
Fig. 10.
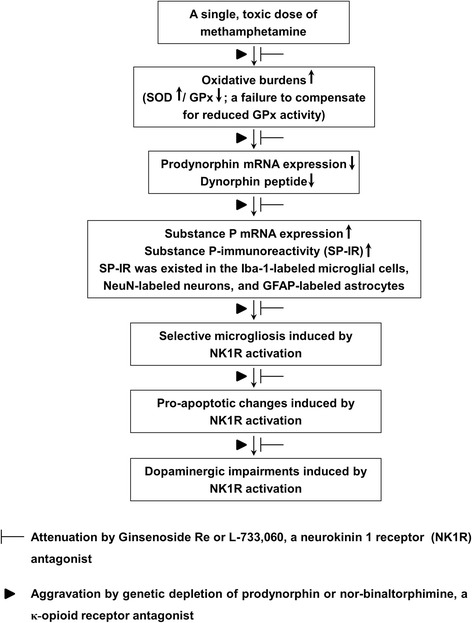
A schematic depiction ginsenoside Re (GRe)-mediated dopaminergic neuroprotective effects against MA insult. Treatment with a toxic dose of MA resulted in initial oxidative burdens, a perturbed redox status (a failure to compensate for reduced GPx) followed by the decreases in dynorphin level and prodynorphin mRNA expression. This decrease led to imbalance between dynorphin and substance P and to enhance substance P mRNA and substance P-immunoreactivity (SP-IR). At that time, SP-IR was co-localized in the Iba-1-immunoreactive microglia, NeuN-immunoreactive neurons, and GFAP-immunoreactive astrocytes. More importantly, GRe did not affect MA-induced astrocytic expression (i.e., GFAP expression), but GRe significantly attenuated against MA-induced microgliosis (i.e., Iba-1-immunoreactive-immunoreactivity and Iba-1 expression). Neurokinin 1 receptor activation, genetic depletion of dynorphin, or κ-opioid receptor antagonism (i.e., nor-binaltorphimine) potentiated this microgliosis, as well as pro-apoptotic changes. This morbid phenomenon contributed to dopaminergic impairment. Therefore, we propose that GRe attenuates MA-induced neurotoxicity via upregulation of prodynorphin-mediated κ-opioid receptor and downregulation of substance P-mediated NK1 receptor