG, Bouma BN, Gebbink MF. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118:3208–18. doi: 10.1172/JCI35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gajsiewicz JM, Smith SA, Morrissey JH. Polyphosphate and RNA Differentially Modulate the Contact Pathway of Blood Clotting. J Biol Chem. 2017;292:1808–14. doi: 10.1074/jbc.M116.754325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gandhi PK, Gentry WM, Bottorff MB. Thrombotic events associated with C1 esterase inhibitor products in patients with hereditary angioedema: investigation from the United States Food and Drug Administration adverse event reporting system database. Pharmacotherapy. 2012;32:902–9. doi: 10.1002/j.1875-9114.2012.01126. [DOI] [PubMed] [Google Scholar]
- 26.Tans G, Janssen-Claessen T, Rosing J, Griffin JH. Studies on the effect of serine protease inhibitors on activated contact factors. Application in amidolytic assays for factor XIIa, plasma kallikrein and factor XIa. Eur J Biochem. 1987;164:637–42. doi: 10.1111/j.1432-1033.1987.tb11174.x. [DOI] [PubMed] [Google Scholar]
- 27.Levy JH, O'Donnell PS. The therapeutic potential of a kallikrein inhibitor for treating hereditary angioedema. Expert Opin Investig Drugs. 2006;15:1077–90. doi: 10.1517/13543784.15.9.1077. [DOI] [PubMed] [Google Scholar]
- 28.Kenniston JA, Faucette RR, Martik D, Comeau SR, Lindberg AP, Kopacz KJ, Conley GP, Chen J, Viswanathan M, Kastrapeli N, Cosic J, Mason S, DiLeo M, Abendroth J, Kuzmic P, Ladner RC, Edwards TE, TenHoor C, Adelman BA, Nixon AE, et al. Inhibition of plasma kallikrein by a highly specific active site blocking antibody. J Biol Chem. 2014;289:23596–608. doi: 10.1074/jbc.M114.569061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanaka KA, Szlam F, Katori N, Vega JD, Levy JH. Evaluation of a novel kallikrein inhibitor on hemostatic activation in vitro. Thromb Res. 2004;113:333–9. doi: 10.1016/j.thromres.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 30.Bird JE, Smith PL, Wang X, Schumacher WA, Barbera F, Revelli JP, Seiffert D. Effects of plasma kallikrein deficiency on haemostasis and thrombosis in mice: murine ortholog of the Fletcher trait. Thromb Haemost. 2012;107:1141–50. doi: 10.1160/th-11-10-0682. [DOI] [PubMed] [Google Scholar]
- 31.Long SB, Long MB, White RR, Sullenger BA. Crystal structure of an RNA aptamer bound to thrombin. RNA. 2008;14:2504–12. doi: 10.1261/rna.1239308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bompiani KM, Monroe DM, Church FC, Sullenger BA. A high affinity, antidote-controllable prothrombin and thrombin-binding RNA aptamer inhibits thrombin generation and thrombin activity. J Thromb Haemost. 2012;10:870–80. doi: 10.1111/j.1538-7836.2012.04679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rusconi CP, Roberts JD, Pitoc GA, Nimjee SM, White RR, Quick G, Jr, Scardino E, Fay WP, Sullenger BA. Antidote-mediated control of an anticoagulant aptamer in vivo. Nat Biotechnol. 2004;22:1423–8. doi: 10.1038/nbt1023. [DOI] [PubMed] [Google Scholar]
- 34.Oney S, Nimjee SM, Layzer J, Que-Gewirth N, Ginsburg D, Becker RC, Arepally G, Sullenger BA. Antidote-controlled platelet inhibition targeting von Willebrand factor with aptamers. Oligonucleotides. 2007;17:265–74. doi: 10.1089/oli.2007.0089. [DOI] [PubMed] [Google Scholar]
- 35.Layzer JM, Sullenger BA. Simultaneous generation of aptamers to multiple gamma-carboxyglutamic acid proteins from a focused aptamer library using DeSELEX and convergent selection. Oligonucleotides. 2007;17:1–11. doi: 10.1089/oli.2006.0059. [DOI] [PubMed] [Google Scholar]
- 36.Xu Y, Cai TQ, Castriota G, Zhou Y, Hoos L, Jochnowitz N, Loewrigkeit C, Cook JA, Wickham A, Metzger JM, Ogletree ML, Seiffert DA, Chen Z. Factor XIIa inhibition by Infestin-4: in vitro mode of action and in vivo antithrombotic benefit. Thromb Haemost. 2014;111:694–704. doi: 10.1160/TH13-08-0668. [DOI] [PubMed] [Google Scholar]
- 37.Buddai SK, Layzer JM, Lu G, Rusconi CP, Sullenger BA, Monroe DM, Krishnaswamy S. An anticoagulant RNA aptamer that inhibits proteinase-cofactor interactions within prothrombinase. J Biol Chem. 2010;285:5212–23. doi: 10.1074/jbc.M109.049833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sullenger B, Woodruff R, Monroe DM. Potent anticoagulant aptamer directed against factor IXa blocks macromolecular substrate interaction. J Biol Chem. 2012;287:12779–86. doi: 10.1074/jbc.M111.300772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woodruff RS, Xu Y, Layzer J, Wu W, Ogletree ML, Sullenger BA. Inhibiting the intrinsic pathway of coagulation with a factor XII-targeting RNA aptamer. J Thromb Haemost. 2013;11:1364–73. doi: 10.1111/jth.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samuel M, Pixley RA, Villanueva MA, Colman RW, Villanueva GB. Human factor XII (Hageman factor) autoactivation by dextran sulfate. Circular dichroism, fluorescence, and ultraviolet difference spectroscopic studies. J Biol Chem. 1992;267:19691–7. [PubMed] [Google Scholar]
- 41.Larsson M, Rayzman V, Nolte MW, Nickel KF, Bjorkqvist J, Jamsa A, Hardy MP, Fries M, Schmidbauer S, Hedenqvist P, Broome M, Pragst I, Dickneite G, Wilson MJ, Nash AD, Panousis C, Renne T. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci Transl Med. 2014;6:222ra17. doi: 10.1126/scitranslmed.3006804. [DOI] [PubMed] [Google Scholar]
- 42.Page MJ, Macgillivray RT, Di Cera E. Determinants of specificity in coagulation proteases. J Thromb Haemost. 2005;3:2401–8. doi: 10.1111/j.1538-7836.2005.01456.x. [DOI] [PubMed] [Google Scholar]
- 43.Wynne Jones D, Russell G, Allford SL, Burdon K, Hawkins GA, Bowden DW, Minaee S, Mumford AD. Severe prekallikrein deficiency associated with homozygosity for an Arg94Stop nonsense mutation. Br J Haematol. 2004;127:220–3. doi: 10.1111/j.1365-2141.2004.05180.x. [DOI] [PubMed] [Google Scholar]
- 44.Turi DC, Peerschke EI. Sensitivity of three activated partial thromboplastin time reagents to coagulation factor deficiencies. Am J Clin Pathol. 1986;85:43–9. doi: 10.1093/ajcp/85.1.43. [DOI] [PubMed] [Google Scholar]
- 45.Golas A, Yeh CH, Pitakjakpipop H, Siedlecki CA, Vogler EA. A comparison of blood factor XII autoactivation in buffer, protein cocktail, serum, and plasma solutions. Biomaterials. 2013;34:607–20. doi: 10.1016/j.biomaterials.2012.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hooley E, McEwan PA, Emsley J. Molecular modeling of the prekallikrein structure provides insights into high-molecular-weight kininogen binding and zymogen activation. J Thromb Haemost. 2007;5:2461–6. doi: 10.1111/j.1538-7836.2007.02792.x. [DOI] [PubMed] [Google Scholar]
- 47.Soule EE, Bompiani KM, Woodruff RS, Sullenger BA. Targeting Two Coagulation Cascade Proteases with a Bivalent Aptamer Yields a Potent and Antidote-Controllable Anticoagulant. Nucleic Acid Ther. 2016;26:1–9. doi: 10.1089/nat.2015.0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.