

Abstract
Solute carrier family 26, member 3 (Slc26a3), also termed downregulated-in-adenoma (DRA) is a member of the Slc26 family of anion transporters and is mutated in congenital chloride diarrhea. Our previous study demonstrated that DRA deficiency is associated with severely reduced colonic HCO3− secretion, a loss of colonic fluid absorption, a lack of a firmly adherent mucus layer and a severely reduced colonic mucosal resistance to dextran sodium sulfate (DSS) damage. However, the direct effect of mediators that trigger intestinal inflammatory factors on DRA has not been fully investigated. Tumor necrosis factor (TNF)-α is a central mediator of intestinal inflammation in inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn’s disease. However, to the best of our knowledge, whether TNF-α acts reciprocally with DRA leading to the development of gut inflammation in IBD has not been reported. The present study identified that the expression level of DRA was reduced in active UC patients and DSS-induced colitis mice with high expression levels of TNF-α identified in the peripheral blood serum. In addition, TNF-α may affect the expression level of DRA in human colonic Caco2BBE cells in a dose-dependent manner, including in DRA overexpressed Caco2BBE cells. Furthermore, knockdown of TNF-α in Caco2BBE cells led to a higher expression level of DRA and a markedly reduced secretion of TNF-α in the culture media. In addition, knockdown of DRA in Caco2BBE cells led to a higher secretion of TNF-α in the culture media compared with the control cells, which could be reversed by overexpression of DRA. Overall, these results indicate that TNF-α may act reciprocally with DRA, leading to the development of intestinal inflammation. Based on the pivotal position of TNF-α in IBD, DRA is hypothesized to have therapeutic potential against colitis serving as an important target.
Keywords: tumor necrosis factor-α, solute carrier family 26, member 3, downregulated-in-adenoma, Caco2BBE cells, colitis
Introduction
Ulcerative colitis (UC) and Crohn’s disease, collectively termed inflammatory bowel disease (IBD), are two separate clinical entities characterized by intestinal inflammation with a chronic relapsing course and, in recent years, the incidence of IBD is increasing worldwide (1–3). Though the pathogenesis of IBD is not totally clear, it is known that IBD occurs due to the role of complex interactions between genetic, environmental and immunological factors (4–6). Exogenous and endogenous triggers and multiple mediators are involved in the pathogenetic mechanisms of IBD, and their interactions lead to abnormalities of multiple immunological signaling pathways in genetically susceptible individuals.
Solute carrier family 26, member 3 (Slc26a3) ion transporter, also termed downregulated-in-adenoma (DRA), is a Cl−/HCO3− exchanger. Slc26a3 absorbs Cl− and exports HCO3−, contributing to intestinal fluid absorption and enterocyte acid/base balance (7). DRA predominantly locates at the mid-distal colon and its mutation may lead to congenital chloride diarrhea, an autosomal recessive disorder, which occurs worldwide (8). In addition, the risk of acute and chronic intestinal inflammation was significantly increased, presenting as clinical manifestations of IBD (9). Our previous study indicated that Slc26a3 deficiency is associated with the absence of a firmly adherent mucus layer and HCO3−/mucus barrier impairment in mice; thus, Slc26a3−/− mice are susceptible to dextran sulfate sodium (DSS)-induced colitis (10). Farkas et al (11) demonstrated a robust reduction in Cl− absorption with a corresponding decrease of ~50% in DRA mRNA expression levels in the surface cells of the colonic crypts in UC patients. Furthermore, the modulation of DRA in the inflammation environment remains unknown. Tumor necrosis factor (TNF)-α, which is predominantly synthesized by lymphocytes and activated macrophages, promotes the activation and recruitment of immune cells, induces apoptosis of intestinal epithelial cells and abnormal expression levels of tight junction molecules, thus finally leading to disruption of the intestinal mucosal barrier and increased permeability (12–14). TNF-α is a central mediator of intestinal inflammation in IBD, and the clinical efficacy of anti-TNF-α drugs indicates that TNF-α performs an important role in the pathogenesis of IBD. A recent study indicated that loss of DRA is associated with decreased mucosal HCO3− secretion and may occur due to the effect of proinflammatory cytokines on epithelial ion transporters (15). In addition, a study by Juric et al (16) demonstrated that DRA mRNA expression levels are significantly higher in the distal colon, and the expression levels differ significantly between TNF+/+ and TNF+/ΔARE mice. Based on these data, the present study hypothesizes that TNF-α may act reciprocally with DRA leading to the development of gut inflammation in IBD.
In the present study, the expression levels of DRA and TNF-α were examined in UC patients and DSS-induced colitis mice. Subsequently, the expression level of DRA was evaluated following treatment with TNF-α or silencing of TNF-α in colonic epithelial Caco2BBE cells. In addition, DRA was overexpressed or depleted to observe the expression level of TNF-α in Caco2BBE cells and supernatant. The aim of the present study was to reveal the association between DRA and TNF-α, and establish a novel therapeutic target for IBD.
Materials and methods
Ethics statement
All animal experiments were performed in accordance with the relevant national guidelines. The protocols were approved by the Committee on the Ethics of Animal Experiments of Tongji Medical School, Huazhong University of Science and Technology (Wuhan, China).
Patients and specimens
Endoscopic biopsy specimens from the sigmoid colon of active UC patients and healthy control patients were obtained from the endoscopy ward of the Department of Gastroenterology, Tongji Hospital, Huazhong University of Science and Technology. The specimens were obtained during routine diagnostic sample collection and patient data were made anonymous. The collection of samples was approved by the local ethical committee and the institutional review board of Huazhong University of Science and Technology and each patient provided written informed consent.
DSS-induced colitis model and evaluation of the severity of clinical colitis
Male C57BL/6 mice (age, 6–8 weeks; weight, 19–23 g) were purchased from the Department of Experimental Animals of Tongji Medical School (Wuhan, China). The mice were maintained on a standard mouse diet with free access to tap water and food, under a 12-h light-dark cycle at room temperature (22±2°C) in relative humidity (55–60%). All C57BL/6 mice were housed together for 2 weeks before commencing studies. The mice were divided randomly into control and experimental groups, and each group contained 8 mice. Mice in the treatment group were administered with 3% DSS (MP Biomedicals, Solon, OH, USA) drinking solution ad libitum for 7 days, followed by regular water for another 2 days. The animals were weighed daily and monitored for signs of distress, as well as rectal bleeding, weight loss, stool consistency, water content and rectal bleeding. To evaluate the colitis severity, the disease activity index (DAI) was, measured as described previously (17). Briefly, stool samples from individual mice were evaluated using a test for occult blood, whereas the macroscopic examination was performed by two independent researchers under blinded conditions: 0=Normal, no occult blood; 1=loose stool, no occult blood; 2=loose stool, positive for occult blood; 3=diarrhea, positive for occult blood; and 4=bleeding. Following oral DSS for 7 days and regular water for 2 days, the blood was collected from the angular vein and the mice were sacrificed. Colons were collected for haematoxylin and eosin (H&E) staining, immunohistochemistry (IHC), immunofluorescence, quantitative polymerase chain reaction (qPCR) and western blot analysis.
IHC and immunofluorescence staining
In order to detect the location and expression level of DRA in colitis mice and healthy mice, histological analysis was performed on colon tissue samples that were fixed with 4% phosphate-buffered paraformaldehyde at room temperature for 24 h, embedded in paraffin and sectioned at a thickness of 4 μm. Following hydration in a decreasing ethanol gradient, all sections were deparaffinized and stain in Harris hematoxylin solution for 5 min at 37°C. For IHC, the sections were routinely dewaxed and rehydrated followed by endogenous peroxidase blocking. Antigen retrieval was performed by pressurized steam cooking. A DRA antibody (1:50, sc-34942; Santa Cruz Biotechnology, Heidelberg, Germany) served as the primary antibody and was used at 4°C overnight. Following incubation with horseradish peroxidase-conjugated polyclonal goat anti-rabbit secondary antibody (Dako REAL™ EnVision™ kit; cat. no. K5007; Dako, Agilent Technologies GmbH, Waldbronn, Germany) for 30 min at 37°C, the expression of the proper antigen in cells was detected with Dako 3,3′-diaminobenzidine (Agilent Technologies GmbH).
For immunofluorescent analysis, frozen sections of the distal colon were washed in phosphate-buffered saline (PBS; pH 7.2) and stained to detect the expression level of DRA. Sections were blocked with 5% horse serum for 30 min at room temperature and incubated at 4°C overnight with a primary antibody against DRA, followed by incubation with Cy3-labeled donkey anti-goat secondary antibody (1:200, #705-165-003; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) for 1 h at room temperature. After washing with PBS for three times, sections were stained with 4′,6-diamidino-2-phenylindole at room temperature for 10 min (1:1,000, C1002) for nuclear staining and mounted with antifade medium (P0126) (both from Beyotime Institute of Biotechnology, Jiangsu, China). Images were acquired under an Olympus 1 X71 fluorescence microscope.
Cell culture and lentiviral-mediated DRA overexpressed in Caco2BBE cells
Caco2BBE cells were provided by Professor Seidler U (Hannover Medical School, Germany) and all the experiments were performed within 25 passages. The cells were cultured in Gibco Dulbecco’s modified Eagle’s medium (DMEM) medium supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1% penicillin and streptomycin, 1% non-essential amino acid and 1% L-glutamine, and incubated in a 5% CO2 incubator at 37°C as previously described (18). Recombinant lentivirus overexpressing DRA was constructed by Lenti-Easy Packaging System (Shanghai GeneChem Co., Ltd., Shanghai, China). Infection cells were obtained by selection in puromycin conditioned culture. Thus, Caco2BBE cell proliferation occurred following transduction with recombinant lentivirus, and infected cells were used for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blot analysis.
RNA interference
For RNA interference, synthesis of DRA siRNA (sense, GAACCAACATCTATAAGAA) and TNF-α siRNA (sense, GCGTGGAGCTGAGAGATAA) was performed by Guangzhou RiboBio Co., Ltd. (Guangzhou, China) and transfected into cells using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) as previously described (19). Cells were transfected at a density of 80–90% and used for experiments 24 h after transfection. When necessary, the DMEM medium of cell culture was collected.
RNA extraction and RT-qPCR
Total RNA was extracted using TRIzol reagent (Life Technologies; Thermo Fisher Scientific, Inc.). Reverse-transcribed complementary DNA was synthesized using the PrimeScript RT reagent kit (RR036A; Takara Bio, Inc., Otsu, Japan). qPCR was performed for 40 cycles of 95°C for 30 sec and 60°C for 30 sec using SYBR Premix ExTaq (Takara Bio, Inc.) on an ABI StepOne Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.). The value of 2−ΔΔCq (20) was used to determine the fold difference between samples. The primer sequences used for PCR are presented in Table I.
Table I.
Primer sequences used for polymerase chain reaction.
| Name | Sequence 5′-3′
|
|
|---|---|---|
| Sense | Antisense | |
| Mouse GAPDH | TGACCTCAACTACATGGTCTACA | CTTCCCATTCTCGGCCTTG |
| Human GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG |
| Mouse Slc26A3 | GCCGTGGTTGGGAACATGA | GCAAATCCTTTGAATGCTCCAG |
| Human Slc26A3 | GTTGCAGAAGCTCCTCACAGA | GGCTGTGGCAAGTTGAAGAC |
| Mouse TNF-α | CATCTTCTCAAAATTCGAGTGACAA | TGGGAGTAGACAAGGTACAACCC |
| Human TNF-α | CCTCTCTCTAATCAGCCCTCTG G | AGGACCTGGGAGTAGATGAG |
Slc26A3, solute carrier family 26, member 3 (also termed downregulated-in-adenoma); TNF-α, tumor necrosis factor-α.
Western blot analysis
For western blot analysis, colon tissue samples from mice were added to RIPA lysis buffer. These tissue samples were homogenized at 4°C, centrifuged at 12,000 g for 30 min and the supernatant was retained. Cultured cells were grown in 6-well plates and lysed using RIPA lysis buffer. Following degeneration, 30 μg samples were separated by 10% SDS-PAGE. The gel was run at 70 V (at constant voltage) until separation began. Subsequently the voltage was increased to 110 V. The gel was transferred to polyvinylidene difluoride membranes at 4°C and 250 mA for 1.5 h. Membranes were blocked with 5% non-fat milk (5 g non-fat dry milk powder in 100 ml Tris-buffered saline with Tween 20) at room temperature for 1 h, then incubated with specific DRA antibody (1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) at 4°C overnight. Finally, membranes were incubated with Peroxidase AffiniPure Rabbit Anti-Goat IgG (1:5,000, #305-035-003; Jackson ImmunoResearch Laboratories, Inc.), followed by detection with enhanced chemiluminescence reagents (Pierce; Thermo Fisher Scientific, Inc.). GAPDH served used as a loading control.
ELISA
Expression levels of TNF-α were determined in the serum of UC patients and DSS-induced colitis mice, as well as in the DMEM medium of Caco2BBE cell lines using a Mouse/Human TNF-α ELISA kit (EMC102a.96 and EHC103a.96; NeoBioscience Technology, Shenzhen, China) according to the manufacturer’s instructions.
Statistical analysis
Data were presented as the mean ± standard deviation. One-way analysis of variance or Student’s t tests were performed to assess the significant differences between treatment groups. P<0.05 was considered to indicate a statistically significant difference.
Results
UC patients demonstrated decreased expression levels of DRA and higher expression levels of TNF-α in peripheral blood serum
The expression level of DRA protein was determined in sigmoid colonic samples obtained from active UC patients and healthy control subjects. In addition, to observe the change of proinflammatory cytokines, the expression level of TNF-α was detected in the serum of UC patients and healthy control subjects at the same time. In total, specimens from 11 UC patients were obtained. In the 11 patients (5 males and 6 females; mean age, 42±3 years), the presence of UC was confirmed by routine histopathological evaluation. Nine control samples (5 males and 4 females; mean age, 36±4 years) were obtained from patients who did not suffer from any inflammatory conditions during the physical examination.
During western blot analyses, DRA was significantly reduced in UC patients when compared with the control subjects, whereas the level of TNF-α serum was significantly increased in UC patients compared with the control subjects (Fig. 1).
Figure 1.
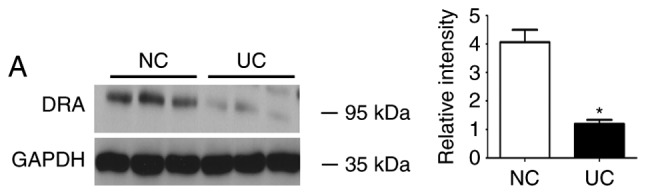
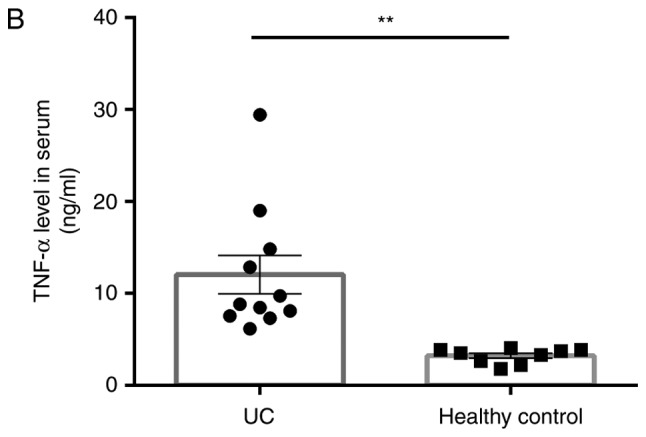
Expression levels of DRA protein and TNF-α level in the serum of UC patients and NC subjects. (A) Western blot analysis demonstrating that DRA protein expression was significantly decreased in active UC patients compared with NC subjects. (B) Compared with healthy control subjects, the TNF-α expression level in the serum markedly increased (12.03±5.09 ng/ml; n=11 vs. 3.23±0.67 ng/ml; n=9) *P<0.05 and **P<0.01. DRA, downregulated-in-adenoma; TNF-α, tumor necrosis factor-α; UC, ulcerative colitis; NC, normal control.
Mice suffering from DSS-induced colitis exhibit reduced levels of DRA expression as well as a higher expression level of TNF-α in peripheral blood serum
Subsequently, whether the expression level of DRA correlates with gut inflammation in a mouse model of UC was investigated (21). Mice were treated with DSS for 7 days before being exposed to regular water for another 2 days. In contrast to the untreated mice, DSS-treated mice exhibited significant body weight loss and bloody stools that were similar to the symptoms of UC on day 4 of DSS treatment (Fig. 2A). Additionally, the water content of stools in DSS-treated mice was significantly more than that in untreated mice following 3 days of DSS treatment (Fig. 2B). Macroscopic analysis of colons in the two groups of mice revealed a marked shortening of colon length in the DSS-treated mice on day 7 of DSS treatment (Fig. 2C). DAI results demonstrated an increased DAI score on day 3 of DSS treatment and continuous elevation during the process of DSS treatment (Fig. 2D). Consistent with these findings, histological analysis of H&E-stained colonic tissue samples demonstrated that intestinal crypts in the colon of DSS-treated mice were deformed (similar to the clinical symptom of human UC), exhibited epithelial sloughing and were even destroyed (Fig. 2E).
Figure 2.


In vivo assessment of colitis severity and histological characteristics in DSS-induced colitis mice. (A) Body weight curves for different groups. Compared with the control group, the DSS group mice exhibited obviously decreased weight over time, particularly from day 4 of oral DSS treatment (13.98±2.2 vs. 19.82±1.5 g on day 8). (B) Stool water content curves for the different groups. Compared with the control group, the DSS group mice demonstrated significantly increased stool water content (60.2±4.2 vs. 72.1±3.2% on day 6), and eventually experienced serious diarrhea and bleeding from day 6 of oral DSS treatment, so the stools could not be collected. (C) Length of colon curves in the different groups. Compared with the control group, the DSS group mice demonstrated markedly shortened lengths of colon (7.65±0.2 vs. 4.34±0.2 cm). (D) DAI score curves for the different groups. Compared with the control group, the DSS group mice exhibited marked increases in their DAI score. (E) Hematoxylin and eosin staining of the different groups (magnification, x200). Compared with the control group, the crypts in the colon samples of the DSS group were deformed and even destroyed in line with clinical signs of human ulcerative colitis. *P<0.05 vs. NC. DSS, dextran sodium sulfate; DAI, Disease Activity Index.
Furthermore, the mRNA and protein expression levels of DRA in the distal colon of mice, as well as the TNF-α expression level in the blood serum of mice were analyzed after 7 days of DSS treatment. Compared with untreated mice, DSS-treated mice displayed a significantly higher expression level of cytokine TNF-α in blood serum while a significantly reduced expression level of DRA was observed (Fig. 3A and B). This finding was further confirmed by immunostaining of colonic tissue samples with a DRA antibody. According to IHC and immunofluorescence staining, DRA was predominantly located in the colonic surface and its expression level was significantly decreased in the DSS-treated mice (Fig. 3C and D). In addition, the colonic crypts in the DSS group were distorted and the colonic surface was destroyed on day 7 of DSS treatment (Fig. 3C).
Figure 3.
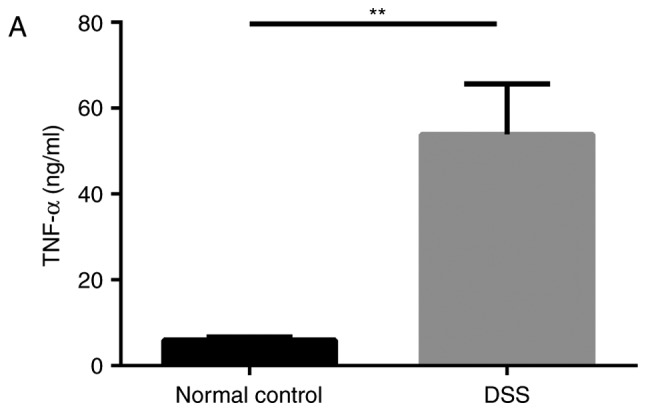



DRA expression levels were decreased in DSS-induced colitis mice, which was associated with changes in TNF-α expression in vivo. (A) Compared with the normal control mice, TNF-α expression level in the serum markedly increased. (5.98±0.63 vs. 53.89±8.98 ng/ml). (B) In DSS-induced colitis mice, DRA mRNA and protein expression levels were significantly decreased compared with the control mice. *P<0.05 as indicated. (C and D) DRA expression levels are predominantly observed in the colon crypt surface, and rarely observed in the cytoplasm. Similarly, in DSS-induced colitis mice, DRA expression levels were significantly decreased when compared with control mice, according to immunohistochemistry and immunofluorescence staining. **P<0.01. DRA, downregulated-in-adenoma; DSS, dextran sodium sulfate; TNF-α, tumor necrosis factor-α; NC, normal control; DAPI, 4′,6-diamidino-2-phenylindole.
TNF-α affects the expression level of DRA in Caco2BBE cells following stimulation with or silencing of TNF-α
Whether TNF-α affects DRA expression levels was investigated during the present study. Colonic epithelial cells, Caco2BBE and lentiviral-mediated DRA overexpressed Caco2BBE cells were used. As presented in Fig. 4A and B, the expression levels of DRA mRNA and protein were significantly increased following infection with recombinant lentivirus overexpressing DRA. Caco2BBE cells were treated with TNF-α at different dosages (50 and 100 ng/ml) for 24 h. TNF-α treatment reduced the expression level of DRA protein and this effect was apparent at a dosage of 100 ng/ml (Fig. 4C). In addition, DRA overexpressing cells were treated with TNF-α according to the above-mentioned method. It was found that while the expression level of DRA in DRA overexpressing cells was markedly higher than that in control cells, the expression level of DRA was decreased in response to TNF-α treatment, particularly at a dosage of 100 ng/ml (Fig. 4D).
Figure 4.

TNF-α affects the expression level of DRA in Caco2BBE cells following stimulation with or silencing of TNF-α. (A and B) DRA mRNA and protein expression levels were significantly increased in lentiviral-mediated DRA overexpressed Caco2BBE cells. (C) The expression level of DRA protein correspondingly decreased in the TNF-α-treated Caco2BBE cells. (D) Similarly, following 24 h of treatment with different dosages (50 and 100 ng/ml) of TNF-α treatment, the expression level of DRA protein was also correspondingly decreased in the overexpression DRA Caco2BBE cells. (E) After transfecting siTNF-α into the Caco2BBE cells, the TNF-α mRNA expression level was markedly decreased and, accordingly, the DRA mRNA expression level was upregulated. (F) Subsequent to transfecting siTNF-α in the Caco2BBE cells, the TNF-α expression level was obviously reduced in the cell supernatant. (G) The DRA protein expression level was significantly increased after transfecting siTNF-α into Caco2BBE cells. *P<0.05 and **P<0.01. TNF-α, tumor necrosis factor-α; DRA, downregulated-in-adenoma; NC, normal control.
Conversely, whether silencing TNF-α would affect the expression level of DRA was evaluated. Notably, the TNF-α mRNA level in Caco2BBE cells and the expression level of TNF-α in the supernatant of Caco2BBE cells following transfection with TNF-α siRNA were significantly decreased when compared with control cells and cell supernatant (Fig. 4E and F). Furthermore, the expression levels of DRA mRNA and protein were significantly increased when compared with the control cells (Fig. 4E and G).
Therefore, it was hypothesized that TNF-α may down-regulate DRA, which may affect intestinal fluid absorption, enterocyte acid/base balance and barrier function (4–5), and ultimately exacerbate colon inflammation.
Silencing or overexpressing DRA may affect TNF-α expression and secretion in Caco2BBE cells
Whether interference with DRA expression levels may influence TNF-α expression levels was analyzed. As presented in Fig. 5A and B, the expression levels of DRA mRNA and protein were significantly decreased following transfection of DRA siRNA in Caco2BBE cells. Additionally, TNF-α mRNA and protein expression levels of TNF-α in the cell culture medium in the DRA-silenced Caco2BBE cells were significantly increased compared with the control cells (Fig. 5A and C).
Figure 5.

DRA silencing and overexpression modulated TNF-α expression levels in Caco2BBE cells. (A) Knockdown of DRA increased TNF-α mRNA expression levels in Caco2BBE cells. (B) The level of DRA protein expression was markedly decreased following transfection with siDRA. (C) DRA silencing correspondingly increased TNF-α expression levels in the supernatant of Caco2BBE cells. (D) The TNF-α mRNA expression level was significantly decreased in DRA overexpression Caco2BBE cells. *P<0.05. DRA, downregulated-in-adenoma; TNF-α, tumor necrosis factor-α; NC, normal control.
Similarly, the expression level of TNF-α mRNA was determined in DRA overexpression Caco2BBE cells. As expected, the expression level of TNF-α mRNA was significantly decreased in DRA overexpression Caco2BBE cells when compared with control cells (Fig. 5D).
These results indicate that DRA may affect TNF-α expression levels, and the interaction between TNF-α and DRA may lead to inflammation progression.
Discussion
IBD-associated mucosal inflammation and the consequent impaired secretion and absorption of electrolytes often results in electrolytic and acid-base imbalance in IBD patients (22,23). DRA, which is an anion exchanger involved in anion secretion, as well as NaCl absorption, appears to be the major anion exchanger involved in the pathophysiology of IBD-associated and enteropathogenic Escherichia coli-mediated diarrhea disorders (11,24–26). These findings implicated that DRA expression levels may serve as a potential target during inflammatory diarrhea.
While a decrease in DRA expression level is associated with intestinal inflammation, the direct effect of mediators that trigger intestinal inflammatory mediators on DRA has not been fully investigated. A previous study provided evidence for the involvement of hepatocyte nuclear factor 4 and GATA transcription factors in DRA expression (27). Cytokines, such as interleukin-1β (24) and interferon (IFN)-γ (28) also reduce DRA expression levels in Caco-2 cells. In this regard, TNF-α is also a central mediator of gut inflammation in IBD (29–31). Whether TNF-α directly modulates DRA expression remains unknown. It is hypothesized that TNF-α inhibits DRA expression levels in intestinal epithelial cells, and that the interaction between TNF-α and DRA promotes the progression of gut inflammation.
In the present study, the expression level of the DRA protein was markedly reduced whereas the expression level of TNF-α in blood serum was significantly increased in UC patients compared to healthy control subjects. Subsequent studies are required to establish whether this is the case in UC animal models. The DSS-induced colitis is a commonly utilized mouse model, mimicking the clinical pathology of UC (32), and this is characterized by a significant colonic inflammation, reduction of colon length, weight loss and bloody diarrhea, which have been demonstrated in the present study. Furthermore, it was observed that DRA mRNA and protein expression levels were identified to be markedly decreased in DSS-induced colitis compared with the normal control subjects. DRA is predominantly expressed on the apical membrane of intestinal epithelial cells in IHC and immunofluorescence staining, and thus exerts the function of NaCl absorption combined with Na+/H+ exchanger3. Additionally, the present findings suggested that the expression level of TNF-α was apparently elevated in the blood serum of colitis mice. Thus, it is hypothesized that TNF-α may regulate DRA expression levels in colitis. Colonic epithelial cells (Caco2BBE cells) were used to perform further investigations.
Caco2BBE cells express a well-developed brush-border and the cytoskeleton of the microvilli is polarized and structured, as in human enterocytes in vivo. In addition, Caco2BBE cells are more homogenous with respect to brush-border expression than the parental cell line, and the expression level of proteins associated with the brush- border was similar to that identified in human enterocytes (33). To avoid the heterogeneity of different passages of cells, the experiments were performed in early passages of Caco2BBE cells (between 15 and 25 passages). It was demonstrated that subsequent to stimulating with TNF-α, the expression level of DRA markedly decreased. Furthermore, this effect was dose-dependent; when stimulated with 100 ng/ml TNF-α, the decrease in DRA expression levels was more pronounced than when stimulated with 50 ng/ml TNF-α. Then, whether overexpression of DRA may block this effect was investigated. Caco2BBE cells with lentiviral-mediated DRA overexpression were used and treated with different dosages of TNF-α. The expression level of DRA decreased following stimulation with 100 ng/ml TNF-α in DRA overexpression Caco2BBE cells. These results indicate that TNF-α is a negative regulator of DRA expression in Caco2BBE cells. Conversely, in order to investigate whether suppression of TNF-α has an effect on the expression level of DRA, Caco2BBE cells transfected with siTNF-α were used to perform further experiments. The expression level of TNF-α was identified to be reduced in the cell culture medium after silencing TNF-α in the Caco2BBE cells. Simultaneously, a marked increase in the expression levels of DRA mRNA and protein was detected, indicating that DRA was a target site of TNF-α. Based on the results, it was confirmed that TNF-α downregulates DRA expression. Furthermore, DRA promoter activity was inhibited by IFN-γ via the Janus kinase/signal transducers and activators of transcription signaling pathway (28). The overall decrease in DRA expression may be involved in the process of IBD.
In the development of gut inflammation, many factors, including cytokines, diet, the microbiome and immunity are involved in the pathogenetic mechanisms of IBD, and their interactions lead to abnormalities in individuals (34–36). As TNF-α may regulate ion transporter DRA, it is proposed that DRA may also affect TNF-α. Notably, the present results indicated the knockdown of DRA increased the mRNA expression level of TNF-α and the protein expression level of TNF-α in the cell culture medium. One hypothesis is that DRA may function as an important mediator that drives the negative cycle of TNF-α-induced gut inflammation.
However, a limitation of the present study is that the result did not indicate the direct regulation of TNF-α on DRA. Further investigation is required to identify the underlying molecular mechanisms. Overexpression of DRA was demonstrated to be beneficial in the inflammatory environment; however, the signaling pathways have not been well defined.
In conclusion, the current in vitro and in vivo data demonstrate that DRA is involved in intestinal inflammation and TNF-α may reduce DRA expression levels; however, DRA also affects TNF-α expression and secretion. In the clinical setting, the TNF-α monoclonal antibody has been widely used to treat IBD patients with favorable results (37,38). The present study increases knowledge of the underlying mechanisms of TNF-α leading to the progression of intestinal inflammation and reveals a novel perspective of the interaction between TNF-α and ion transporters.
Acknowledgments
The authors would like to thank Professor Ursula Seidler (Hannover Medical School, Hannover, Germany) for providing the human colon epithelial cell Caco2BBE cell line. The present study was supported by a grant from the National Natural Science Foundation of China (grant no. 81400593).
References
- 1.Chi KR. Epidemiology: Rising in the East. Nature. 2016;540:S100–S102. doi: 10.1038/540S100a. [DOI] [PubMed] [Google Scholar]
- 2.Nq SC, Tang W, Ching JY, Wong M, Chow CM, Hui AJ, Wong TC, Leung VK, Tsang SW, Yu HH, et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-pacific Crohn’s and colitis epidemiology study. Gastroenterology. 2013;145:158–165.e2. doi: 10.1053/j.gastro.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Wang YF, Ouyang Q, Hu RW. Progression of inflammatory bowel disease in China. J Dig Dis. 2010;11:76–82. doi: 10.1111/j.1751-2980.2010.00421.x. [DOI] [PubMed] [Google Scholar]
- 4.Dixon LJ, Kabi A, Nickerson KP, McDonald C. Combinatorial effects of diet and genetics on inflammatory bowel disease pathogenesis. Inflamm Bowel Dis. 2015;21:912–922. doi: 10.1097/MIB.0000000000000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park JH, Peyrin-Biroulet L, Eisenhut M, Shin JI. IBD immunopahogenesis: A comprehensive review of inflammatory molecules. Autoimmun Rev. 2017;16:416–426. doi: 10.1016/j.autrev.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 6.Ng SC, Bernstein CN, Vatn MH, Lakatos PL, Loftus EV, Jr, Tysk C, O’Morain C, Moum B, Colombel JF, Epidemiology and Natural History Task Force of the International Organization of Inflammatory Bowel Disease (IOIBD) Geographical variability and environmental risk factors in inflammatory bowel disease. Gut. 2013;62:630–649. doi: 10.1136/gutjnl-2012-303661. [DOI] [PubMed] [Google Scholar]
- 7.Xia W, Yu Q, Riederer B, Singh AK, Engelhardt R, Yeruva S, Song P, Tian DA, Soleiman M, Seidler U. The distinct roles of anion transporters Slc26a3 (DRA) and Slc26a6 (PAT-1) in fluid and electrolyte absorption in the murine small intestine. Pflugers Arch. 2014;466:1541–1556. doi: 10.1007/s00424-013-1381-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wedenoja S, Pekansaari E, Höglund P, Mäkelä S, Holmberg C, Kere J. Update on SLC26A3 mutations in congenital chloride diarrhoea. Hum Mutat. 2011;32:715–722. doi: 10.1002/humu.21498. [DOI] [PubMed] [Google Scholar]
- 9.Wedenoja S, Höglund P, Holmberg C. Review article: The clinical management of congenital chloride diarrhoea. Aliment Pharmacol Ther. 2010;31:477–485. doi: 10.1111/j.1365-2036.2009.04197.x. [DOI] [PubMed] [Google Scholar]
- 10.Xiao F, Yu Q, Li J, Johansson ME, Singh AK, Xia W, Riederer B, Engelhardt R, Montrose M, Soleimani M, et al. Slc26a3 deficiency is associated with loss of colonic HCO3 (−) secretion, absence of a firm mucus layer and barrier impairment in mice. Acta Physiol (Oxf) 2014;211:161–175. doi: 10.1111/apha.12220. [DOI] [PubMed] [Google Scholar]
- 11.Farkas K, Yeruva S, Rakonczay Z, Jr, Ludolph L, Molnar T, Nagy F, Szepes Z, Schnur A, Wittmann T, Hubricht J, et al. New therapeutic targets in ulcerative colitis: The importance of ion transporters in the human colon. Inflamm Bowel Dis. 2011;17:884–898. doi: 10.1002/ibd.21432. [DOI] [PubMed] [Google Scholar]
- 12.Peake ST, Bernardo D, Mann ER, Al-Hassi HO, Knight SC, Hart AL. Mechanisms of action of anti-tumor necrosis factor-α agents in Crohn’s disease. Inflamm Bowel Dis. 2013;19:1546–1555. doi: 10.1097/MIB.0b013e318281333b. [DOI] [PubMed] [Google Scholar]
- 13.Günther C, Neumann H, Neurath MF, Becker C. Apoptosis, necrosis and necroptosis: Cell death regulation in the intestinal epithelium. Gut. 2013;62:1062–1071. doi: 10.1136/gutjnl-2011-301364. [DOI] [PubMed] [Google Scholar]
- 14.Fries W, Muja C, Crisafulli C, Cuzzocrea S, Mazzon E. Dynamics of enterocyte tight junctions: Effect of experimental colitis and two different anti-TNF strategies. Am J Physiol Gastrointest Liver Physiol. 2008;294:G938–G947. doi: 10.1152/ajpgi.00469.2007. [DOI] [PubMed] [Google Scholar]
- 15.Xiao F, Juric M, Li J, Riederer B, Yeruva S, Singh AK, Zheng L, Glage S, Kollias G, Dudeja P, et al. Loss of downregulated in adenoma (DRA) impairs mucosal HCO3(−) secretion in murine ileocolonic inflammation. Inflamm Bowel Dis. 2012;18:101–111. doi: 10.1002/ibd.21744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Juric M, Xiao F, Amashen S, May O, Wahl K, Bantel H, Manns MP, Seidler U, Bachmann O. Increased epithelial permeability is the primary cause for bicarbonate loss in inflamed murine colon. Inflamm Bowel Dis. 2013;19:904–911. doi: 10.1097/MIB.0b013e3182813322. [DOI] [PubMed] [Google Scholar]
- 17.Sutherland LR, Martin F, Greer S, Robinson M, Greenberger N, Saibil F, Martin T, Sparr J, Prokipchuk E, Borgen L. 5-Aminosalicylic acid enema in the treatment of distal ulcerative colitis, proctosigmoiditis, and proctitis. Gastroenterology. 1987;92:1894–1898. doi: 10.1016/0016-5085(87)90621-4. [DOI] [PubMed] [Google Scholar]
- 18.Yeruva S, Chodisetti G, Luo M, Chen M, Cinar A, Ludolph L, Lünnemann M, Goldstein J, Singh AK, Riederer B, et al. Evidengce for a causal link between adaptor protein PDZK1 downregulation and Na+/H+ exchanger NHE3 dysfunction in human and murine colitis. Pflugers Arch. 2015;467:1795–1807. doi: 10.1007/s00424-014-1608-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Q, Liu ZY, Chen Q, Lin JS. Mcl-1 as a potential therapeutic target for human hepatocellular carcinoma. J Huazhong Univ Sci Technolog Med Sci. 2016;36:494–500. doi: 10.1007/s11596-016-1614-7. [DOI] [PubMed] [Google Scholar]
- 20.Liva KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Kolios G. Animal models of inflammatory bowel disease: How useful are they really? Curr Opin Gastroenterol. 2016;32:251–257. doi: 10.1097/MOG.0000000000000287. [DOI] [PubMed] [Google Scholar]
- 22.Priyamvada S, Gomes R, Gill RK, Saksena S, Alfefai WA, Dudeja PK. Mechanisms underlying dysregulation of electrolyte absorption in inflammatory bowel disease associated diarrhea. Inflamm Bowel Dis. 2015;21:2926–2935. doi: 10.1097/MIB.0000000000000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barkas F, Liberopoulos E, Kei A, Elisaf M. Electrolyte and acid-base disorders in inflammatory bowel disease. Ann Gastroenterol. 2013;26:23–28. [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H, Jiang W, Furth EE, Wen X, Katz JP, Sellon RK, Silberg DG, Antalis TM, Schweinfest CW, Wu GD. Intestinal inflammation reduces expression of DRA, a transporter responsible for congential chloride diarrhea. Am J Physiol. 1998;275:G1445–G1453. doi: 10.1152/ajpgi.1998.275.6.G1445. [DOI] [PubMed] [Google Scholar]
- 25.Gill RK, Borthakur A, Hodges K, Turner JR, Clayburgh DR, Saksena S, Zaheer A, Ramaswamy K, Hecht G, Dudeja PK. Mechanisms underlying inhibition of intestinal apical Cl/OH exchange following infection with enteropathogenic E. coli. J Clin Invest. 2007;117:428–437. doi: 10.1172/JCI29625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar A, Anbazhagan AN, Coffing H, Chatterjee I, Priyamvada S, Gujral T, Saksena S, Gill RK, Alrefai WA, Borthakur A, Dudeja PK. Lactobacillus acidphilus counteracts inhibition of NHE3 and DRA expression and alleviates diarrheal phenotype in mice infected with Citrobacter rodentium. Am J Physiol Gastrointest Liver Physiol. 2016;311:G817–G826. doi: 10.1152/ajpgi.00173.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alrefai WA, Wen X, Jiang W, Katz JP, Steinbrecher KA, Cohen MB, Williams IR, Dudeja PK, Wu GD. Molecular cloning and promoter analysis of downregulated in adenoma (DRA) Am J Physiol Gastrointest Liver Physiol. 2007;293:G923–G934. doi: 10.1152/ajpgi.00029.2007. [DOI] [PubMed] [Google Scholar]
- 28.Saksena S, Singla A, Goyal S, Katyal S, Bansal N, Gill RK, Alrefai WA, Ramaswamy K, Dudeja PK. Mechanisms of transcriptional modulation of the human anion exchanger SLC26A3 gene expression by IFN-{gamma} Am J Physiol Gastrointest Liver Physiol. 2010;298:G159–G166. doi: 10.1152/ajpgi.00374.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olesen CM, Coskun M, Peyrin-Biroulet L, Nielsen OH. Mechanisms behind efficacy of tumor necrosis factor inhibitors in inflammatory bowel disease. Pharmacol Ther. 2016;159:110–119. doi: 10.1016/j.pharmthera.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 30.Soufli I, Toumi R, Rafa H, Touil-Boukoffa C. Overview of cytokines and nitric oxide involvement in immune-pathogenesis of inflammatory bowel diseases. World J Gastrointest Pharmacol Ther. 2016;7:353–360. doi: 10.4292/wjgpt.v7.i3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strober W, Fuss IJ. Proinflammatory cytokine in the pathogenesis of inflammatory bowel disease. Gastroenterology. 2011;140:1756–1767. doi: 10.1053/j.gastro.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.te Velde AA, de Kort F, Sterrenburg E, Pronk I, ten Kate FJ, Hommes DW, van Deventer SJ. Comparative analysis of colonic gene expression of three experimental colitis models mimicking inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:325–330. doi: 10.1002/ibd.20079. [DOI] [PubMed] [Google Scholar]
- 33.Sambuy Y, De Angelis I, Ranaldi G, Scarino ML, Stammati A, Zucco F. The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol Toxicol. 2005;21:1–26. doi: 10.1007/s10565-005-0085-6. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Chao K, Ng SC, Bai AH, Yu Q, Yu J, Li M, Cui Y, Chen M, Hu JF, Zhang S. Pro-inflammatory miR-223 mediates the cross-talk between the IL23 pathway and the intestinal barrier in inflammatory bowel disease. Genome Biol. 2016;17:58. doi: 10.1186/s13059-016-0901-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaubeck M, Haller D. Reciprocal interaction of diet and microbiome in inflammatory bowel disease. Curr Opin Gastroenterol. 2015;31:464–470. doi: 10.1097/MOG.0000000000000216. [DOI] [PubMed] [Google Scholar]
- 36.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 37.Sandborn WJ, van Assche G, Reinisch W, Colombel JF, D’Haens G, Wolf DC, Kron M, Tighe MB, Lazar A, Thakkar RB. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012;142:257–265.e1-3. doi: 10.1053/j.gastro.2011.10.032. [DOI] [PubMed] [Google Scholar]
- 38.Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, Rachmilewitz D, Wolf DC, Olson A, Bao W, et al. Maintenance infliximab for Crohn’s disease: The ACCENT 1 randomized trial. Lancet. 2002;359:1541–1549. doi: 10.1016/S0140-6736(02)08512-4. [DOI] [PubMed] [Google Scholar]